
Early feature extraction drives model performance in high-resolution chromatin accessibility prediction
- Aayush Grover1,2,
- Till Muser3,
- Liine Kasak1,
- Lin Zhang3,
- Ekaterina Krymova3 and
- Valentina Boeva1,2,4
- 1Department of Computer Science, ETH Zurich, 8092 Zurich, Switzerland;
- 2SIB Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland;
- 3Swiss Data Science Center, EPF Lausanne and ETH Zurich, 8092 Zurich, Switzerland;
- 4Institut Cochin, Inserm U1016, CNRS UMR 8104, Université Paris Cité, 75014 Paris, France
Abstract
Fine-grained prediction of chromatin accessibility from DNA sequence is a foundational step in modeling gene expression changes resulting from sequence variants. Yet, few methods operate at the resolution necessary to capture subtle effects of single-nucleotide changes. Furthermore, it remains unclear which architectural components, such as residual connections, normalization strategies, or attention mechanisms, drive performance in these high-resolution predictions. To address these knowledge gaps, we systematically evaluate classic architectural choices and introduce ConvNeXt V2 blocks, originally developed for computer vision, as high-resolution feature extractors in deep learning models for genomic data. Integrated into diverse architectures such as convoluted neural networks (CNNs), long short-term memory (LSTM), dilated CNNs, and transformers, ConvNeXt V2 blocks consistently improve performance, leading to similar prediction accuracy across these different model types. This reveals that early feature extraction, rather than downstream architecture, is the primary determinant of prediction accuracy. A comprehensive evaluation of these models on ATAC-seq signal prediction at 4-bp resolution in a cell type–specific manner identifies the ConvNeXt-based dilated CNN as the most robust performer, better preserving the signal’s shape. Our codebase and benchmarks provide practical tools for high-resolution chromatin modeling.
Predicting chromatin accessibility from DNA sequence is one of the central challenges in regulatory genomics, with applications including the interpretation of noncoding genetic variants and the decoding of regulatory element architecture (Zhou and Troyanskaya 2015; Kelley et al. 2018; Avsec et al. 2021a; Pampari et al. 2024). Accurate, high-resolution maps of chromatin accessibility are essential for downstream tasks such as identifying regulatory elements, modeling 3D chromatin organization, and predicting gene expression (Thibodeau et al. 2021; Karbalayghareh et al. 2022; Tan et al. 2023; Zhang et al. 2023; Grover et al. 2024). Although experimental assays such as FAIRE-seq (Giresi et al. 2007), DNase-seq (Song and Crawford 2010), and ATAC-seq (Buenrostro et al. 2015) provide high-resolution measurements of chromatin accessibility in specific cellular contexts, they are resource-intensive and often unavailable for specific genetic variants. In the absence of such experimental assays, neural network–based predictions offer a reliable and efficient alternative for estimating changes in chromatin accessibility induced by the genetic variants in a cell type–specific manner.
Numerous deep learning approaches have been proposed to predict chromatin accessibility from DNA sequence. Earlier methods (Zhou and Troyanskaya 2015; Kelley et al. 2016; Quang and Xie 2016; Min et al. 2017; Guo et al. 2020) treated this as a binary classification task, labeling regions as either open or closed. Some of the later methods, such as Deopen (Liu et al. 2018) demonstrated the performance of their model in predicting the maximum intensity of chromatin accessibility corresponding to the input DNA segment. Subsequently, Basenji (Kelley et al. 2018) and Enformer (Avsec et al. 2021a) showed that it was possible to predict the intensity of the chromatin accessibility signal corresponding to the input DNA sequence with a relatively lower resolution (128bp) by binning the signal tracks. Although these methods are highly accurate, this coarse formulation (i.e., predicting chromatin accessibility profiles at 128bp or lower resolution) is still insufficient to study local changes in chromatin accessibility caused by point mutations. Moreover, it hinders their applicability for downstream tasks, including prediction of transcription factor binding (Cazares et al. 2023), cell type–specific chromatin structure (Tan et al. 2023; Yang et al. 2023; Zhang et al. 2023; Gao et al. 2024; Grover et al. 2024), and gene expression (Karbalayghareh et al. 2022; Zhang et al. 2023). Therefore, there is a need for the development of deep learning methods that can accurately predict high-resolution chromatin accessibility signals.
