
High-fidelity bidirectional translation between single-cell transcriptomes and DNA methylomes with scBOND
- 1School of Mathematical Sciences and LPMC, Nankai University, Tianjin 300071, China;
- 2College of Electronic Information and Optical Engineering, Nankai University, Tianjin 300350, China;
- 3Academy for Advanced Interdisciplinary Studies, Nankai University, Tianjin 300071, China
-
↵4 These authors contributed equally to this work.
Abstract
Single-cell multiomic sequencing technologies have offered unprecedented insights into cellular heterogeneity by jointly profiling gene expression and epigenetic landscapes at single-cell resolution. However, the application of these technologies remains limited owing to technical challenges and high costs. Computational approaches for cross-modality translation provide a promising solution to these limitations by enabling the inference of one modality from another. However, existing methods for cross-modality translation between single-cell RNA sequencing (scRNA-seq) and single-cell DNA methylation (scDNAm) data face limitations, including unidirectionality, inadequate modeling of context-specific DNA methylation–expression associations, neglect of biological relevance in evaluation, and poor performance in limited paired training data. To fill these gaps, we introduce scBOND, a bidirectional cross-modal translation framework tailored for scRNA-seq and scDNAm profiles. scBOND leverages a mixture-of-experts block to capture context-dependent regulatory patterns, while implementing self-attention mechanism and a feature recalibration module to enhance biological signal fidelity. Extensive experiments demonstrate scBOND consistently outperforms baseline methods in both translation directions, yielding high-accuracy translation while preserving cellular structure. In mouse embryonic data, scBOND preserves subtle, functionally significant differences between closely related cell types, which are undetected in the original data. Downstream analyses confirm that scBOND effectively recovers tissue-specific signals in human brain neurons. Moreover, using RNA-only data, we reconstruct scDNAm profiles and identify cell-type- and stage-specific regulatory mechanisms in oligodendrocyte lineage. To further improve model generalization in paired data-scarce scenarios, we propose scBOND-Aug, a variant of scBOND equipped with a biologically informed data augmentation strategy, which demonstrates superior results with limited paired data.
The development of single-cell sequencing technologies has enabled high-resolution profiling of cellular heterogeneity (Papalexi and Satija 2018). Among various single-cell omic sequencing technologies, single-cell RNA sequencing (scRNA-seq) has emerged as a fundamental technique for characterizing the functional state of individual cells. Complementary to scRNA-seq, single-cell DNA methylation (scDNAm) sequencing provides genome-wide maps of epigenetic modifications at single-cell resolution, offering insights into the regulatory mechanisms underlying gene expression (Clark et al. 2017). In particular, DNA methylation has proven valuable in cancer research, in which it uncovers intratumoral heterogeneity and aids in the discovery of early diagnostic biomarkers and personalized therapeutic targets (Uzun et al. 2021). However, such single-modality sequencing offers only a limited dimension of cellular regulatory complexity, as it measures either gene expression or epigenetic modification in isolation (Wu et al. 2021), which is insufficient to resolve the multilayered regulatory architecture that orchestrates cellular identity, lineage commitment, and dynamic state transitions.
To gain a more comprehensive understanding of cellular states and gene regulatory mechanisms, researchers have developed single-cell multiomic sequencing technologies (Demetci et al. 2022; Li et al. 2024b). These technologies allow the simultaneous profiling of transcriptomic and epigenomic layers from the same cell, providing unprecedented opportunities to reconstruct regulatory networks and elucidate the relationships between transcriptional and epigenetic regulation. Nevertheless, their widespread application remains constrained by critical challenges including experimental complexity, low detection sensitivity, limited sequencing throughput, significant data noise, and high costs (Yang et al. 2021; Zhang et al. 2022). Because of these technical constraints, most publicly available single-cell data sets remain unimodal, which limits the discovery of cross-modality regulatory relationships that are essential for comprehensive biological interpretation. Consequently, establishing mappings between different modalities and predicting or reconstructing information from one modality to another, known as cross-modality translation, is of paramount importance (Demetci et al. 2022).
Several computational methods have been developed for cross-modality translation between scRNA-seq data and single-cell ATAC sequencing (scATAC-seq) data, with each approach introducing distinct modeling strategies (Wu et al. 2021; Zhang et al. 2022; Cao et al. 2024). However, these algorithms are challenging to apply for cross-modality translation between scRNA-seq and scDNAm data. This is because of the unique challenges presented by scDNAm data, including its lower coverage, continuous numerical nature (with DNA methylation levels ranging between zero and one), and the complex dynamics between DNA methylation and gene expression (Saripalli et al. 2020; He et al. 2022b). These intrinsic properties make cross-modality translation between scRNA-seq and scDNAm data even more challenging than between other modalities.
Despite these challenges, a few computational methods have been developed to enable cross-modality translation between scRNA-seq and scDNAm data. For example, scCross leverages variational autoencoders (VAEs), generative adversarial networks, and the mutual nearest neighbors technique for modality alignment (Yang et al. 2024), whereas MAPLE utilizes an ensemble supervised learning strategy to infer gene activity scores from DNA methylation features (Uzun et al. 2021). However, these methods also have notable limitations: (1) Some existing methods are constrained by their unidirectional prediction capability, limiting their applicability in scenarios that require reverse cross-modality translation; (2) existing methods fall short in modeling the intricate and highly cell-type-specific associations between DNA methylation and gene expression, which exhibit substantial variability across diverse genomic contexts; (3) most existing methods evaluate model performance primarily by measuring the numerical correlation between translated data and ground truth, neglecting the biological relevance and interpretability of the translated data; and (4) the limited availability of paired scRNA-seq and scDNAm profiles substantially impedes the effective training of these models, thereby constraining their performance in real-world applications.
To fill these gaps, we propose scBOND, a sophisticated framework for bidirectional cross-modality translation between scRNA-seq and scDNAm profiles with broad biological applicability. scBOND adopts a dual-channel VAE architecture to project scRNA-seq and scDNAm data into latent space, enabling effective cross-modality alignment and translation. To adaptively capture the context-dependent DNA methylation patterns related to gene regulation, we implement a mixture-of-experts (MoE) block, which utilizes a gating network to dynamically assign input cells to specialized expert subnetworks. Given the complex interactions in DNA methylation and gene expression across different genomic regions, we further incorporate a self-attention mechanism and a feature recalibration module to effectively model long-range dependencies. Additionally, considering the limited availability of paired scRNA-seq and scDNAm profiles for training, we further propose scBOND-Aug, a variant of scBOND based on a biologically guided data augmentation strategy, which enables the model to better capture the inherent biological relationships and patterns within the limited samples. The aim of this study is to introduce scBOND as a powerful tool for bridging the gap between single-cell transcriptomes and DNA methylomes, enabling the reconstruction of both RNA and DNA methylation profiles to uncover novel biological insights.
Results
Overview of the scBOND framework
scBOND is a dual-aligned VAE framework designed for bidirectional cross-modality translation between scRNA-seq and scDNAm profiles, facilitating the recovery of missing omics and enhancing biological understanding of regulatory mechanisms at the single-cell level. scBOND features a modular architecture composed of modality-specific encoders and decoders, along with cross-modality translators and adversarial discriminators, designed symmetrically for both scRNA-seq and scDNAm data (Fig. 1A). Specifically, the encoders project high-dimensional omic inputs into a shared latent space, serving as the foundation for integrative modeling: The RNA encoder employs a multilayer perceptron to capture complex transcriptomic patterns, whereas the DNAm encoder leverages chromosome-wise feature partitioning combined with a parameter-sharing multilayer perceptron to efficiently encode genomic regulatory signals. Additionally, to enhance model generalization and mitigate overfitting, both encoders incorporate a masking strategy inspired by masked autoencoders (He et al. 2022a) and apply dropout regularization (Methods).
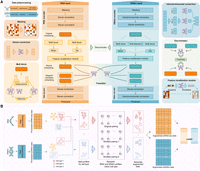
Overview of the scBOND framework and its variant scBOND-Aug. (A) The model features a dual-stream VAE design, with modality-specific encoders and decoders for translation. RNA and DNAm inputs undergo separate preprocessing steps, followed by masking and dense feature extraction. The RNA and DNAm translators perform the translation of each modality. A discriminator is employed to differentiate between real and translated embeddings. (B) The augmentation strategy of scBOND-Aug. scRNA-seq and scDNAm profiles are first preprocessed and then grouped by annotated cell type. Within each group, original cross-modal pairings are disrupted by randomly shuffling scDNAm profiles to generate biologically coherent synthetic pairs. These augmented data are then used to expand the training set for improved generalization under limited sample scenarios.
