
Ataxia-Telangiectasia Locus: Sequence Analysis of 184 kb of Human Genomic DNA Containing the Entire ATM Gene
Abstract
Ataxia-telangiectasia (A-T) is an autosomal recessive disorder involving cerebellar degeneration, immunodeficiency, chromosomal instability, radiosensitivity, and cancer predisposition. The genomic organization of the A-T gene, designatedATM, was established recently. To date, more than 100 A-T-associated mutations have been reported in the ATM gene that do not support the existence of one or several mutational hotspots. To allow genotype/phenotype correlations it will be important to find additional ATM mutations. The nature and location of the mutations will also provide insights into the molecular processes that underly the disease. To facilitate the search for ATMmutations and to establish the basis for the identification of transcriptional regulatory elements, we have sequenced and report here 184,490 bp of genomic sequence from the human 11q22–23 chromosomal region containing the entire ATM gene, spanning 146 kb, and 10 kb of the 5′-region of an adjacent gene named E14/NPAT.The latter shares a bidirectional promoter with ATM and is transcribed in the opposite direction. The entire region is transcribed to ∼85% and translated to 5%. Genome-wide repeats were found to constitute 37.2%, with LINE (17.1%) and Alu (14.6%) being the main repetitive elements. The high representation of LINE repeats is attributable to the presence of three full-length LINE-1s, inserted in the same orientation in introns 18 and 63 as well as downstream of the ATM gene. Homology searches suggest that ATM exon 2 could have derived from a mammalian interspersed repeat (MIR). Promoter recognition algorithms identified divergent promoter elements within the CpG island, which lies between the ATM andE14/NPAT genes, and provide evidence for a putative secondATM promoter located within intron 3, immediately upstream of the first coding exon. The low G + C level (38.1%) of theATM locus is reflected in a strongly biased codon and amino acid usage of the gene.
[The sequence data described in this paper have been submitted to the GenBank data library under accession no. U82828.]
Ataxia telangiectasia (A-T) is an autosomal recessive disorder with a remarkable range of clinical manifestations affecting different tissues. It has a frequency of 1:40,000–100,000 live births worldwide. A-T patients suffer from progressive neurological degeneration, immune deficiency, lymphoreticular malignancies, chromosomal instability, growth retardation, gonadal dysgenesis, telangiectases (dilated blood vessels) appearing in the eyes and face, and premature aging of skin and hair (for review, see Shiloh 1995; Lavin and Shiloh 1997). Epidemiological studies of A-T heterozygotes have suggested an elevated risk for cancer, particularly breast cancer (Swift et al. 1991). Cultured cells from these individuals show an increased sensitivity to ionizing radiation (Weeks et al. 1991).
The gene mutated in A-T patients (ATM) was mapped to chromosome 11q22–23 (Gatti et al. 1988) and has been identified recently by positional cloning (Savitsky et al. 1995a). It contains an open reading frame (ORF) of 9168 nucleotides. The predicted protein of 3056 amino acids belongs to a family of large proteins that share sequence homologies to the catalytic domain of phosphatidylinositol-3 (PI-3) kinases (Savitsky et al. 1995b). Among these proteins are TEL1p and MEC1p in budding yeast, Rad3 in fission yeast, the TOR proteins in yeast and their mammalian counterpart, FRAP (RAFT1), mei-41 inDrosophila melanogaster, and the catalytic subunit of DNA-dependent protein kinase in mammals. These proteins are involved in signal transduction, meiotic recombination, and control of cell cycle (for review, see Savitsky et al. 1995b; Zakian et al. 1995).
More than 100 mutations have been identified so far among A-T patients and these are spread over the entire coding region of the ATMgene. The vast majority of the mutations are expected to inactivate the ATM protein by truncation or large deletions. Most of the patients were found to be compound heterozygotes (Baumer et al. 1996; Byrd et al. 1996a; Gilad et al. 1996; Telatar et al. 1996; Wright et al. 1996).