Recently introduced ChromBPNet (Pampari et al. 2024) predicts chromatin accessibility at base-pair resolution using dilated convolutional networks, representing an advance in resolution capabilities. However, it is not extensively compared against other model architectures, like recurrent neural networks and transformers, leaving questions about optimal architectural choices unanswered. GOPHER (Toneyan et al. 2022) was introduced as an extensive benchmark that compared various deep learning methods, such as Basenji (Kelley et al. 2018) and BPNet (Avsec et al. 2021b), in their ability to predict chromatin accessibility for different experimental setups. Both of these methods use dilated convolutional neural networks (dCNNs) to capture long-range information from the input DNA sequence. These methods were evaluated against a convolutional neural network (CNN) baseline, highlighting the predictive prowess of dCNN-based methods over CNN-based methods. However, recurrent neural network-based methods and transformer-based methods are not explored in this benchmark, creating a gap in understanding which architectural paradigms perform best for high-resolution chromatin accessibility prediction.
In this work, we present a systematic evaluation of deep learning models for predicting ATAC-seq signals at 4-bp resolution from DNA sequence. We begin by extensively tuning three commonly used architectures—CNNs, recurrent neural networks (specifically, long short-term memory, LSTM), and dCNNs, for high-resolution chromatin accessibility prediction. Building on this, we incorporate ConvNeXt V2 (Woo et al. 2023) blocks, originally developed for vision tasks, as feature extractors across all architectures and introduce a novel transformer-based model tailored for this task. We evaluate each model’s performance on accessible regions and genome-wide, assess robustness to input shifts, examine interpretability, and test sensitivity to single-nucleotide variants (SNVs) using cancer patient data. This benchmark reveals how architectural choices impact predictive accuracy, robustness, and variant interpretation, providing a practical foundation for future development of deep learning methods in regulatory genomics.
Results
Designing a deep-learning model benchmark for high-resolution ATAC-seq prediction
We evaluated four categories of deep learning architectures—CNNs, recurrent neural networks (specifically, LSTMs), dilated CNNs (dCNNs), and transformers, based on their ability to predict ATAC-seq signal at high resolution. To ensure a fair comparison, we standardized the experimental setup across models: each received a 2048-bp one-hot encoded DNA sequence as input and was trained to predict the chromatin accessibility signal at 4-bp resolution across the central 1024-bp region (Fig. 1). This resolution was chosen because transcription factor binding motifs typically span more than 4 bp (Stewart et al. 2012), making it sufficient to capture relevant regulatory variation. The 2048-bp input window also provides adequate genomic context for the models to learn both local and distal sequence features.
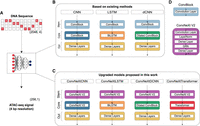
Overview of the benchmarking setup. (A) Models are compared based on their ability to predict experimental ATAC-seq at 4-bp resolution from an input of 2048-bp long DNA sequences. (B) Models based on existing methods include CNN, LSTM, and dCNN-based architectures. Each model has a convolution-based stem to extract genomic features. (C) Models proposed in this work, including a transformer-based architecture. The new models use ConvNeXt V2 blocks to effectively extract features from the input DNA sequence. (D) ConvBlock uses a single convolutional layer whereas ConvNeXt V2 block additionally uses dense layers, layer normalization and Global Response Normalization (GRN).
We began by benchmarking architectures used in existing methods, broadly categorized into CNNs (Zhou and Troyanskaya 2015), LSTMs (Min et al. 2017; Liu et al. 2018), and dilated CNNs (Kelley et al. 2018; Pampari et al. 2024) (Fig. 1A,B). Because these models were originally developed for different prediction tasks and resolutions, we conducted an extensive hyperparameter search to adapt each to our standardized 4-bp resolution setup. Next, to improve feature extraction, we replaced standard convolutional layers in the stem of all three architectures with transformer-inspired ConvNeXt V2 blocks, originally designed for vision tasks but well-suited for capturing local and global genomic features (Woo et al. 2023) (Fig. 1C,D).
Previous studies have shown that transformer architectures can outperform convolution-based models in capturing long-range dependencies in genomic sequences (Avsec et al. 2021a; Ji et al. 2021; Yang and Ma 2023; Dalla-Torre et al. 2025). However, their application to high-resolution chromatin accessibility prediction has not been explored. To address this limitation, we designed a novel transformer-based model incorporating rotary positional embeddings (Su et al. 2024) (Fig. 1C).