Subsequently, scBOND implements a MoE block consisting of multiple specialized subnetworks, each focusing on a different aspect of the data, enabling the model to capture complex, nonlinear relationships in RNA and DNAm modalities. Then two complementary mechanisms are designed after the MoE block: A self-attention mechanism is employed to model complex intramodality dependencies by dynamically weighing feature interactions based on contextual relevance, thereby improving the capacity to capture subtle regulatory relationships. Moreover, a feature recalibration module is applied to adaptively recalibrate feature importance, selectively amplifying biologically informative dimensions while suppressing irrelevant noise. This dual refinement strategy ensures that the latent embeddings retain both global contextual coherence and localized biological specificity, which are critical for effective decoding and modality alignment. However, there still exist substantial distributional discrepancies across modalities, for instance, scRNA-seq captures gene expression profiles, whereas scDNAm data reflects DNA methylation levels. These biologically rooted representational differences can lead to misaligned embeddings for the same cell type across modalities. To enforce modality-consistent and translatable latent representations, scBOND incorporates modality-specific adversarial discriminators that are trained to differentiate between original and cross-modality translated embeddings. This adversarial framework encourages the translated representations to capture the statistical and biological characteristics of the target modality with high fidelity, thereby bridging domain discrepancies between scRNA-seq and scDNAm profiles. Finally, decoders are employed to reconstruct the original high-dimensional inputs from their corresponding latent embeddings, with loss functions specifically tailored to each data modality, ensuring biologically coherent reconstructions (Methods).
To mitigate the challenges posed by the scarcity of paired single-cell multiomic data, which often results from high experimental costs and technical limitations, we further propose scBOND-Aug, which introduces a biologically informed data augmentation strategy as an extension of the scBOND framework (Fig. 1B). This method leverages the biological assumption that cells belonging to the same annotated type share similar regulatory characteristics across different modalities. Based on this assumption, scBOND-Aug generates synthetic paired scRNA-seq and scDNAm profiles by recombining features from scRNA-seq and scDNAm data within the same cell type through random permutations (Methods). By expanding the training data set in a biologically coherent manner, this strategy enables the model to learn a broader range of cross-modality correspondences while preserving intraclass consistency.
scBOND preserves cellular heterogeneity and improves the discrimination of similar cell types in early embryonic development
It is of great importance in cross-modality analysis that a translation method preserves the intrinsic biological structure and cellular heterogeneity of the original data, as these properties are critical for accurate downstream biological interpretation. To quantitatively assess whether the data translated by scBOND retain such heterogeneity, we performed clustering on the translated outputs and evaluated the results using four widely adopted metrics: adjusted mutual information (AMI), adjusted Rand index (ARI), homogeneity (HOM), and normalized mutual information (NMI; Methods) (Supplemental Text S1). We first accessed a MouseEmbryo data set, which contains 1082 mouse embryonic cells across four key developmental stages (Argelaguet et al. 2019). To ensure robust performance evaluation, we conducted fivefold cross-validation on the data set, using 80% of the cells for training the model and holding out the remaining 20% for translating in each fold. We benchmarked scBOND against two existing methods, scCross (Yang et al. 2024) and MAPLE (Uzun et al. 2021), which are tailored to cross-modality translation between scRNA-seq and scDNAm data. Notably, MAPLE supports only one-direction cross-modality translation (i.e., from DNAm to RNA), thereby limiting its general applicability. The results clearly demonstrate that scBOND consistently outperforms both MAPLE and scCross across all evaluation metrics (Fig. 2A). Specifically, in the DNAm-to-RNA direction, scBOND achieves average improvements of 97.20% in AMI, 182.19% in ARI, 40.48% in HOM, and 38.93% in NMI compared with MAPLE, which is the second-best method. In the RNA-to-DNAm direction, scBOND shows even greater performance, with average increases of 474.26% in AMI, 407.01% in ARI, 254.95% in HOM, and 208.53% in NMI compared with scCross, the baseline method that is limited to directional translation, highlighting its superior accuracy and robustness in cross-modality translation.
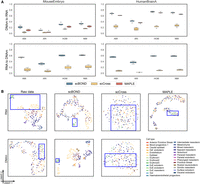
Benchmarking the cross-modal translation performance of scBOND. (A) Quantitative comparison of scBOND, scCross, and MAPLE on the MouseEmbryo data set and the HumanBrainA data set. Boxplots show four clustering evaluation metrics for both translation directions (DNAm to RNA and RNA to DNAm). (B) UMAP visualizations of the raw and translated profiles from the first test fold of the MouseEmbryo data set.
To further explore the biological relevance of scBOND's translated profiles, we visualized the translated profiles from the first test fold of the MouseEmbryo data set using uniform manifold approximation and projection (UMAP) (Fig. 2B) and t-distributed stochastic neighbor embedding (t-SNE) (Supplemental Fig. S1). We specifically examined three biologically important yet similar cell types, including epiblast, extraembryonic cells (ExEs), and notochord (Fig. 2B). These populations are known to co-occur during early embryonic development and engage in complex signaling interactions (Shahbazi et al. 2019; Xu et al. 2023). However, because of their close developmental origins, they are notoriously difficult to distinguish in raw data. In both the original scRNA-seq and scDNAm data, these cell types exhibited substantial overlap, making precise downstream analyses challenging (Fig. 2B). Following cross-modality translation with scBOND, however, these cell types became markedly more distinguishable, particularly in the RNA-to-DNAm direction, in which all three populations formed distinct and well-separated clusters (Fig. 2B). In the DNAm-to-RNA direction, the cells belonging to ExEs were clearly segregated from epiblast and notochord, suggesting that scBOND is capable of recovering biologically relevant boundaries even in highly noisy or sparse input conditions. From a developmental biology perspective, this enhanced separation is particularly meaningful: Epiblast gives rise to the entire embryo proper; notochord emerges from the epiblast-derived mesoderm and plays a key role in axis formation; and ExEs originate from the trophectoderm and contribute to extraembryonic structures such as the placenta (Huang et al. 2024). Properly resolving these lineages in single-cell multiomic data is essential for accurately reconstructing developmental trajectories and understanding cell fate decisions. In contrast, scCross struggled to disentangle these populations, especially in the DNAm-to-RNA translation, in which the three cell types remained heavily intermixed. Although scCross achieved partial separation between ExEs and notochord in the RNA-to-DNAm direction, the clustering boundaries were notably less defined than those observed with scBOND. MAPLE, which only supports DNAm-to-RNA translation, performed the worst: All three cell types, particularly epiblast, remained highly entangled in the latent space, suggesting the limited capacity of MAPLE to preserve fine-grained biological distinctions.
These results collectively demonstrate that scBOND not only excels at generating accurate cross-modality predictions but also preserves subtle but functionally significant differences among closely related cell types. This capability is crucial for downstream analyses such as lineage tracing and the inference of gene regulatory mechanisms during early development.
Cross-modality translation with scBOND recovers functional and tissue-specific signals in human brain
Following scBOND's demonstrated effectiveness in capturing subtle cell-type differences, we next investigated its capacity to recover functional and tissue-specific signals in more complex data set. Here, we additionally obtained the HumanBrainA data set, which comprises 4013 nuclei isolated from postmortem human frontal cortex, offering a complex and heterogeneous adult tissue context with a broad diversity of neuronal and glial cell types (Luo et al. 2022). We first performed fivefold cross-validation on the data set and found that scBOND is superior to other baseline methods (Fig. 2A; Supplemental Fig. S2). Quantitatively, in the DNAm-to-RNA direction, scBOND outperformed MAPLE by an average of 10.46% in AMI, 16.12% in ARI, 3.90% in HOM, and 7.91% in NMI. In the RNA-to-DNAm direction, scBOND achieved significantly higher performance than scCross, with average improvements of 611.28% in AMI, 1271.40% in ARI, 523.65% in HOM, and 422.56% in NMI, highlighting its superior accuracy in cross-modality translation. To assess the biological relevance of the cross-modality translation results and to gain deeper insights into cell-type-specific regulatory functions, we further conducted downstream analyses on the translated scRNA-seq and scDNAm profiles. These analyses aimed to determine whether the translated profiles preserve meaningful transcriptional and epigenetic signals and whether such signals can be used to uncover regulatory pathways that are specific to individual cell types.