To enable screening of A-T mutations based on genomic DNA as the resource, we have determined previously the entire genomic organization of the ATM gene. It is composed of 66 exons spread over a genomic region of ∼150 kb (Uziel et al. 1996). Other groups have confirmed our findings (Rasio et al. 1996; Vorechovsky 1996). Analysis of the promoter region and mapping of cDNAs to the ATM locus revealed a second gene, designated E14 (Byrd et al. 1996a,b) or NPAT (nuclear protein mapped to the AT -locus; Imai et al. 1996), 0.5 kb upstream of ATM. The E14/NPAT gene is transcribed in the opposite direction and codes for a 1421-amino-acid protein.ATM and E14/NPAT are both ubiquitously expressed and probably regulated by a bidirectional promoter (Byrd et al. 1996b).
Because of the importance of the ATM gene in biomedical research, we carried out a large-scale sequencing effort of the entireATM genomic locus. By sequencing five cosmids derived from a cosmid contig spanning most of the D11S384–D11S1818 interval (Savitsky et al. 1995b), we have determined a contiguous genomic stretch of 184,490 bp containing the entire ATM gene as well as the 5′ region of the E14/NPAT gene. In addition, we have obtained a comprehensive map of repeated elements and predicted several putative promoters. One potential secondary promoter region is located within intron 3 of the ATM gene, immediately upstream of the first coding exon. Together with the recently determined 5′ and 3′ untranslated regions (UTRs), which display large variability, these data suggest a complex posttranscriptional regulation of theATM gene (Savitsky et al. 1997). The complete genomic sequence of the ATM gene is a valuable resource for detection of all A-T mutations and for carrier diagnostics. The sequence also provides new insights into the organization and the evolution of theATM locus.
RESULTS
Genomic Sequencing of 184 kb Spanning the ATM Locus
A cosmid contig between chromosomal markers D11S384 and D11S535 spanning the coding region of the ATM gene (Savitsky et al. 1995b) served as the starting point for sequencing. Five cosmids, B10, A12, C7, C12, and E3 (Fig. 1), were completely sequenced using the M13 random shotgun method. A region of ∼10 kb between cosmids A12 and C7 was bridged by PCR products generated from exons 24–27.
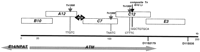
Cosmid contig across the sequenced region in 11q22–23. Horizontal arrows indicate orientation and coverage of ATM andE14/NPAT genes. Vertical arrows point to insertion sites ofE. coli transposons, and the sequence of the duplications at the insertion sites are shown.
Sequencing revealed that four of the five cosmids (A12, C7, C12, E3) carry the Escherichia coli transposon Tn1000 (5981 bp) (Broom et al. 1995) as a cloning artifact. In cosmids A12, C7, and C12 the transposon was located within the human insert, whereas in cosmid E3 it was inserted in the cosmid vector. In addition, a previously unknown composite transposon of 4447 bp was identified in the human insert of cosmid C12. It comprises two insertion elements IS10, together with an E. coli sequence derived from the 81.5–84.5 min region (V.M. Platzer, unpubl.). Examination of the insertion sites of the Tn1000 and the composite transposon revealed 5- and 9-bp duplications of the host DNA, respectively. To confirm that these duplications were artifacts caused by the transposon insertions and to exclude the possibility of additional alterations in the cosmid DNA, the integration sites were amplified from human genomic DNA (primers given in Table 1) and sequenced. No differences were found between the cosmid and genomic sequences except for the duplication of the insertion site.
Exon–Intron Organization of the ATMgene
The final genomic contig of 184,490 bp was derived from a total of 4659 sequencing reads with an average redundancy of 8.03 for the whole project. The sequence is completely double-stranded. We found that the use of dye-terminator chemistry in the shotgun phase significantly speeded up contig assembly and editing because it virtually eliminated compressions, frequently observed with dye-primers. Each position of the contig is represented by at least one dye-terminator read. The entire sequence was deposited in the GenBank database under accession no. U82828.
Recently, partial sequences of the ATM gene have been published (accession nos. U40887–U40918: Rasio et al. 1996;U55702–U55757: Vorechovsky et al. 1996) comprising 46,204 bp. GAP alignment of this data with the genomic contig of 184,490 bp revealed 188 discrepancies. After revision of the primary data, we were able to exclude errors at these positions in our sequence. Because many of the divergent positions in database entries U55702–U55757 are represented by N’s or are found at the ends of the entries deep within introns, these data probably represent regions of poor quality rather than polymorphic sites. We also compared the 184 kb genomic sequence with 4841 bp of the 5′-half of the ATM transcript (accession no. X91169: Byrd et al. 1996a) and with 2193 bp of the intergenic region between ATM and E14/NPAT (accession no.D83244: Imai et al. 1996). Although 14 discrepancies were detected, no sequencing errors within the primary data at the respective positions of our sequence could be identified. We therefore conclude, that at least 53,238 of the presented 184,490 bp were definitely obtained without any errors. This indicates an exceptional high sequencing accuracy for the database entry U82828.