The detailed architecture of each model can be found in Supplemental Fig. S1. Together, these models form a comprehensive benchmark across four deep learning architecture families.
Improving model prediction accuracy with ConvNeXt-based feature extraction
Each model was extensively trained and tested on data from eight distinct data sets: four established human cell lines (GM12878, K562, IMR-90, and HepG2) and four cancer patient samples (two colon adenocarcinomas [COAD] and two kidney renal papillary cell carcinomas [KIRP]). We implemented a cell type–specific training approach to account for the distinct regulatory mechanisms that govern chromatin accessibility in different cellular contexts. Specifically, separate models were trained and evaluated independently for each cell line or cancer patient sample, without sharing parameters across cell types. To ensure statistical robustness, we implemented a 5-fold cross-validation strategy for each model within each cell type or tumor sample. The validation folds were stratified by chromosomes, allowing us to evaluate model performance on entirely unseen genomic regions while maintaining the unique characteristics of each cell type.
Within the test chromosomes, we evaluated model performance using two complementary measurements: prediction accuracy on the reported ATAC-seq peaks and prediction accuracy on entire genomic regions. This dual assessment allowed us to benchmark both the models’ ability to accurately characterize highly accessible chromatin regions (peak-focused evaluation) and their capacity to distinguish between accessible and inaccessible regions across the complete chromosomal landscape (genome-wide evaluation).
Among the architectures employed in existing methods (CNNs, LSTMs, and dCNNs), dCNNs consistently outperformed others in both peak-focused and genome-wide settings (Fig. 2A, Supplemental Fig. S2). However, upon incorporating ConvNeXt V2 blocks in the stem of these architectures, all models demonstrated similar high correlations with the experimental ATAC-seq values in the accessible regions as well as on whole genome (average Pearson’s R ≥ 0.7) (Fig. 2B, Supplemental Fig. S2).
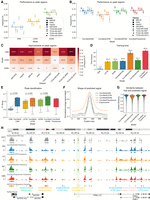
Model comparison across data sets. (A) Pearson’s correlation between true and predicted ATAC-seq signals in peak regions across eight distinct data sets for state-of-the-art models. (B) Performance comparison among the four models proposed in this work (ConvNeXtCNNs, ConvNeXtLSTMs, ConvNeXtDCNNs, and ConvNeXtTransformers) for the ATAC-seq peak regions stratified by cell lines and primary tumor samples. Each point depicts a test chromosome. The black dashed line shows the average performance of a model across all data sets and chromosomes. (C) Improvements of the new ConvNeXt-based methods proposed in this work for the ATAC-seq peaks as compared to existing methods. The significance is calculated with a two-sided Mann–Whitney U test on Pearson’s R calculated for each test chromosome. (****) P ≤ 0.0001, (***) P ≤ 0.001, (**) P ≤ 0.01, (*) P ≤ 0.05, ns: P > 0.05. The Δ Pearson’s R is calculated as the difference between mean Pearson’s R across all chromosomes for a ConvNeXt-based method and the corresponding existing method. (D) The total training time (in hours) of each method on a single RTX2080Ti GPU averaged across eight data sets. The error bar shows 95% confidence interval. Median training time is mentioned over each bar. (E) The F1 score is calculated for all the methods to evaluate the retrieval of ATAC-seq peak calls from the predictions. Each point represents one of the eight data sets. P-values are calculated using the Wilcoxon signed-rank test. (F) The average shape of the predicted ATAC-seq signal is compared against the experimental shape in the ATAC-seq peak regions of the GM12878 cell line. (G) Cosine similarity between experimental ATAC-seq and predicted ATAC-seq in the peak regions of the GM12878 cell line. (H) An example of predicted signal versus the experimental signal around the FMNL1 gene in the HepG2 cell line. A distal enhancer, discovered as a peak only based on the predictions of the ConvNeXtDCNN method, is highlighted.
The novel transformer-based architecture, ConvNeXtTransformer, that we also introduced in this evaluation, performed comparably to the ConvNeXtCNN and ConvNeXtDCNN models. We also explored the xLSTM architecture, a more recent development than the transformer, which offers improved memory efficiency for sequential data processing (Beck et al. 2024). However, when tested in our framework, ConvNeXt-xLSTM was outperformed by ConvNeXtLSTM, suggesting that its recurrent, long-range dependency modeling impaired the capture of local context critical for ATAC-seq prediction compared to ConvNeXtTransformer’s attention-based approach (Supplemental Fig. S3). Overall, incorporating ConvNeXt blocks into the stem consistently improved the performance of the existing models across all data sets, although the effect size was modest when comparing ConvNeXtDCNN to the dilated CNN (Fig. 2C, Supplemental Fig. S2). This underscores the better effectiveness of ConvNeXt blocks in capturing genomic features with local and global context.