Firstly, we conducted Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (Kanehisa and Goto 2000) and Gene Ontology (GO) (Gene Ontology Consortium 2004) enrichment analyses based on the translated scRNA-seq profiles. Specifically, we focused on the Exc_L4-5_FOXP2 cell type from the first test fold of the HumanBrainA data set to investigate whether biologically meaningful signals are preserved in the translated data. Note that Exc_L4-5_FOXP2 refers to excitatory neurons in layers 4 and 5 of the neocortex that express the FOXP2 gene. Using the SCANPY pipeline (Wolf et al. 2018), we identified two sets of differentially expressed genes (DEGs) specific to this excitatory neuronal subtype, one derived from the original scRNA-seq profiles and the other from the translated scRNA-seq profiles. Each set of DEGs were used for KEGG pathway and GO enrichment analyses, respectively, to further evaluate the biological relevance of the translated scRNA-seq profiles. As shown in Figure 3A, KEGG pathway analysis reveals a marked difference between the original and translated profiles. The DEGs from the translated profiles exhibit significantly broader and more functionally coherent pathway enrichment, including key neuronal processes such as oxidative phosphorylation, synaptic signaling pathways (e.g., GABAergic, cholinergic, and calcium signaling), and neuroactive ligand–receptor interaction. These pathways are closely aligned with the physiological roles of excitatory neurons in cortical information processing (Supplemental Text S2; Koh et al. 2023; Wang et al. 2023). In contrast, enrichment derived from the original data was weaker and less specific, indicating limited ability to capture functional signatures. Figure 3B further supports this observation at the GO level. In the translated profiles, enriched biological processes such as regulation of membrane potential, synaptic transmission, and axon development are highly relevant to the cellular identity and function of Exc_L4-5_FOXP2 neurons. The cellular component terms, including postsynaptic membrane, glutamatergic synapse, and dendritic tree, accurately reflect the anatomical and functional specialization of these neurons. Moreover, molecular function enrichment, such as ion channel activity and neurotransmitter receptor binding, further underscores the translated data's ability to recover meaningful regulatory programs. Collectively, these results demonstrate that the scRNA-seq profiles translated by scBOND not only retain cell-type identity but also enhance the biological interpretability of downstream analyses. Compared with the original profiles, the translated profiles more clearly expose regulatory and functional pathways consistent with the known roles of Exc_L4-5_FOXP2 neurons.
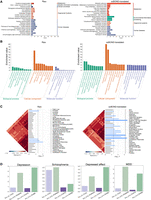
Biological interpretability of scBOND-translated scRNA-seq and scDNAm profiles. (A) KEGG pathway enrichment analysis of DEGs from the original (left) and scBOND-translated (right) scRNA-seq profiles for the Exc_L4-5_FOXP2 cell type in the HumanBrainA data set. (B) GO enrichment results based on DEGs from original (left) and scBOND-translated (right) scRNA-seq profiles. (C) Tissue-specific enrichment analysis of original (left) and scBOND-translated (right) scDNAm profiles for the Inh_CGE_VIP cell type using SNPsea. (D) LDSC-based partitioned heritability analysis comparing enrichment of scBOND-translated scDNAm profiles in four psychiatric disorder-associated traits: depression, schizophrenia, depressed affect, and major depressive disorder (MDD).
Building upon these transcriptomic findings, we next asked whether the scBOND-translated epigenetic profiles (i.e., scDNAm profiles) would similarly enhance biological interpretability except for excitatory neurons. To this end, we turned to another inhibitory neuronal subtype and carried out parallel enrichment analyses at the genetic level. Specifically, using the Inh_CGE_VIP cell type (an inhibitory neuron subtype expressing the VIP gene) of the first test fold of the HumanBrainA data set as an example, we first identified two sets of differentially methylated regions (DMRs) from the original scDNAm profiles and translated scDNAm profiles, using the EpiScanpy pipeline (Danese et al. 2021), and then, we performed tissue-specific enrichment analyses on the two sets of DMRs using SNPsea, respectively (Supplemental Text S2; Slowikowski et al. 2014). The results showed that the translated scDNAm profiles exhibited stronger enrichment significance among the top 30 enriched tissues compared with the raw data, with markedly improved enrichment in several brain-related tissues such as the prefrontal cortex, cerebellum, and hippocampus (Fig. 3C). This suggests that the translated scDNAm data more accurately capture biologically meaningful associations with the cells’ tissue of origin. We further applied linkage disequilibrium score regression (LDSC) (Finucane et al. 2015) to evaluate enrichment of the translated scDNAm profiles in neuropsychiatric disorder–related pathways. As shown in Figure 3D, the translated data demonstrated notable enrichment signals across four representative psychiatric pathways, including depression, schizophrenia, depressed affect, and major depressive disorder (MDD). Collectively, these SNPsea and LDSC analyses further validate the enhanced biological relevance of the translated scDNAm data, demonstrating that scBOND not only enables effective structural reconstruction at the data level but also preserves meaningful functional and genetic associations, thereby achieving superior biological consistency.
scBOND reconstructs epigenetic landscapes from scRNA-seq data to uncover stage-specific regulatory mechanisms in the oligodendrocyte lineage
Building on the ability of scBOND to perform cross-modal translation between RNA and DNAm modalities, we further employed the scBOND model to infer scDNAm profiles from single-modality RNA data, thereby constructing a pseudopaired multiomic data set. This strategy enables the exploration of transcriptional and epigenetic regulatory relationships in the absence of experimentally paired measurements. Specifically, we trained the model using the aforementioned paired single-cell multiomic data set HumanBrainA and subsequently applied the trained model to predict scDNAm profiles from a newly accessed RNA-only human brain data set, referred to as HumanBrainB-RNA (Siletti et al. 2023). As shown in Figure 4A, the predicted scDNAm profiles successfully discriminated between different cell types, highlighting the model's capacity to capture biologically relevant variation. To further explore transcriptional–epigenetic interactions, we quantified the relationship between gene expression and DNA methylation within each cell type. Specifically, we computed Spearman's correlation coefficients between gene expression levels and promoter-proximal DNA methylation levels for individual cells (Methods) (Supplemental Text S1). The resulting distributions exhibited marked differences across cell types, suggesting that transcriptional–epigenetic regulation is highly cell-type-specific, likely reflecting underlying differences in cellular function and differentiation status (Supplemental Fig. S3).
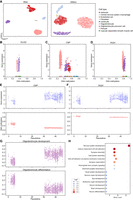
Transcriptomic–epigenetic dynamics in the oligodendrocyte lineage. (A) UMAP visualization of RNA and predicted DNAm profiles on the HumanBrainB data set. (B–D) Scatter plots showing the relationship between gene expression and promoter-proximal DNA methylation levels across cell types for three key oligodendrocyte lineage genes: OLIG2, CNP, and FA2H. (E,F) Pseudotime dynamics of CNP and FA2H gene expression and promoter-proximal DNA methylation along the oligodendrocyte lineage trajectory. (G) Pseudotime-based module scores of two biological processes: oligodendrocyte development and oligodendrocyte differentiation. (H) Bubble plot showing enriched GO terms among genes with significant pseudotime-associated promoter-proximal DNA methylation.
We next investigated the regulatory relationship between gene expression and DNA methylation during the differentiation of oligodendrocyte precursor cells (OPCs) into mature oligodendrocytes (OLs). OPCs are neural glial progenitors with the capacity for both proliferation and differentiation, serving as the developmental origin of OLs. In contrast, OLs are fully differentiated glial cells responsible for forming the myelin sheath, representing the terminal stage of the oligodendrocyte lineage (Rowitch and Kriegstein 2010). To characterize molecular changes accompanying this differentiation process, we selected three well-established OL marker genes from the CellMarker database (Zhang et al. 2019): OLIG2, CNP, and FA2H. We first confirmed that all three genes exhibit high expression specifically in OLs, consistent with their known roles in oligodendrocyte identity and function (Supplemental Fig. S4). We then examined how their gene expression levels relate to promoter-proximal DNA methylation across different cell types. As shown in Figure 4B, OLIG2 showed consistently high expression and stable promoter-proximal DNA methylation level in both OPCs and OLs, indicating a constitutively active state. This is in line with its function as a core transcription factor that is essential for maintaining oligodendrocyte lineage identity and is expressed continuously from OPCs to early OLs (Ligon et al. 2004; Wegener et al. 2015). In contrast, CNP and FA2H displayed a different regulatory pattern (Fig. 4C,D). Both genes showed significantly increased expression in OLs compared with OPCs, accompanied by substantial demethylation at their promoter regions, particularly for CNP. These findings suggest that CNP and FA2H are transcriptionally activated during OL differentiation and that this activation is likely driven by promoter demethylation (Moyon et al. 2021). Importantly, to assess whether this inverse methylation–expression relationship is a general principle or is specific to the oligodendrocyte lineage, we extended our analysis to marker genes of other major cell types (Supplemental Fig. S5). Together, these results highlight a shift from transcriptional maintenance in OPCs to epigenetically regulated activation of functional effector genes in OLs, reflecting distinct regulatory modes underlying lineage specification and terminal differentiation.