Exon–Intron Structure of the ATM Gene with Single Base Pair Resolution
GAP alignment of the 184-kb sequence contig with the ATM mRNA containing the complete ORF (accession no. U33841; Savitsky et al. 1995b) and recently obtained cDNA clones representing alternative 5′ and 3′ ends of the ATM mRNA (accession nos. U67092and U67093; Savitsky et al. 1997) revealed the exon–intron structure of the ATM gene at single base resolution. The ATMgene has 66 exons (Tab. 1; Fig. 2A). Donor and acceptor splice sites of the ATM gene follow the GT–AG consensus (Shapiro and Senapathy 1987). The only exception is the GC 5′ splice site of intron 52. This GC variant is by far the most common nonconsensus mRNA splice site (Jackson 1991). It is the only alternative splice site known to allow accurate cleavage in vitro, allthough more slowly than the usual GT sequence (Aebi 1987). The genomic structure is consistent with our previous results from cDNA sequencing and long-distance PCR using human DNA as the template (Uziel et al. 1996). It also proves the colinearity of the cosmid and genomic sequence.

Schematic representation of the sequenced ATM locus. Boxes above the line represent features oriented toward the telomere; below the line, toward the centromere. (A) Exons identified by cDNAs. (B) CpG islands and predicted promoters (CpG islands are 0.5-high peaks; promoter regions are indicated by F and R in respect to their orientation). (C) SINEs. (D) LINEs, DNA transposons, and unclassified repeats.
The introns vary considerably in size from 77–10,207 bp. Homology search algorithms confirmed that the first exon of E14/NPAT(accession nos. D83243, U58852, and X97186) is located at a distance of 468 bp 5′ of the first ATM exon, extending in the opposite orientation (Byrd et al. 1996a; Imai et al. 1996). We found no further match of the E14/NPAT mRNA sequence with the proximal 10,200 bp. This is consistent with the recently reported exon/intron structure of the gene (Byrd et al. 1996b), where intron 1 was reported to be >12 kb.
Using BLAST searches, we identified 20 expressed sequence tags (ESTs) that map to the ATM gene. The ESTs are highly redundant. Eighteen ESTs were from the 3590-bp long 3′ UTR and of these, nine represent the extreme 3′ end of the ATM mRNA. Only two ESTs (accession no. H43382 and H45943) were aligned to the coding region and mapped to the region of exons 62–65. Identical start points of both of these ESTs suggest that they were derived independently from the same cDNA clone.
We have used several gene prediction programs to predict exons in the 184-kb ATM locus. Their performance, however, was quite poor. Initially, XPOUND and XGRAIL 1.2 did not predict any of theATM exons, whereas XGRAIL version 1.3 predicted 41 out of the 66 ATM exons almost correctly but falsely predicted another 28. This stands in contrast to our own experience (accession nos.U52111 and U52112) and that of others (Lopez et al. 1994; Chen et al. 1996) who have noticed excellent performance of these programs in G + C-rich regions.
Repeat Analysis Reveals That ATM Exon 2 Is Related to a Genome-Wide Repetitive Element
Repeat analysis was performed to identify several types of simple-sequence repeats, microsatellites, and genome-wide repeats. Runs of five or more consecutive dinucleotides and trinucleotides are shown in Table 2. The dinucleotide repeat CA/TG was found at 12 sites in the region including marker D11S2179, 180 bp downstream to exon 62. Computer prediction revealed three copies of a 106-bp tandem repeat in intron 61, each repeat copy consisting of two head-to-tail arranged 53-bp units (position: 135,843; 136,024 and 136,209).
Runs of More than Five Consecutive Dinucleotide and Trinucleotide Repeats
Genome-wide repeats were identified by CENSOR and divided into the four major classes (1) short interspersed nucleotide elements (SINE) [Alu and mammalian-wide interspersed repeats (MIRs)] (Smit and Riggs 1995; Batzer et al. 1996); (2) long interspersed nucleotide elements (LINEs) (Smit et al. 1995); (3) long terminal repeats [LTRs; and mammalian apparent LTR retrotransposons (MaLRs)] (Smit 1993); and (4) DNA transposons (Smit and Riggs 1996) (Table 3). Taken together, all genome-wide repeats together constitute 37.2% of the ATM locus, with Alu (14.6%) and LINE-1 (17.1%) being the major contributors (Fig. 2C,D).