Adding ConvNeXt blocks to the stem, however, increased training times, with ConvNeXtDCNN requiring approximately 1.7 more hours as compared to dCNN (Fig. 2D). Despite this, all models exhibited fast inference, predicting ATAC-seq signal across the entire Chromosome 17 in under 2 min, underscoring their practical utility for variant-effect prediction tasks (Supplemental Fig. S2).
Assessing peak detection and signal shape fidelity in predicted ATAC-seq tracks
Accurate identification of ATAC-seq peaks is essential for deep learning models to generate outputs suitable for downstream tasks. We performed peak calling on the predicted ATAC-seq signals from each model (Methods) and compared the resulting peaks against experimental data. For robust evaluation, we used the F1 score, which accounts for class imbalance. ConvNeXt-based models outperformed existing baselines, with ConvNeXtDCNN achieving the highest F1 score (Fig. 2E).
To assess how well these models capture signal shapes around the peak maxima, we plotted the min-max normalized predicted ATAC-seq signal averaged across all peaks, data sets, and training folds (Fig. 2F). Whereas ConvNeXtLSTM tended to overpredict the signal at peak boundaries, producing smoother profiles, other ConvNeXt-based methods closely matched the experimental ATAC-seq signal, accurately capturing nucleosome positioning around peak centers. This qualitative observation was further supported quantitatively, as the other ConvNeXt-based methods showed higher cosine similarity with the experimental ATAC-seq signal than ConvNeXtLSTM (Fig. 2G).
Finally, we visualized the chromatin accessibility signals generated by each ConvNeXt-based model on test chromosomes and their corresponding peaks (Fig. 2H). The predicted signals and peaks co-localized with many known promoters and enhancers (Moore et al. 2020), corroborating quantitative overlap analyses with ground-truth accessible regions and confirming the high fidelity of predicted peak shapes (Fig. 2E–H). Notably, some enhancers were uniquely identified by specific methods; for example, an enhancer in the gene body of FMNL1-DT in HepG2 cells was marked as accessible only by ConvNeXtDCNN (Fig. 2H). Overall, ConvNeXtDCNN was the top-performing method based on its peak-calling accuracy and correlation with experimental data, with other ConvNeXt-based models also showing strong performance.
Evaluating model robustness to shifts in the DNA sequence inputs
A critical property of reliable genomic models is robustness to input shifts, that is, the ability to generate consistent predictions regardless of whether the input sequence is precisely centered on a peak maximum or slightly offset. To quantify this property, we performed a systematic robustness test on all ConvNeXt-based models, measuring the variation in predicted ATAC-seq signal across common genomic regions when the input window is shifted (Toneyan et al. 2022) (Supplemental Fig. S4). For each ATAC-seq peak across all test chromosomes and training folds, we generated 17 different input shifts and calculated the coefficient of variation (CoV) across each 4-bp bin in the commonly predicted region. Lower CoV corresponded to higher robustness, indicating more consistent predictions despite slight shifts in input positioning.
The position-stratified CoV, averaged across all peaks in different data sets, showed that all methods displayed higher robustness at peak centers (Fig. 3A). The periodic variation pattern observed in ConvNeXtCNN can be attributed to the inherent sensitivity of convolutional networks to random input shifts (Zhang 2019).

Evaluation of model robustness to input shifts. (A) Position-stratified CoV is calculated for all the methods for the ATAC-seq peak regions. The variation is averaged across all the peaks in unseen chromosomes of the eight data sets. (B) The mean CoV measured for ATAC-seq peak regions across the eight data sets used in this study. Each point represents mean CoV for a chromosome unseen by the model during training.
Among all the methods, ConvNeXtDCNN consistently emerged as the most robust method across all data sets, followed by ConvNeXtLSTM (Fig. 3B). The robustness of ConvNeXtDCNN was further confirmed by a low variation score observed using the entire unseen chromosomes of each data set (Supplemental Fig. S4).