To further validate the observed relationship between transcriptional activation and promoter demethylation, we incorporated single-cell trajectory analysis to explore the temporal dynamics of this regulatory mechanism during OPC-to-OL differentiation. Specifically, we extracted OPC and OL cells from the HumanBrainB-RNA data set and constructed a pseudotime trajectory using Monocle3 (Supplemental Fig. S6; Trapnell et al. 2014). We found that the aforementioned three genes exhibited distinct expression and methylation dynamics along this trajectory (Fig. 4E,F; Supplemental Fig. S7). OLIG2, a core transcription factor critical for maintaining oligodendrocyte lineage identity, displayed stable expression and persistently low promoter methylation throughout the trajectory, indicating a constitutively active state (Supplemental Fig. S7). In contrast, CNP underwent promoter demethylation during the early phase of differentiation, followed by a marked increase in expression, suggesting that epigenetic derepression facilitated its transcriptional activation and functional involvement in myelination (Fig. 4E). A similar pattern was observed for FA2H, whose expression increased progressively with differentiation, accompanied by a gradual decrease in promoter methylation (Fig. 4F). Together, these results highlight a consistent trend in which promoter demethylation precedes transcriptional activation, underscoring the key role of epigenetic regulation in driving cell fate transitions from OPCs to OLs. Moreover, to examine whether this regulatory mechanism is reflected at the pathway level, we conducted gene set scoring using SCANPY (Wolf et al. 2018) to evaluate the temporal activity of two key biological pathways: oligodendrocyte development and oligodendrocyte differentiation. As shown in Figure 4G, the activity of the developmental pathway increased significantly along pseudotime, indicating sustained activation during the late stages of OL maturation. In contrast, the differentiation pathway showed higher activity at early pseudotime and declined thereafter, suggesting its predominant role in the initial stages of lineage commitment during the OPC phase (Fig. 4G). These pathway-level results are well aligned with the dynamics observed at individual gene loci and provide further evidence that promoter demethylation serves as a key epigenetic switch to activate gene expression programs essential for oligodendrocyte development and maturation.
Encouraged by these gene-level observations, we next sought to systematically identify additional genes exhibiting similar patterns of dynamic epigenetic regulation across the OPC-to-OL trajectory. Specifically, we performed a regression analysis using pseudotime as the independent variable and DNA methylation levels at gene promoter regions as the response (Supplemental Fig. S8). This analysis aimed to detect genes exhibiting significant changes in promoter methylation along the differentiation trajectory, which may represent candidate targets of stage-specific epigenetic modulation. We then carried out GO enrichment analysis on the identified differentially methylated genes. As shown in Figure 4H, the enriched terms were primarily associated with biological processes relevant to neuron development and cellular organization, including nervous system development, calcium-dependent cell–cell adhesion, and synapse assembly. These findings further support the idea that promoter demethylation events during OL differentiation not only activate cell-type-specific functional genes but may also coordinate broader neuron developmental regulatory programs, providing potential insight into how oligodendrocyte maturation is functionally coupled to the surrounding neural environment.
In summary, by leveraging the cross-modal translation capability of scBOND, we reconstructed scDNAm profiles from RNA-only data and uncovered cell-type-specific and stage-specific patterns of transcriptional–epigenetic regulation in human brain development, establishing scBOND as a powerful framework for dissecting transcriptional–epigenetic interplay from single-modality data and providing new insights into the epigenetic logic of brain cell fate decisions.
scBOND demonstrates resilience to DNA methylation noise in low-quality scDNAm profiles
Because of the immature state of scDNAm sequencing technologies, the scDNAm data often suffer from low coverage, high sparsity, and substantial technical noise (Kremer et al. 2024). These limitations arise from factors such as limited DNA input, bisulfite conversion inefficiencies, and sequencing dropouts, which together compromise the reliability and consistency of scDNAm measurements. In practice, this results in high rates of missing or erroneous methylation calls, which pose a significant challenge for cross-modality translation methods.
To assess the robustness of scBOND against noise-induced perturbations, we simulated common sources of error observed in scDNAm data. To emulate these conditions, we randomly selected varying proportions of genomic regions (1%, 2%, 3%, 4%, and 5%) from scDNAm data of the HumanBrainA data set and applied a value flipping operation (Supplemental Text S3) mimicking DNA methylation–level inversions caused by sequencing artifacts or experimental biases. This noise-injection strategy enabled us to systematically evaluate the model's ability to maintain reliable performance under corrupted input scenarios. We assessed clustering consistency using fivefold cross-validation on the simulated data sets with different noise levels. The results demonstrate that model performance remains largely stable as the noise ratio increases from 1% to 5%, with only minimal fluctuations observed across most evaluation metrics (Fig. 5A). The performance under RNA sequencing error was also tested by utilizing the dropout strategy (Supplemental Text S3), adding noise rate from 1% to 5%. The robustness of the model is also largely maintained with merely marginal variations in the majority of metrics (Supplemental Fig. S9). This indicates that scBOND exhibits strong resilience to noise in both translation directions. Notably, the RNA-to-DNAm translation task under DNA purtubation demonstrated particularly robust performance, consistently achieving high clustering scores across all levels of simulated noise. This suggests that the model effectively captures the regulatory signatures embedded in scRNA-seq profiles, allowing accurate reconstruction of DNA methylation landscapes even under perturbed conditions. In contrast, the DNAm-to-RNA direction exhibited slight decreases in AMI and ARI at higher noise levels (e.g., 4% and 5%), indicating that gene expression predictions are somewhat more sensitive to inaccuracies in DNAm input. Overall, the model maintains stable performance in both tasks, despite showing slight fluctuations when subjected to perturbations within the source data domain.
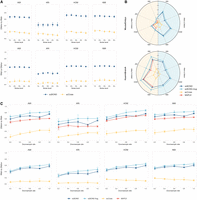
scBOND and scBOND-Aug demonstrates robustness and superior performance under data perturbations. (A) Robustness evaluation of scBOND and baseline methods under increasing DNA methylation noise levels (1%–5%) on the HumanBrainA data set. (B) Radar plots comparing the translation performance of scBOND, scBOND-Aug, scCross, and MAPLE across four clustering metrics on two paired data sets. (C) Downsampling experiments on the HumanBrainA data set to evaluate robustness under varying training sample sizes, comparing methods of scBOND, scBOND-Aug, scCross, and MAPLE. The x-axis indicates the proportion of training cells sampled.
Together, scBOND demonstrates robust resilience to noise-induced perturbations in scDNAm data, which is often compromised by low coverage, sparsity, and technical noise. These findings highlight scBOND's robustness in handling noisy and sparse scDNAm data, making it a powerful tool for cross-modality translation in challenging scenarios.
scBOND-Aug enables accurate cross-modality translation with limited training data
One of the major challenges in training cross-modality translation models for single-cell multiomic data lies in the limited availability of paired data sets, especially those involving scRNA-seq and scDNAm profiles. Because of experimental and technical constraints, such paired profiles are often sparse and small in scale, which can lead to underfitting and suboptimal model performance. To address this limitation, we construct an augmented version of scBOND, referred to as scBOND-Aug, by incorporating cell-type-level information from available paired profiles. Specifically, we introduce a cell-type-guided data augmentation strategy, wherein we randomly pair scRNA-seq and scDNAm profiles from different cells that are annotated as the same cell type (Methods). This process generates synthetic paired profiles that preserve biological coherence while effectively expanding the training data set, enabling the model to capture cell-type-specific patterns and improve its performance under data-scarce conditions.