Distribution of Genome-Wide Repeats into the Main Four Classes of Human Transposable Elements
Three full-length LINE-1s (6017, 6031, and 6116 bp) reside in introns 18 and 63, as well as 4.5 kb downstream of the polyadenylation site. They are oriented in the same direction as the ATM gene. Of the three LINE-1 repeats, only the two located in the introns are flanked by target-site duplications of 9 and 13 bp, respectively. The LINE-1s show more than 93% homology to LRE-1 (LINE-1retransposable element; Dombroski et al. 1991); however, the presence of at least one premature termination codon in both LINE-1 ORFs suggests that none of them represents a transpositionally active element. A fourth LINE-1 repeat of 4212 bp was found in intron 24 in the opposite orientation. This repeat element is truncated at its 3′-end and shows only 74% homology to LRE-1.
All repeats, censored out using the initial conservative parameter set, are located deeply in the introns with the exception, that a MIR overlaps the intron 2 donor site (Fig. 3). The initial match obtained with CENSOR started at the last 3 bases of exon 2 and extended over a distance of 152 bp into intron 2 [score value 270 with P(270)=3.4 × 10−15]. This region is related to the 5′ bases 12–165 of the 262-bp MIR consensus (Smit and Riggs 1995) and contains the entire box B and the 3′ part of box A of the polymerase III promoter. Use of parameter settings at a higher sensitivity detected a similarity between the 3′end of the MIR consensus and a region spanning 29 bp of the intron 1b acceptor site and the first 11 bp of exon 2 [score value 63 with P(63)=0.0059].

Sequence comparison of ATM exon 2 and the consensus of the genome-wide MIR repeat. Dark shaded regions indicate exons, lightly shaded arrows characteristic elements of the repeats, (Vbr) Identities; (:) base transitions. Homology region withP(270) = 3.4 × 10−15 is located in the thick-lined box, the region with P(65) = 0.0059 in the thin-lined box.
Local Content Analysis Reveals Large DNA Segments With Distinct G + C Content and a Major CpG Island in Between ATM andE14/NPAT Genes
Using a ±2-kb-window in steps of 1 kb, the G + C distribution along the 184 kb was estimated to be 38.1 ± 2.9% (mean ± standard deviation).
As shown in Figure 4, we found two large regions fluctuating around distinct G + C average values (1) The central region from 16–136 kb: 37.0 ± 2.3% and (2) the 3′region from 136–184 kb: 40.2 ± 2.4%. If all genome-wide repeats are removed from the 184-kb ATM locus, the G + C content of the remaining 115,767 bp is 34.0%. Moreover, we found that the intron length of the central region correlates moderately with the G + C content. Shorter introns exhibit a lower G + C content regardless of their repeat content (Fig. 5).
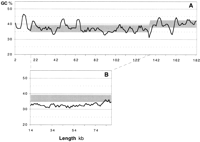
(A) G + C content of the sequenced ATM locus across the entire region 1–184 kb. Several distinct peaks above the G + C average represent genome-wide repeats (Alu: 61.38%; LINE: 41.73% G + C). (B) The region 14–136 kb relieved from genome-wide repeats. (A,B) Obtained with a moving window of ±2 kb, step 1 kb.
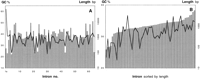
Correlation of intron length and G + C content. (A) Introns sorted by number. (B) Introns 3–61 relieved from genome-wide repeats sorted by length. (Shaded bars) Intron length; (solid line) G + C content.
Screening for CpG islands (Gardiner and Frommer 1987) in the 184-kb sequence contig identified a major CpG island from positions 10,186 to 10,943 (61.6% G + C; 0.88 CpG observed/expected). The first exons of ATM and E14/NPAT are contained within this CpG island. A BLAST search of this region identified a sequence (accession no. Z66089; six mismatches) that was obtained during construction of a human CpG island library (Cross et al. 1994). Two smaller CpG regions, predicted from positions 48,327 to 48,693; (59.4% G + C; 1.04 CpG observed/expected) and from 161,545 to 161,761 (62.2%; 1.00 CpG observed/expected), fall into the 5′ region of LINE-1s.