Interpreting sequence features underlying model predictions
To gain mechanistic insights into ConvNeXt-based architectures, we performed feature attribution using integrated gradients (Sundararajan et al. 2017) followed by motif discovery (Methods). This analysis revealed that the ConvNeXt-based models consistently learned sequence motifs corresponding to transcription factors with known roles in regulating chromatin accessibility in each cellular context (Fig. 4A–D). For example, in K562, motifs of erythroid regulators such as GATA1 and MAFK were highly enriched, whereas in HepG2, motifs of hepatic factors, including HNF4G and FOXA2 predominated. Similar enrichment patterns were observed in GM12878 and IMR-90, where motifs of transcription factors such as SP1 and CTCF emerged prominently. The majorly identified transcription factors for each cell type were consistent with the findings of ChromBPNet (Pampari et al. 2024) (Supplemental Fig. S5). These findings indicate that the ConvNeXt V2 block effectively captures cell type–specific regulatory features that underlie chromatin accessibility.
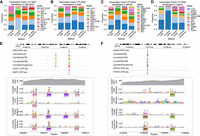
Interpreting sequence features underlying model predictions. (A–D) Proportion of identified transcription factor motifs in different cell lines: (A) GM12878, (B) K562, (C) IMR-90, and (D) HepG2. (E) GATA1 and MAFK motifs identified by all ConvNeXt-based methods for an ATAC-seq peak region in the K562 cell line. (F) Similarly, HNF4G and FOXA2 motifs identified in an ATAC-seq peak region in the HepG2 cell line.
We next examined attribution profiles at individual genomic loci to evaluate how these motifs contribute to local predictions. At a particular locus on Chromosome 4 in K562 cells, ConvNeXtCNN, ConvNeXtDCNN, and ConvNeXtTransformer strongly highlighted GATA1 and MAFK motifs in close agreement with experimental ChIP-seq binding, whereas ConvNeXtLSTM produced weaker and less localized attributions (Fig. 4E). Similarly, in HepG2, ConvNeXt-based methods accurately recovered HNF4G and FOXA2 motifs at regulatory elements validated by ChIP-seq, with sharper correspondence between predicted and experimental accessibility profiles compared to ConvNeXtLSTM (Fig. 4F). These examples illustrate that ConvNeXt architectures not only identify relevant transcription factor motifs but also integrate them into coherent accessibility predictions at individual regulatory sites.
Together, these global and locus-specific analyses demonstrate that the ConvNeXt V2 models learn biologically meaningful sequence features, including transcription factor motifs that are central to cell type–specific chromatin accessibility.
Evaluating model sensitivity to genetic point variants
Experimentally profiling chromatin accessibility for every genetic variant is prohibitively resource-intensive, highlighting the need for accurate in silico prediction of variant effects. To evaluate the ability of benchmarked models to capture the effects of genomic variants, we obtained SNVs from four cancer patients, two with colon adenocarcinoma and two with kidney renal papillary cell carcinoma, using data from the ICGC (Zhang et al. 2019a), and estimated variant-induced changes on chromatin accessibility based on ATAC-seq read counts (Fig. 5A). A total of 1025 SNVs were analyzed after filtering out variants with minimal effect, defined as those with odds ratios between 0.67 and 1.33. For this task, all models were retrained on the full genome, excluding only regions containing SNVs. Model predictions were then generated using 2048-bp SNV-centered input sequences, where the reference and variant sequences differed by a single nucleotide.
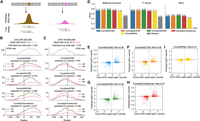
Evaluating model ability to accurately predict effects of single-nucleotide variants on chromatin accessibility. (A) Experimental setup to test each model’s ability to predict the change in chromatin accessibility between the reference DNA sequence (left) and a genomic variant (right). (B,C) Examples of allele-specific chromatin accessibility predictions for the reference allele and a genomic variant in Chromosomes 2 and 5, respectively. WGS VAF: Whole genome sequencing variant allele frequency; R, V: ATAC-seq total reference reads and variant reads, respectively. The model predictions are smoothed using a 1D Gaussian kernel with σ = 8. (D) ConvNeXt-based models are compared against ChromBPNet based on their ability to predict the correct directionality of change in accessibility between reference and variant. A total of 1025 variants are considered across all cancer patients included in this study. Balanced accuracy, F1 score, and Matthews correlation coefficient (MCC) are used as metrics. A random baseline is included to demonstrate the improvements in metrics across all models proposed in this work. (E–H) Scatter plots showing the quantitative change in accessibility by each of the ConvNeXt-based models. (I) Performance of ChromBPNet is also shown for reference. Log odds ratio based on ATAC-seq read counts for reference and variants are compared against log fold change (FC) between model predictions with reference and variant as model inputs and Spearman’s correlation (Rho) is calculated.