We evaluate the effectiveness of the proposed scBOND-Aug model on the two paired data sets introduced above (MouseEmbryo and HumanBrainA) by comparing its performance with the scBOND and baseline methods in cross-modality translation tasks. As shown in Figure 5B, scBOND-Aug consistently outperforms the baseline methods across the two data sets, indicating that the cell-type-guided augmentation strategy brings substantial performance improvements. Specifically, on the MouseEmbryo data set, scBOND-Aug improves AMI by 3.06% and 9.09%, ARI by 9.43% and 13.77%, HOM by 5.74% and 9.03%, and NMI by 3.13% and 7.39% in DNAm-to-RNA and RNA-to-DNAm directions, respectively; similarly, on the HumanBrainA data set, it achieves significant enhancements including an increase of 19.97% in AMI, 42.54% in ARI, 16.05% in HOM, and 17.39% in NMI for DNAm-to-RNA translation and 10.16% in AMI, 17.24% in ARI, 10.24% in HOM, and 9.04% in NMI for RNA-to-DNAm translation. These results demonstrated that translated data generated by scBOND-Aug exhibits more accurate and biologically coherent clustering, suggesting enhanced preservation of cell-type structure and heterogeneity during translation.
To further investigate the robustness of scBOND-Aug under varying levels of data availability, we conducted an additional experiment designed to simulate low-resource conditions. Specifically, we took the HumanBrainA data set as an example and simulated varying levels of data availability by uniformly downsampling the data set within each cell type. For each cell type, a fixed percentage of cells was randomly sampled to construct simulated single-cell multiomic data sets at different scales, ranging from 20% to 100% of the original data set size. For each simulated data set, we performed fivefold cross-validation and evaluated the clustering results on the translated data. The results demonstrate that scBOND-Aug consistently outperforms scBOND and baseline methods across all levels of data availability, with particularly notable improvements in the direction of DNAm to RNA under different data-scarce conditions, highlighting the effectiveness of data augmentation (Fig. 5C). To further assess the practical utility of this improvement beyond clustering, we evaluated the performance of scBOND and scBOND-Aug in a downstream cell-type identification task (Supplemental Text S4; Supplemental Fig. S10) and the robustness of scBOND-Aug under cell-type perturbation (Supplemental Text S5; Supplemental Fig. S11), in which its downstream outlook and stable performance were confirmed.
In conclusion, scBOND-Aug effectively addresses the critical limitation of data scarcity in paired single-cell multiomic data sets by leveraging biologically coherent augmentation strategies. Through systematic evaluations on multiple data sets and under varying data availability conditions, scBOND-Aug demonstrates consistently superior performance compared with the baseline methods. These results highlight its robustness and generalizability, making it a promising approach for improving cross-modality translation in real-world applications in which paired single-cell data are limited.
Discussion
In this work, we present scBOND, a dual-aligned VAE framework that enables accurate and biologically meaningful translation between scRNA-seq and scDNAm data at the single-cell level. By integrating modality-specific encoders, MoE block, self-attention mechanism, and a feature recalibration module, scBOND effectively models complex regulatory relationships and preserves cell-type identity across modalities. The results collectively demonstrate that scBOND not only excels in generating accurate cross-modality predictions but also preserves subtle, yet functionally significant, differences among closely related cell types in the mouse embryo. Additionally, scBOND effectively captures and reveals biologically meaningful transcriptional and epigenetic signals, thereby enhancing our understanding of cell-type-specific regulatory pathways and their implications in human brain and neuropsychiatric diseases. It is important to emphasize that the translated profiles are not intended to replace experimentally observed data but rather serve as a computationally inferred complement in scenarios in which one modality is missing. By leveraging scBOND's cross-modal translation capabilities, we successfully reconstructed scDNAm profiles from RNA-only data, uncovering cell-type-specific and stage-specific patterns of transcriptional–epigenetic regulation in the oligodendrocyte lineage. We have also demonstrated the resilience of scBOND to DNA methylation noise in low-quality scDNAm profiles. To address the common challenges of limited paired data sets in the oligodendrocyte lineage, we further developed scBOND-Aug, which introduces a biologically guided data augmentation strategy. This extension significantly improves model robustness and generalization under limited training samples, making it a practical and effective solution for single-cell multiomic translation. Although scBOND-Aug currently requires annotated cell types, it can be adapted to unlabeled data by first generating “pseudolabels” via unsupervised methods such as pseudotime binning, metacell clustering, or graph-based partitioning and using these as augmentation groups. This enables the application on continuous processes like differentiation trajectories, extending its utility beyond discrete cell types. We anticipate scBOND will empower cost-effective multiomic studies and serve as a foundation for future methodological innovations in single-cell data integration.
In addition to the above findings, our framework also addresses several critical limitations observed in existing cross-modality translation methods. Approaches such as scCross and MAPLE suffer from inherent architectural and modeling constraints. For example, scCross relies heavily on adversarial training and mutual nearest-neighbor alignment, which often leads to unstable optimization and restricts its ability to capture complex, cell-type-specific relationships between scDNAm and gene expression. MAPLE, on the other hand, only supports one-directional prediction and depends on ensemble supervised learning strategies that require large amounts of high-quality labeled data, conditions that are rarely met in practical single-cell applications. In contrast, scBOND effectively overcomes these shortcomings. The MoE block enables the model to automatically disentangle context-dependent regulatory patterns across diverse cellular states, whereas self-attention and feature recalibration mechanisms enhance the capture of key biological signals even under noisy or sparse scDNAm inputs (Li et al. 2025b). Moreover, although scBOND is designed for translating between scRNA-seq and scDNAm data, the core components of the model, particularly the MoE block, are highly adaptable for cross-modality tasks involving other modalities, such as scATAC-seq (Wu et al. 2024; Zhou and Wu 2024; Zhu et al. 2025; Shi et al. 2026). This flexibility allows scBOND to be generalized to other types of single-cell multiomic integration, enabling broader applications in biological research.
Despite these advances, scBOND may still be subject to general limitations such as susceptibility to overfitting, dependence on hyperparameter tuning, and limited interpretability of latent representations. Although attention mechanisms and feature recalibration partially mitigate these issues, further efforts are needed to enhance model transparency and to ensure that the cross-modality mappings align with experimentally validated biological mechanisms (Li et al. 2025a). Moreover, our evaluation is conducted on specific data sets from mouse embryonic development and the human cerebral cortex. Although these data sets span diverse cell types, they cannot fully represent the broad range of biological systems. The scalability and robustness of scBOND across additional tissues, species, perturbation experiments, and disease conditions warrant further investigation. The following are some possible improvement methods. First, integrating prior knowledge from bulk omic atlases or curated gene regulatory networks could improve the biological plausibility and precision of cross-modality mappings (Chen et al. 2021; Cui et al. 2024). Such incorporation of established biological context may help guide the model toward more accurate and interpretable translation outcomes. In addition, adopting continual learning strategies (Wang et al. 2024) would enable the model to adapt incrementally as new single-cell data sets become available, thereby enhancing its scalability and flexibility in real-world applications. Furthermore, expanding the model to incorporate other omic data, such as single-cell proteomics or metabolomics, could provide a more comprehensive understanding of cellular functions and regulatory networks, thus further improving the robustness and versatility of scBOND in complex multiomic studies (Cao et al. 2024).
Methods
Basic architecture of scBOND
The basic architecture of scBOND is built upon a dual-aligned VAE framework, comprising seven core components: RNA encoder
Φr and DNAm encoder Φm, RNA decoder Ψr and DNAm decoder Ψm, an integrative translator Γ , and discriminators for two modalities Δr and Δm (Fig. 1A). For paired scRNA-seq and scDNAm data, we first perform preprocessing and obtain the preprocessed data matrices 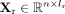 and
and 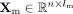 , in which n denotes the number of cells, lr denotes the number of genes in the preprocessed scRNA-seq data set, and lm denotes the number of genomic regions in the preprocessed scDNAm data set. Leveraging these inputs, scBOND enables bidirectional
omic translation: predicting scRNA-seq data from scDNAm data through Ψr(Γm→r(Φm(Xm))) and predicting scDNAm data from scRNA-seq data through Ψm(Γr→m(Φr(Xr))). Detailed specifications of each architectural component are elaborated below.
, in which n denotes the number of cells, lr denotes the number of genes in the preprocessed scRNA-seq data set, and lm denotes the number of genomic regions in the preprocessed scDNAm data set. Leveraging these inputs, scBOND enables bidirectional
omic translation: predicting scRNA-seq data from scDNAm data through Ψr(Γm→r(Φm(Xm))) and predicting scDNAm data from scRNA-seq data through Ψm(Γr→m(Φr(Xr))). Detailed specifications of each architectural component are elaborated below.