The ATM Gene Shows a Biased Codon and Amino Acid Usage
The ATM gene resides at an A + T-rich portion of the human genome that can be classified as an L1 isochore (Bernardi et al. 1985). The G + C content of the first, second, and third codon positions is 47.5%, 34.2%, and 34.8%, respectively. The ATMcodon and amino acid usage were compared with the human average values (9465 genes, GenBank release 96). Figure 6 shows a strong underrepresentation of codons with G or C in third position and of amino acids of the G/C class (i.e., amino acids with G and/or C in the first two codon positions: Arg, Ala, Gly, and Pro).
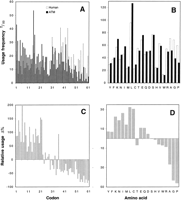
Absolute and relative usage of codons (A,C) and amino acids (B,D) of the ATM gene in comparison with the human average. Codons are ordered from left to right with primary sort criterion “3rd codon position A or T” and secondary criterion “increasing G + C content.” The order of columns is (1) AAA; (2) AAT; (3) ATA; (4) ATT; (5) TAT; (6) TTA; (7) TTT; (8) AGA; (9) AGT; (10) ACA; (11) ACT; (12) TGT; (13) TCA; (14) TCT; (15) GAA; (16) GAT; (17) GTA; (18) GTT; (19) CAA; (20) CAT; (21) CTA; (22) CTT; (23) GGA; (24) GGT; (25) GCA; (26) GCT; (27) CGA; (28) CGT; (29) CCA; (30) CCT; (31) AAG; (32) AAC; (33) ATG; (34) ATC; (35) TAC; (36) TTG; (37) TTC; (38) GAG; (39) GAC; (40) GTG; (41) GTC; (42) CAG; (43) CAC; (44) CTG; (45) CTC; (46) AGG; (47) AGC; (48) ACG; (49) ACC; (50) TGG; (51) TGC; (52) TCG; (53) TCC; (54) GGG; (55) GGC; (56) GCG; (57) GCC; (58) CGG; (59) CGC; (60) CCG; (61) CCC. Amino acids are tentatively arranged from left to right order of increasing G + C content of their codons. Columns are labeled with the single letter code.
Promoter Prediction Reveals a Bidirectional Promoter in the CpG Island and Additional Promoter Elements for ATM
Two independent algorithms (TSSG/W, PSII) were used to identify potential promoter regions on both strands of the 184-kb sequence. Thirteen high-scoring promoter regions were predicted, eight in the direction of ATM transcription (F1–F8) and five in the direction of E14/NPAT (R1–R5) (Table 4; Fig.2).
Potential Promoter Elements Predicted by TSSG, TSSW, and PSII Algorithms
The promoter regions F2 and R1 are located within the CpG island covering the first exons of ATM and E14/NPAT,respectively. The intergenic region of 468 bp contains two CCAAT boxes and four SP1-binding sites (Byrd et al. 1996a,b). Several other regulatory elements were predicted, including three potential γ-interferon response elements (IREs; Yang et al. 1990).
The putative F3 promoter is of particular interest as it is located within intron 3 of the ATM gene, just upstream of the first coding exon. The predicted promoter region overlaps with twoAlu repeats. Interestingly, when the repetitive elements were removed from the analyzed region, the TSSG/W promoter prediction failed. One of the repeats, the AluSg, shows high homology to a functionally active Alu estrogen response element (ERE;Norris et al. 1995). The F3 putative promoter region contains potential binding sites for Sp1, AP1, AP2, CF1, GCF, and three TBP sites, one of which was identified as a TATA-box by TSSG/W (Fig.7). Four additional putative promoter regions (F1, F5, F7, and R5) overlap with Alu repeats.

Nucleotide sequence of the putative promoter located in intron 3. The darkly shaded region indicates exon 4, lightly shaded arrows the twoAlu repeats. Potential binding sites for transcription factors are boxed. The predicted transcription start site is marked by a bold arrow; the ATM start codon by a hatched arrow. Numbers along the right side of the figure indicate the nucleotide numbering of the database entry (accession no. U82828).