We first qualitatively observed the predicted ATAC-seq profiles by each of the ConvNeXt-based models using reference and alternate alleles as input. Although all models generally produced consistent changes in accessibility between the two alleles, ConvNeXtDCNN most accurately recapitulated the fine-grained shape of the predicted signal (Fig. 5B, Supplemental Fig. S6). An exception was observed at an SNV near 149.6 Mb on Chromosome 5, where only the ConvNeXtLSTM and ConvNeXtDCNN models correctly predicted the direction of change, with ConvNeXtDCNN providing a prediction closer to the ground-truth odds ratio (Fig. 5C).
We then assessed the ability of each model to correctly predict the directionality of change in chromatin accessibility between the reference and variant alleles across all SNVs. All models outperformed the state-of-the-art method ChromBPNet (Pampari et al. 2024) and the random baseline, where for each SNV, a value of 0 or 1 was randomly assigned according to the proportion of SNVs with positive and negative effects on experimental ATAC-seq (Fig. 5D). Moreover, ConvNeXtDCNN achieved the highest performance with balanced accuracy of 0.77, F1 score of 0.76, and Matthews correlation coefficient (MCC) of 0.55 (Fig. 5D). Both F1 score and MCC account for class imbalance and are, therefore, more robust metrics for this task.
We next tackled the more challenging task of quantitatively predicting the change in chromatin accessibility induced by an SNV. For this evaluation, the estimated log odds ratio from ATAC-seq read counts for the reference and variant alleles served as the ground truth. This was compared against the log fold change between the ATAC-seq signal predicted by each model for the variant and the reference nucleotide (Methods). ConvNeXtLSTM and ConvNeXtDCNN achieved the highest Spearman’s correlation with the true odds ratio (Rho = 0.48) (Fig. 5E–H), closely followed by ConvNeXtTransformer (Rho = 0.47) and ConvNeXtCNN (Rho = 0.46). All methods outperformed the ChromBPNet method (Fig. 5I), demonstrating that incorporating ConvNeXt-based feature extraction improves sensitivity to sequence variation.
In conclusion, although all models performed well on the task of predicting the effect of SNVs on chromatin accessibility, ConvNeXtDCNN had a slight edge over the other methods.
Discussion
In this work, we compared existing deep learning architectures—CNNs, recurrent neural networks (specifically, LSTMs), and dCNNs, on their ability to predict high-resolution chromatin accessibility signals from genome sequences (Fig. 1B). To improve model performance, we proposed new ConvNeXt-based models: ConvNeXtCNN, ConvNeXtLSTM, and ConvNeXtDCNN. Additionally, we introduced a transformer-based model, ConvNeXtTransformer, to create a more comprehensive set of architectures for this task (Fig. 1C,D).
The ConvNeXt-based models outperformed their traditional counterparts in accurately predicting ATAC-seq signals at 4-bp resolution across eight different cell types and primary tumor samples. Among them, ConvNeXtCNN, ConvNeXtDCNN, and ConvNeXtTransformer most accurately captured the shape of the ATAC-seq peaks (Fig. 2). We further evaluated the robustness of these models to shifts in the DNA sequence inputs. ConvNeXtDCNN emerged as the most robust method, efficiently integrating local and global sequence features through its dilated CNN layers (Fig. 3).
Using the feature attribution measures, we observed that the ConvNeXt-based models (ConvNeXtCNN, ConvNeXtLSTM, ConvNeXtDCNN, and ConvNeXtTransformer) learn biologically meaningful sequence features (Fig. 4). The previously observed Tn5 insertion bias in signal profile prediction was observed only in the ConvNeXtDCNN model when trained and evaluated on the ATAC-seq from HepG2 cells (Pampari et al. 2024). In this experiment, out of all motifs that were considered important by the model based on the attribution scores (Methods), only 3.5% corresponded to the Tn5 motif (Fig. 4D).