The encoders in scBOND
Encoders Φr and Φm are designed to embed scRNA-seq and scDNAm data into a low-dimensional space. To enhance model robustness and generalization,
a masking strategy inspired by masked autoencoders (He et al. 2022a) is adopted: During training, 50% of the input elements in Xr and 30% of those in Xm are randomly set to zero. Both encoders also incorporate a dropout rate of 0.1 in their latent layers to prevent overfitting.
A LeakyReLU activation function is employed in the encoders (Xu et al. 2020), defined as
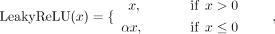 where the negative slope coefficient a is set to 0.01, which helps mitigate the vanishing gradient problem by allowing a small, nonzero gradient when the unit is
inactive. More specifically, in Φr, the gene expression vector of the kth cell
where the negative slope coefficient a is set to 0.01, which helps mitigate the vanishing gradient problem by allowing a small, nonzero gradient when the unit is
inactive. More specifically, in Φr, the gene expression vector of the kth cell 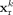 is successively transformed to 256 and then to 128 dimensions through a multilayer perceptron. In Φm, DNA methylation features are first partitioned by chromosome, based on the biological rationale that intrachromosomal interactions
are generally more functionally relevant than interchromosomal ones. Each chromosome-specific feature set is independently
processed using a shared neural network module, which projects the high-dimensional inputs into 32-dimensional latent representations.
This weight-sharing strategy not only reduces the number of trainable parameters but also enables the model to learn consistent
feature extraction patterns across different chromosomes. The resulting embeddings are then concatenated and passed through
multilayer perceptron to obtain a unified 128-dimensional latent representation. With the above encoder architecture, we obtain
low-dimensional representations for both modalities,
is successively transformed to 256 and then to 128 dimensions through a multilayer perceptron. In Φm, DNA methylation features are first partitioned by chromosome, based on the biological rationale that intrachromosomal interactions
are generally more functionally relevant than interchromosomal ones. Each chromosome-specific feature set is independently
processed using a shared neural network module, which projects the high-dimensional inputs into 32-dimensional latent representations.
This weight-sharing strategy not only reduces the number of trainable parameters but also enables the model to learn consistent
feature extraction patterns across different chromosomes. The resulting embeddings are then concatenated and passed through
multilayer perceptron to obtain a unified 128-dimensional latent representation. With the above encoder architecture, we obtain
low-dimensional representations for both modalities, 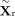 and
and 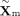 , serving as the basis for subsequent cross-modality translation.
, serving as the basis for subsequent cross-modality translation.
The translator in scBOND
After encoding both modalities into a unified latent space, the translator Γ is introduced to enable effective modality alignment and translation. Taking translation of DNAm to RNA, for instance, inspired by the idea of MoE (Jacobs et al. 1991; Eigen et al. 2013), we developed a MoE block that combines the outputs of multiple specialized expert networks, each focusing on different expression patterns such as cell-type-specific signals or context-dependent regulatory elements. A gating network is used to dynamically assign different weights to each expert's contribution, allowing the model to select the most relevant pathways for each input sample. This dynamic expert routing mechanism significantly enhances the model's ability to generalize across diverse biological conditions and facilitates more accurate reconstruction of the target modality.
In our implementation, the MoE block consists of six expert networks, each of which independently transforms the DNAm latent
input 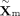 into a 128-dimensional representation. Each expert is composed of a multilayer perceptron, batch normalization, and a nonlinear
activation. For each input, the model computes both the mean vector
into a 128-dimensional representation. Each expert is composed of a multilayer perceptron, batch normalization, and a nonlinear
activation. For each input, the model computes both the mean vector  and the log-variance vector
and the log-variance vector 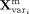 from every expert i ∈ 1, 2, …, 6. A shared gating network, composed of a two-layer perceptrons with ReLU and Softmax activation, produces the
expert assignment probability vector P = [p1, p2, …, p6]. The final mean and log-variance are calculated as weighted sums across experts:
from every expert i ∈ 1, 2, …, 6. A shared gating network, composed of a two-layer perceptrons with ReLU and Softmax activation, produces the
expert assignment probability vector P = [p1, p2, …, p6]. The final mean and log-variance are calculated as weighted sums across experts:
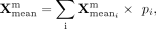
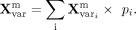
After the weighted combination, the MoE block yields a unified latent representation. This representation is then passed to
the translator module Γ, which performs stochastic sampling from a multivariate Gaussian distribution parameterized by the
final mean 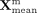 and log-variance
and log-variance 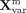 to generate the refined latent embedding
to generate the refined latent embedding 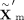 for downstream processing and analysis. This architecture enables the model to adapt to diverse input data types and to automatically
integrate the most relevant expert outputs, thereby enhancing translation accuracy and robustness.
for downstream processing and analysis. This architecture enables the model to adapt to diverse input data types and to automatically
integrate the most relevant expert outputs, thereby enhancing translation accuracy and robustness.
To further refine the latent representation obtained through the MoE block and improve the model's ability to capture intramodality
dependencies, we implement a self-attention mechanism (Vaswani et al. 2017) within the translator (Supplemental Text S6). This allows the model to dynamically recalibrate feature interactions by attending to informative positions within the
same modality. Specifically, given the representation 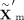 obtained from the MoE block, we calculate query, key, and value matrices as follows:
obtained from the MoE block, we calculate query, key, and value matrices as follows:
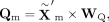
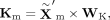
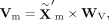 where WQ, WK, and WV are learnable projection matrices. The attention output is then calculated as
where WQ, WK, and WV are learnable projection matrices. The attention output is then calculated as
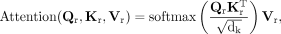 where dk is set to 128. Then we can derive the temporary latent representation as
where dk is set to 128. Then we can derive the temporary latent representation as
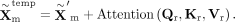
Although self-attention mechanism enables dynamic, context-aware recalibration by modeling pairwise interactions between features, it treats all features equally when computing attention outputs. However, not all feature dimensions contribute equally to downstream decoding. To further enhance the representation and emphasize informative features, we apply a feature recalibration module to the latent representation (Hu et al. 2018). The feature recalibration module adaptively recalibrates feature-wise responses through a two-step process. First, a global average pooling operation is applied to aggregate contextual information across each feature, producing a compact feature descriptor. Second, this descriptor is passed through a lightweight gating mechanism composed of two-layer perceptrons with nonlinear activations, generating a set of per-feature modulation weights. These weights are then used to rescale the original feature map via feature-wise multiplication. By selectively highlighting informative features and suppressing less relevant ones, the feature recalibration module enhances the expressiveness of the latent embedding passed to the downstream decoders.
Specifically, given the input latent representation 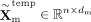 , we first apply a squeeze operation by performing global average pooling to obtain a feature-wise descriptor
, we first apply a squeeze operation by performing global average pooling to obtain a feature-wise descriptor 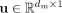 :
:
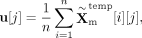 where u[j] stands for the squeeze result of the jth feature, dm is the number of features, and n represents the number of cells.
where u[j] stands for the squeeze result of the jth feature, dm is the number of features, and n represents the number of cells.
Then we put the squeezed feature vector into a two-layer perceptron and an activation function to receive the weight of each
channel:
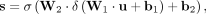 in which
in which 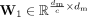 and
and 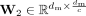 are the weight matrix of two-layer perceptron, c is the reduction rate set to 32, and δ and σ represents the ReLU excitation function and Sigmoid excitation function.
are the weight matrix of two-layer perceptron, c is the reduction rate set to 32, and δ and σ represents the ReLU excitation function and Sigmoid excitation function. 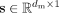 is the obtained feature modulation weight vector. Then, we can obtain the final result by
is the obtained feature modulation weight vector. Then, we can obtain the final result by
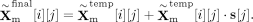 Here, we designed four translators Γr→m, Γr→r, Γm→r, and Γm→m, each adopting the above architecture but using different training approaches. Specifically, the self-attention mechanism
was applied exclusively to the DNAm data. We called Γr→m(Φr(Xr)) and Γr→r(Φr(Xr)) as the translated DNAm embeddings and the mapped RNA embeddings. Correspondingly, the translated RNA embeddings and the
mapped DNAm embeddings could be obtained through Γm→r(Φm(Xm)) and Γm→m(Φm(Xm)).
Here, we designed four translators Γr→m, Γr→r, Γm→r, and Γm→m, each adopting the above architecture but using different training approaches. Specifically, the self-attention mechanism
was applied exclusively to the DNAm data. We called Γr→m(Φr(Xr)) and Γr→r(Φr(Xr)) as the translated DNAm embeddings and the mapped RNA embeddings. Correspondingly, the translated RNA embeddings and the
mapped DNAm embeddings could be obtained through Γm→r(Φm(Xm)) and Γm→m(Φm(Xm)).