DISCUSSION
To gain further insight into the organization and function of theATM gene, and to develop diagnostic reagents, we sequenced a cosmid contig spanning 184,490 bp containing the entire gene. With 66 exons, including the two alternatively spliced leader exons 1a and 1b, the ATM gene contains one of the largest number of exons reported to date for any human locus. The ATM exons are distributed over a genomic region of 146,182 bp. Therefore, the genomic organization of ATM is comparable with that of the Huntington disease gene with its 67 exons spread over 180 kb (Ambrose et al. 1994), but differs from that of giant genes such as DMD with 79 exons spread over 2.4 Mb (Roberts et al. 1993).
The human genome is a mosaic of long, compositionally homogeneous regions characterized by different G + C levels, called isochores. The G + C level of a genomic region has an impact on major genetic processes such as replication, transcription and recombination (for review, see Bernardi 1995). The 184-kb ATM contig shows a low G + C level of 38.1% and can be classified as part of a L1 isochore (Bernardi et al. 1985). This is consistent with the location of the ATM locus in the chromosomal region 11q22-23, a late-replicating G band that mainly consists of L1 and L2 isochores (for review, see Holmquist 1992). Analysis revealed a 3% shift in the local G + C average at position 136 kb between the proximal and the distal parts of the ATM gene and may represent an L1/L2 boundary. It is not as pronounced as the predicted isochore boundary between G6PD and F8C in human Xq28 (Ikemura et al. 1990) or the L/H and H2/H3 transitions in the human MHC locus of 6q21.3 (Fukagawa et al. 1995). The G + C shift separates the main body of the ATM gene from the 3′ end where the predicted PI-3 kinase activity resides (Savitsky et al. 1995b). This may suggest that these two parts of the gene evolved and/or exist under different compositional constraints.
A + T-rich genes coding for long proteins are presently underrepresented in the databases (Duret et al. 1995). Because of this fact we have experienced considerable difficulties in exon prediction within the ATM contig. Although we failed to predict anyATM exon with XGRAIL 1.2 and XPOUND, XGRAIL 1.3 finally predicted 41 exons, but on the expense of 28 false positives. Our data highlight a current problem of gene prediction in A + T-rich isochores.
A considerable portion of the ATM locus (37.2%) represents genome-wide repeats. Removal of all these repeats from the ATMlocus resulted in a more uniform, lower level G + C profile. Therefore it can be assumed that an ancient precursor of the humanATM locus exhibited an even lower G + C content before mammalian repeat expansion. There are three full-length LINE-1s in introns 18 and 63, as well as 4.5 kb downstream of ATM. The LINE-1s are highly conserved among each other, arranged in the same orientation and may represent hotspots for homologous recombination (Bollag et al. 1989). The number of mutations disrupting the ORFs 1 and 2 of the three LINE-1 repeats suggests that the ATM locus was first invaded by the downstream LINE-1 followed by the element residing in intron 63. The LINE-1 repeat in intron 18 is probably of most recent origin, as its ORF1 is only once truncated by a single G/T substitution.
An interesting evolutionary aspect of the ATM gene structure is the homology between exon 2, its adjacent intronic sequences, and the genome-wide MIR repeat. During evolution, a large gene likeATM most certainly underwent processes like exon shuffling, exon skipping or intron shifting. We hypothize that a MIR repeat has transposed into the ATM gene early in mammalian evolution and was later adopted as exon 2. Part of the MIR repeat functions as exon 2 splice donor site without major changes. However, the exon sequence itself and the acceptor site of intron 1b diverge from the MIR consensus, probably to fulfil requirements of mRNA secondary structure and stability, as well as of the splicing process.