Finally, we assessed the model’s ability to predict changes in chromatin accessibility caused by single-nucleotide genomic variants. ConvNeXtLSTM and ConvNeXtDCNN achieved the highest accuracy in predicting the log odds ratio of ATAC-seq reads between variant and reference alleles. ConvNeXtDCNN also best captured the directionality of accessibility changes, accurately distinguishing between increases and decreases in chromatin accessibility upon SNV introductions (Fig. 5). This highlights the sensitivity of the ConvNeXtDCNN model to genomic variants and suggests its potential utility in studying the functional effects of noncoding genetic variants.
Whereas our ConvNeXt-based models demonstrated strong performance in predicting chromatin accessibility and the effects of SNVs, our models were trained in a cell type–specific manner. Therefore, they were optimized for individual cellular contexts rather than generalized across different cell types. Although this design ensures high accuracy within a given cell type, it also means that a new model must be trained for each additional cell type of interest. Future work could explore data-efficient fine-tuning approaches to accelerate model adaptation to new cellular contexts without requiring full retraining.
Overall, our study highlighted the advantages of ConvNeXt-based models in predicting high-resolution chromatin accessibility and regulatory effects of genetic variants. By combining ConvNeXt blocks as genomic feature extractors with CNNs, LSTMs, dilated CNNs, and transformers, we established a comprehensive framework of sequence-based chromatin modeling. Our results demonstrated that ConvNeXtDCNN is not only highly accurate but also robust to input shifts, capture biologically meaningful features, and effective at capturing effects of SNVs on chromatin accessibility, making ConvNeXtDCNN a promising method for genome-wide regulatory analysis.
Methods
ATAC-seq bigWig data
We downloaded the ATAC-seq signal P-value tracks for four cell lines—GM12878, IMR-90, HepG2, and K562, from the ENCODE Consortium (Kagda et al. 2025)—where the experimentally measured ATAC-seq has been preprocessed with the open source processing pipeline (https://doi.org/10.5281/zenodo.211733). These cell lines consist of healthy and cancer cells, thereby creating a diverse data set.
To further diversify our validation data sets, we used ATAC-seq data of two COAD and two KIRP tumors from The Cancer Genome Atlas (TCGA) (Corces et al. 2018). These data sets have been made available by the Genome Data Commons (GDC) Data Portal (Grossman et al. 2016). The choice of these patients was made due to the availability of the data on their somatic mutations and single-nucleotide polymorphisms (SNPs) in the International Cancer Genome Consortium (ICGC) and GDC data portals, respectively (Grossman et al. 2016; Zhang et al. 2019b). Moreover, these patients had the high number of combined SNV calls (>175,000) before applying SNV-filtering (described below) and high tumor purity (Consensus Purity Estimate >0.8) (Aran et al. 2015).
The ATAC-seq BAM files for the primary tumor samples were downloaded from National Cancer Institute’s Genomic Data Commons portal (Heath et al. 2021). Signal P-value bigWig files were then generated using the ENCODE’s ATAC-seq processing pipeline (https://doi.org/10.5281/zenodo.211733). All data sets used in this study are listed in Supplemental Table S1.
Preprocessing
For the ATAC-seq of each cell line, we composed a data set for each cell line or cancer patient where each item is a pair of one-hot encoded DNA sequence of shape (2048, 4) and the corresponding 4-bp resolution ATAC-seq of shape (512). To reduce the ATAC-seq resolution from 1 to 4 bp in the data set, the values were binned using the maximum value.
Each model was trained on whole-genome data from a single cell line or cancer patient sample, excluding input DNA sequences corresponding to genomic locations with unreliable accessibility measurements to reduce noise and biases. Specifically, we excluded input DNA sequences overlapping with blacklisted regions (https://storage.googleapis.com/basenji_barnyard2/hg38.blacklist.rep.bed) or where less than or equal to 35% of the 2048-bp input overlapped with unmappable regions (https://storage.googleapis.com/basenji_barnyard2/umap_k36_t10_l32_hg38.bed). The annotations for blacklisted and unmappable regions of the GRCh38 (hg38) genome were sourced from Kelley et al. (2018). Reverse-complemented DNA sequences were augmented to the training data to enhance model robustness.
During the evaluation, we imposed a tighter constraint on the mappability and only evaluated the input DNA sequences that did not overlap with any unmappable region. Additionally, we used only the predictions corresponding to the central 1024-bp genomic section. This ensured that there was at least 512 bp of genomic context present on either side of any region that we were trying to predict. Each model was trained on the full 512-length predicted output vector, but for evaluation, we restricted the analysis to the central 256 entries to avoid edge effects.