The discriminators in scBOND
To ensure the translated embeddings align closely with the distribution of the target modality, we introduce the modality-specific
discriminators Δr and Δm. The discriminators exert the effect to differentiate between original embeddings and their translated counterparts, facilitating
adversarial training to enhance the resemblance between the translated embeddings and the original ones. Taking the RNA embedding
discriminator Δr as an example, during the training process, it accepts original RNA embeddings Φr(Xr) and translated embeddings Γm→r(Φm(Xm)) with equally likelihood, whose objective is to produce outputs according to the following criteria:
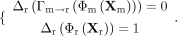
Likewise, the discriminator Δm for DNAm embeddings enables distinguishing between the original DNAm embeddings Φm(Xm) and the translated embeddings Γr→m(Φr(Xr)). The discriminators share an identical architecture: a two-layer network comprising a 128-dimensional perceptron with LeakyReLU activation, followed by a Sigmoid output layer.
The decoders in scBOND
The decoders Ψr and Ψm are designed to reconstruct high-dimensional scRNA-seq and scDNAm profiles from their respective latent representations. Specifically, Ψr mirrors the architecture of the RNA encoder Φr, employing multilayer perceptrons with LeakyReLU activation to progressively upscale the 128-dimensional latent embedding to 256 dimensions and ultimately to the original transcriptomic feature space. In contrast, Ψm, which corresponds to the DNAm encoder Φm, adopts a similar structure but applies a Sigmoid activation function in its final layer. To prevent overfitting, both decoders incorporate a dropout layer with a rate of 0.1 within their latent transformation stages.
The training procedure of scBOND
We develop a two-stage training strategy to optimize the scBOND model: (1) an initial pretraining stage for modality-specific feature learning and (2) a joint integrative stage to align cross-modality dependencies. In the first stage, the RNA encoder–decoder pair (Φr and Ψr) and the DNAm encoder–decoder pair (Φm and Ψm) are trained independently on their respective modalities to capture robust intramodal representations. The pretrained parameters are then used to initialize the second stage, in which data from both modalities are jointly utilized to learn intermodal relationships and facilitate cross-modality translation.
In the pretraining phase, for the RNA modality, input data Xr are sequentially processed by the encoder Φr, the translator Γr→r, and the decoder Ψr, yielding reconstructed output 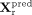 . The reconstruction loss
. The reconstruction loss 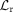 is computed using mean squared error (MSE):
is computed using mean squared error (MSE):
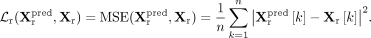
To regularize the latent representation, we further apply the evidence lower bound (ELBO) loss, which penalizes the Kullback–Leibler
divergence between the latent RNA embeddings 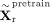 and a standard Gaussian
and a standard Gaussian 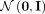 ,
,
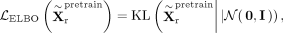 and the total loss for RNA pretraining is a weighted combination of reconstruction and ELBO losses:
and the total loss for RNA pretraining is a weighted combination of reconstruction and ELBO losses:
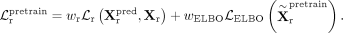
In this work, both loss weights are set to one (i.e., wr = wELBO = 1) to assign equal importance to reconstruction fidelity and latent space regularization, which we found to provide stable and effective pretraining across all data sets. We optimize the model using the Adam optimizer with a learning rate of 0.001. Training proceeds for up to 100 epochs, with early stopping based on validation loss and a patience of 50 epochs.
For DNAm modality, the pretraining procedure mirrors that of RNA, with two key differences: The encoder–decoder pair (Φm, Ψm) and translator Γm→m are used, and the reconstruction loss is replaced by binary cross-entropy (BCE) loss 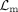 , which can be calculated as
, which can be calculated as
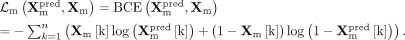
During the integrative training phase, we introduce adversarial training to align cross-modality embeddings. The adversarial
discriminator loss 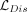 is computed using BCE loss with label smoothing. Specifically, to introduce label smoothing in adversarial training, we replace
hard binary labels with soft labels. For positive examples (i.e., real embeddings), we sample labels uniformly from the range
[0.8, 1.0] instead of assigning a fixed label of one. For negative examples (i.e., translated embeddings), we sample labels
uniformly from the range [0.0, 0.2], instead of using a fixed label of zero (Supplemental Text S7). This technique helps stabilize training by preventing the discriminator from becoming overconfident and encourages better
generalization. The complete discriminator loss combines four components:
is computed using BCE loss with label smoothing. Specifically, to introduce label smoothing in adversarial training, we replace
hard binary labels with soft labels. For positive examples (i.e., real embeddings), we sample labels uniformly from the range
[0.8, 1.0] instead of assigning a fixed label of one. For negative examples (i.e., translated embeddings), we sample labels
uniformly from the range [0.0, 0.2], instead of using a fixed label of zero (Supplemental Text S7). This technique helps stabilize training by preventing the discriminator from becoming overconfident and encourages better
generalization. The complete discriminator loss combines four components:
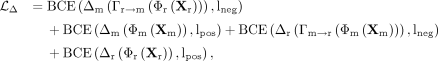 where lneg and lpos represent the smoothed negative and positive labels. Specifically, we adopt an SGD optimizer with a learning rate of 0.001.
where lneg and lpos represent the smoothed negative and positive labels. Specifically, we adopt an SGD optimizer with a learning rate of 0.001.
Following each update of the discriminators, we iteratively train the encoders, translators, and decoders to optimize the full model. In each iteration, we compute both modality-specific reconstructions and cross-modality translations. Specifically, the RNA reconstruction is obtained as Ψr(Γr→r(Φr(Xr))), and the RNA-to-DNAm translation is obtained as Ψm(Γr→m(Φr(Xr))). Similarly, the DNAm reconstruction is given by Ψm(Γm→m(Φm(Xm))), and the DNAm-to-RNA translation is given by Ψr(Γm→r(Φm(Xm))).
To optimize the full model, we define a total loss that integrates four components: reconstruction loss, translation loss,
ELBO loss, and adversarial loss derived from the updated discriminators. The reconstruction loss encourages the model to accurately
reproduce both original and translated data. The translation loss is used to measure the model's performance in the translation
task, ensuring that the translated data are as close as possible to the target data. The ELBO loss regularizes the latent
spaces of RNA and DNAm embeddings by minimizing the KL divergence between the learned distributions and a standard Gaussian
prior. The adversarial loss, based on the output of the updated discriminators, promotes alignment between translated embeddings
and the true data distribution. The overall loss function is formulated as
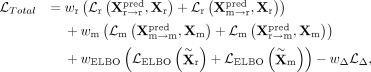 where
where 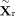 and
and 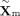 denote the shared embeddings of RNA and DNAm, respectively, and
denote the shared embeddings of RNA and DNAm, respectively, and 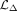 is re-estimated by the updated discriminator for each iteration. wr, wm, wELBO, and
is re-estimated by the updated discriminator for each iteration. wr, wm, wELBO, and 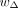 are assigned to one, two, 20/lr + 20/lm, and one, respectively. We train the encoders, translators, and decoders for up to 200 epochs using the Adam optimizer with
a learning rate of 0.0001, employing early stopping with a patience of 50 epochs.
are assigned to one, two, 20/lr + 20/lm, and one, respectively. We train the encoders, translators, and decoders for up to 200 epochs using the Adam optimizer with
a learning rate of 0.0001, employing early stopping with a patience of 50 epochs.
The data augmentation strategy of scBOND-Aug
In real-world scenarios, the number of paired cells is often limited owing to high experimental cost and technical challenges. Such data scarcity can severely degrade model performance and lead to overfitting. To improve the robustness and generalization ability of cross-modality translation under limited training data, we propose scBOND-Aug, an extension of scBOND that incorporates a data augmentation strategy: simulate additional training pairs by recombining features across modalities within the same cell type. The core idea behind this strategy is that two cells from the same cell type share similar biological characteristics, allowing their distinct omics profiles to be aligned with each other (Fig. 1B).