Scanning of the 184-kb contig for potential promoter sites by different computer algorithms revealed several high-scoring regions. Two divergent promoters were predicted within a CpG island (for reviews, see Gardiner and Frommer 1987; Cross and Bird 1995). The island spans the first exons of ATM and the adjacent E14/NPAT and the intergenic region of 468 bp (Byrd et al. 1996a; Imai et al. 1996). This compact arrangement of the ATM and E14/NPATgenes is surprising, because both genes are located in an A + T-rich isochore, for which a very low gene density is expected (Mouchiroud et al. 1991). Their proximity raises the possibility of coordinate gene expression. In humans, ∼60% of genes are associated with CpG islands, including all housekeeping genes analyzed so far (Antequera and Bird 1993). The ubiquitous expression of both theATM and E14/NPAT genes in all tissues examined to date (Savitsky et al. 1995a; Byrd et al. 1996b; Imai 1996) is consistent with the definition of housekeeping genes. Reporter gene constructs showed that the CpG island functions as a bidirectional promoter and that expression directed toward ATM was threefold higher than toward E14/NPAT (Byrd et al. 1996b). In agreement with these studies, 20 hits were found in the public EST databases forATM, whereas only three were found for E14/NPAT. The majority of the ATM-specific ESTs map to the 3′ UTR and only one clone matches the coding region. In the case of E14/NPAT, all three ESTs map to the coding region. This highlights the fact that public ESTs databases are strongly biased toward the 3′ end of mRNA. For this reason, coding regions of genes with very long 3′ UTRs, like that of the ATM gene are significantly underrepresented.
The transcripts of the ATM gene belong to the 5%–10% of vertebrate mRNAs that have long, highly structured 5′ UTRs (Savitsky et al. 1997). These genes often use alternative promoters to generate supplementary transcripts with short leader sequences (Kozak 1992; Ayoubi and Van De Ven 1996). Interestingly, an additional putative ATM promoter, containing a TATA-box, was found within intron 3, immediately upstream of the first coding exon. No transcripts have yet been found that are driven by this promoter, although some of the short bands observed by primer extension might be transcribed from from this promoter (K. Savitsky, unpubl.). Remarkably, the promoter predicted in intron 3 depends on several elements residing within twoAlu repeats. Previously, most Alu sequences have been considered functionally inert. However, recent studies provide strong evidence that significant subsets of Alu repeats can confer hormone responsiveness to a promoter. Two members of differentAlu classes (Sp and Sc) can function as estrogen receptor-dependent transcriptional enhancers (Norris et al. 1995). A unique point mutation in an ERE-like sequence motif (G to A at position 93 of the Alu consensus; Batzer et al. 1996) activates the enhancer. We have found a similar G to A base change in an Alu repeat of ATM intron 3 just downstream of the predicted promoter. This element is therefore identical to the proposed consensus of theAlu ERE except that the half-site (5′-TGACC-3′) is located 7 bp instead of 9 bp downstream from the imperfect ERE (5′-GGTCAnnnTGGTC-3′). The existence of several putative promoter regions containing multiple regulatory motifs, and the extensive structural diversity of the 5′ and 3′ UTRs suggest complex posttranscriptional regulation of the ATM gene. In this respect, the putative promoter within intron 3 could supply the short 5′UTR that will allow the basal levels of ATMtranslation, whereas the different 5′UTRs coming from the upstream promoter, would supply regulative UTRs (Savitsky et al. 1997).
In summary, the presented 184,490 bp of genomic sequence containing the human ATM gene provides a substantial resource for further investigation of ATM regulation, for the detection of mutations and polymorphisms in this gene, and for the development of diagnostic tools. The analysis of the region demonstrates the capability of ongoing large-scale sequencing efforts in addressing questions of organization and evolution in human genes and chromosome regions. Comparative sequencing in model organisms will provide further insights into these processes.
METHODS
Cosmids
A chromosome 11-specific cosmid library, cloned in the vector sCos1, was a gift from Dr. Larry Deaven (Los Alamos National Laboratory, NM). High-density arrayed grids from this library were screened using yeast artificial chromosome (YAC) clones y67 and y41 (Rotman et al. 1994). YAC probes were prepared by fragmenting 20 ng of YAC DNA for 20 min at 100°C, and subsequent labeling by random oligo priming using [α-32P]dCTP. To prevent nonspecific hybridization, YAC probes were blocked by incubation with 30 μg Cot-1 DNA (GIBCO BRL), 3 mg of total human placenta DNA (Sigma), and 4 μg of vector sCos-1 DNA, at 100°C for 10 min in a final volume of 1 ml. NaPO4 (pH 7.2), was added to a final concentration of 120 mm, and the mixture was incubated further at 65°C for 3 hr, before its addition at 1 × 106 cpm/ml to the final hybridization solution (0.25 m NaPO4 at pH 7.2, 0.25 m NaCl, 5% SDS, 10% PEG-8000, 1 mmEDTA). Filters were rinsed in 0.2× SSC, 0.5% SDS at 60°C for 10 to 15 min, and exposed to X-ray film for 24 hr.