Specifically, training cells are first grouped according to biological annotations (e.g., cell types). Within each group,
we generate synthetic paired profiles by utilizing profiles from one modality with unmatched profiles from the other modality
through multiple iterations of random reordering. This augmentation enlarges the training set size by approximately threefold,
enabling the model to learn more robust intermodal mappings while preserving intraclass consistency. Specifically, for each
cell type c, we denote the RNA and DNAm profiles as 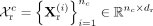 and
and 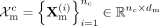 , where nc is the number of cells, and dr, dm are the feature dimensions of RNA and DNAm modalities, respectively. To generate augmented training data, we apply a random
permutation to the cellular indices of scDNAm profiles within each cell type, pairing them with the scRNA-seq profiles from
the same type. This operation permutes the correspondence between individual cells and their DNAm profiles at the cellular
level, preserving the integrity of methylation values across genomic regions within each cell. Formally, a synthetic pair
, where nc is the number of cells, and dr, dm are the feature dimensions of RNA and DNAm modalities, respectively. To generate augmented training data, we apply a random
permutation to the cellular indices of scDNAm profiles within each cell type, pairing them with the scRNA-seq profiles from
the same type. This operation permutes the correspondence between individual cells and their DNAm profiles at the cellular
level, preserving the integrity of methylation values across genomic regions within each cell. Formally, a synthetic pair
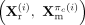 is formed by applying a permutation πc to the DNAm index set {1, 2, …, nc}. This results in a set of label-consistent yet nonaligned synthetic pairs that preserve biological coherence while increasing
the diversity of the training set. Importantly, because all pairings are constrained within the same cell type, the augmented
pairs maintain cell identity fidelity, enabling the model to generalize better without introducing type mismatch noise. This
design ensures that scBOND-Aug remains effective in limited training samples while preserving meaningful biological structure
across modalities.
is formed by applying a permutation πc to the DNAm index set {1, 2, …, nc}. This results in a set of label-consistent yet nonaligned synthetic pairs that preserve biological coherence while increasing
the diversity of the training set. Importantly, because all pairings are constrained within the same cell type, the augmented
pairs maintain cell identity fidelity, enabling the model to generalize better without introducing type mismatch noise. This
design ensures that scBOND-Aug remains effective in limited training samples while preserving meaningful biological structure
across modalities.
Data collection and preprocessing
To comprehensively evaluate the performance and generalizability of scBOND across diverse biological contexts and experimental protocols, we curated single-cell multiomic data sets encompassing different species, tissues, and sequencing technologies. In all data sets, scRNA-seq and scDNAm profiles were jointly measured from the same individual cells, enabling reliable ground truth for cross-modality evaluation. Specifically, we obtained the MouseEmbryo data set generated using single-cell nucleosome, methylome, and transcriptome sequencing (scNMT-seq), which profiles 1082 mouse embryonic cells across four key developmental stages: embryonic day (E) 4.5, E5.5, E6.5, and E7.5 (Argelaguet et al. 2019); this data set is available at the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE121708. Additionally, we obtained the HumanBrainA data set, which was generated using the snmCAT-seq protocol. It comprises 4013 nuclei isolated from postmortem human frontal cortex, offering a complex and heterogeneous adult tissue context with a broad diversity of neuronal and glial cell types (Luo et al. 2022), and is accessible at GEO under the accession number GSE140493. Finally, to reconstruct the missing omics profiles from RNA-only data and investigate transcriptomic and epigenetic regulation, we accessed the HumanBrainB-RNA data set, which comprises 3142 cells from the human cerebral cortex, spanning eight distinct cell types, and is available at https://datasets.cellxgene.cziscience.com/b8bedb33-4f07-41b4-9656-8c428d549c94.h5ad.
We applied modality-specific preprocessing strategies to the scRNA-seq and scDNAm profiles to account for their distinct biological
and technical characteristics. For the scRNA-seq profiles, we first normalized the raw count matrix Xr ∈ ℝn×g, where n is the number of cells and g is the number of genes, to account for sequencing depth differences across cells. For each cell i, we computed the library size 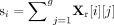 , and normalized the counts as
, and normalized the counts as
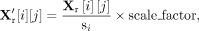 where we set the scaling factor to 1 × 104. We then applied a log1p transformation to stabilize variance:
where we set the scaling factor to 1 × 104. We then applied a log1p transformation to stabilize variance:
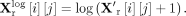
Finally, we selected the top 3000 highly variable genes (HVGs) based on normalized dispersion for analysis. For the scDNAm profiles, we first constructed a cell-by-region scDNAm data matrix from the raw data, in which each entry represents the average methylation level across CpG sites within a 10 kb genomic window for a given cell (Supplemental Text S8; Danese et al. 2021). We then applied feature selection to exclude low-coverage genomic regions, retaining only those genomic regions that were covered in at least seven-tenths of a percent of cells, followed by imputation of missing values using the median to preserve data integrity (Tang et al. 2025). Finally, we performed min-max normalization to scale the features to a common range, facilitating comparability across cells.
Visualization and cell clustering
For data visualization, we first reduced the dimensionality of the raw or translated profiles to 50 using principal component analysis (PCA), which is a widely used dimensionality reduction method in single-cell analysis (Chen et al. 2021; Li et al. 2024a; Tang et al. 2025). We then applied either UMAP or t-SNE to further reduce the dimensionality to two for visualization. In the resulting plots, cells were colored according to their annotated cell-type labels. For cell clustering, we similarly reduced the dimensionality of the translated data to 50 using PCA, followed by Leiden clustering (Traag et al. 2019) with the default resolution to assign cluster labels.
Evaluation metrics
To evaluate the performance of scBOND and baseline methods in preserving cellular heterogeneity during cross-modality translation, we employed four widely adopted metrics (Cao et al. 2024; Li et al. 2024a; Tang et al. 2024): AMI, ARI, HOM, and NMI. Higher values of these metrics indicate better clustering results, reflecting more accurate preservation of cellular heterogeneity. More details are provided in Supplemental Text S1.
Baseline methods
We evaluated the performance of scBOND in comparison with two baseline methods: scCross and MAPLE. For scCross, we followed the preprocessing pipeline and default hyperparameter settings recommended in its official repository (https://github.com/mcgilldinglab/scCross) for cross-modality translation between DNA methylation and gene expression (Yang et al. 2024). MAPLE is a supervised learning framework designed to predict gene activity scores from scDNAm data (Uzun et al. 2021). As such, it supports unidirectional cross-modality translation, specifically from DNA methylation to gene expression. We implemented MAPLE according to the official tutorial provided on GitHub (https://github.com/tanlabcode/MAPLE.1.0). MAPLE offers four pretrained models, and we used the model that demonstrated the best performance in the original MAPLE study for comparison.
Downstream analysis
To assess the biological relevance of the translated scRNA-seq profiles, we performed functional enrichment analyses based on the KEGG (Kanehisa and Goto 2000) and GO (Gene Ontology Consortium 2004) databases. Although these analyses provided insights into transcriptional profiles, additional layers of functional interpretation were necessary for the translated scDNAm profiles. Therefore, to comprehensively evaluate the biological significance of the translated scDNAm profiles, we conducted complementary downstream analyses: expression enrichment analysis using SNPsea (Slowikowski et al. 2014), and partitioned heritability analysis using partitioned LDSC (Finucane et al. 2015). Detailed descriptions of these analyses are provided in Supplemental Text S2.
Calculation of promoter-proximal DNA methylation level
To calculate the promoter-proximal DNA methylation level, we first annotate the chromosomal regions in the data using ChIPseeker (Yu et al. 2015), which helps determine the distance between each chromosomal region and the gene promoter. We then focus on the chromosomal regions within 2000 bp upstream of and downstream from the promoter and compute the average DNA methylation level within these regions as the promoter-proximal DNA methylation for each gene.
Code availability
The MIT-licensed scBOND software, including detailed documents and tutorials, is freely available at GitHub (https://github.com/BioX-NKU/scBOND) and as Supplemental Code. Comprehensive and automatically generated API documentation can be accessed via the read the docs site at https://scbond.readthedocs.io/en/latest/. Additionally, a stable, versioned release of the software is archived on Zenodo (https://zenodo.org/records/17699419).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (62473212, S.C.), the Young Elite Scientists Sponsorship Program by China Association for Science and Technology (CAST) (2023QNRC001, S.C.), the Fundamental Research Funds for the Central Universities (050-63253077, S.C.), and the National Undergraduate Training Program for Innovation and Entrepreneurship.
Author contributions: S.C. conceived and supervised the project. K.L., C.J., and S.L. designed, implemented, and validated scBOND and wrote the manuscript. Y.G. and D.H. helped analyze the results.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.281350.125.
-
Freely available online through the Genome Research Open Access option.
- Received August 24, 2025.
- Accepted March 2, 2026.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