Positive clones were aligned by identifying overlaps between them. DNA blots containing cosmid DNA digested with TaqI andHindIII were hybridized with genetic markers, total cosmid inserts, YACs and cosmid ends, or moderately repetitive elements. Common hybridizing bands for any two cosmids were defined as overlaps.
Sequencing
The cosmids were prepared and sequenced as described previously (Craxton 1993) with several modifications. M13 templates were prepared by the triton method (Mardis 1994) and sequenced using Thermo Sequenase (Amersham). In the shotgun phase of a cosmid sequencing project, identical amounts of samples were sequenced either by dye-primer or dye-terminator chemistries (Perkin Elmer). Data were collected using ABI 373 and 377 automated sequencers and assembled with the XGAP program (Dear and Staden 1991). Gaps were closed using custom-made primers on M13 templates, PCR products, or cosmid DNA in combination with dye terminators. Regions of the final assembly that only consist of dye-primer reads were resequenced using dye-terminator chemistry to resolve all compressions.
Standard PCR conditions for amplification of selected regions of genomic DNA were: 1 min at 94°C, 30 cycles (30 sec at 94°C, 1 min at 55°C, 2 min at 72°C), 4 min at 72°C. Introns 24–26 were amplified using the Expand Long Template PCR System (Boehringer Mannheim) and primer pairs gctgatccttattcaaaatggg and ctctcattccttcctgagctttc, gttccaggacacgaagggag and cacaaggtgaggttctaatcc, and ccatagtgctgagaaccctg and tagaaatcctcaatatttgtgtag, respectively. PCR products appearing as a single clean and distinct band on agarose gels were purified by PEG precipitation (Rosenthal et al. 1993). Otherwise, the appropriate bands were cut out of the agarose gel and purified using the Qiaex Kit (Qiagen). Sequencing was performed using the PCR primers or internal primers using dye-terminator chemistry (Perkin Elmer). Five micrograms of intron 24-specific PCR product were used to prepare a M13 shotgun library that was sequenced as described above.
Computer Analysis
Homology searches against the EMBL database were performed using BLAST (version 1.4) (Altschul et al. 1990) and FASTA (version 2.0) (Pearson and Lipman 1988). Programs XGRAIL (Uberbacher and Mural 1991) and XPOUND (Thomas and Skolnick 1994) were used for exon prediction. Genome-wide repeats were identified using the CENSOR program (Jurka et al. 1996). Local base content was determined with the LPC algorithm (Huang 1994a). The Wisconsin Sequence Analysis Package (Genetics Computer Group, Inc.) was used to determine G + C%, G + C distribution, and codon usage. The window for calculation of the G + C distribution was set at ±2 kb for global and at ±0.2 kb for local analysis and moved in steps of 1 and 0.1 kb, respectively. Statistical analysis was performed by Excel 5.0 (Microsoft Corp.). The identification of CpG islands (V.G. Micklem, pers. comm.) was achieved using the following criteria: G + C > 50%, CpG ratio observed/expected > 0.6, length > 200 bp (Gardiner and Frommer 1987). Sequence alignments were performed using the Global Alignment Program (GAP) (Huang 1994b). To evaluate the significance of sequence similarities we used PRDF (W.R. Pearson, pers. comm.). The human codon usage table was obtained from the Codon Usage Database (Nakamura et al. 1996) compiled from GenBank release 96. Several computer programs were applied for promoter prediction: (1) “Transcription Start Site” using both Ghosh/Prestridge (TSSG) motif data and Wingender (TSSW) motif database (http://dot.imgen.bcm.tmc.edu:9331/gene-finder/help/tssw.html); (2) “Promoter Scan II” (PSII; Prestridge 1995); (3) Neural Network Promoter Prediction (NNPP;http://www-hgc.lbl.gov/projects/promoter.html); (4) Signal Scan (SS;Prestridge 1991); and (5) Transcription Factor Search 1.3 (TFS;http://www.genome.ad.jp/htbin/nph-tfsearch).
Acknowledgments
We thank Diana Wiedemann and Hella Ludewig for the excellent technical assistance.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL arosenth{at}imb-jena.de; FAX 49-3641-656255.
-
- Received February 19, 1997.
- Accepted April 15, 1997.
- Cold Spring Harbor Laboratory Press