For each model trained on a particular cell line or cancer patient sample, we evaluate the model on whole genome data and on a data set composed of peaks by taking input DNA sequences centered on the MACS2-called ATAC-seq peaks to compare how models perform on high signal regions.
Training details
We did a 5-fold cross validation where the genome was randomly split into five sets of Chromosomes—[1, 11, 13, 20], [2, 10, 14, 19, 21], [3, 12, 16, 17, 22], [6, 7, 9, 15], [4, 5, 8, 18]. For each fold, a model was trained for a maximum of 70 epochs with early stopping, with a patience of 5 epochs based on Pearson’s correlation on validation data.
Poisson negative log-likelihood loss was used to train all models with the AdamW optimizer (Loshchilov and Hutter 2019). We began by linearly increasing the learning rate during the first epoch until we reached a maximum of 1 × 10−3. We then used exponential decay coupled with cosine annealing (Loshchilov and Hutter 2017), restarting at the beginning of every epoch.
Model architectures
All models processed one-hot encoded DNA sequences of 2048 bp and predicted ATAC-seq signals corresponding to the central 1024-bp at 4-bp resolution. Each model is illustrated in Supplemental Fig. S1. The runtime and memory requirements of each model are described in Fig. 2D and Supplemental Fig. S2.
Convoluted neural network
The CNN, inspired by DeepSEA (Zhou and Troyanskaya 2015), was adapted for 2048-bp inputs and outputs. It comprised three convolutional layers with 512, 640, and 768 filters (kernel size 8), each followed by max pooling (window 4) and dropout (rate 0.2) to reduce dimensionality and prevent overfitting. The flattened output fed into two dense layers with 1024 and 512 ReLU-activated units. This architecture excels at detecting local motif patterns, such as transcription factor binding sites, indicative of chromatin accessibility.
Long short-term memory
The recurrent model (LSTM), based on DanQ (Quang and Xie 2016), processed inputs through a convolutional stem with 512 filters (kernel size 26), followed by max pooling (window 4) and dropout (rate 0.2). A bidirectional LSTM layer (Schuster and Paliwal 1997) with 512 units captured long-range dependencies in the sequence. Two dense layers (1024 and 512 ReLU-activated units) produced the final output. The LSTM’s memory enables modeling sequential relationships across the DNA.
Dilated CNN
The dCNN, inspired by Basenji (Kelley et al. 2018) and adapted by ChromBPNet (Pampari et al. 2024) for 2048-bp inputs, used 11 residual blocks. Each block contained two convolutional layers, the first with a dilation rate increasing by a factor of 1.5 across blocks, followed by GELU activation (Hendrycks and Gimpel 2016). A dense layer reduced the channel dimension to 1, and a softplus activation ensured positive outputs. This design captures long-range genomic interactions.
To enhance feature extraction, we replaced traditional convolutional blocks with ConvNeXt V2 blocks (Woo et al. 2023), which offer improved efficiency and expressiveness via depthwise convolutions and layer normalization.
ConvNeXtCNN
This model used a ConvNeXt stem with 128 filters (kernel size 15), followed by three ConvNeXt blocks with 256, 512, and 512 filters (kernel size 15), each applying depthwise convolutions (Chollet 2017). Max pooling (window 4) and dropout (rate 0.2) followed each block. Two dense layers (512 and 32 ReLU-activated units) processed the feature matrix, with a softplus activation ensuring positive outputs.
ConvNeXtLSTM
The ConvNeXtLSTM employed a ConvNeXt stem with 256 filters (kernel size 26), followed by a bidirectional LSTM layer (Schuster and Paliwal 1997) with 256 units to model distal genomic interactions. The final LSTM state fed into two dense layers (256 and 512 ReLU-activated units), with a softplus activation for the output.
ConvNeXtDCNN
Similar to dCNN, the ConvNeXtDCNN used 11 dilated residual blocks but replaced the initial convolutional layer with a ConvNeXt stem (256 filters, kernel size 15). The remaining architecture, including dilation rates and softplus output, mirrored dCNN.
ConvNeXtTransformer
We introduced the first transformer-based model for ATAC-seq prediction. Five ConvNeXt blocks (256 filters, kernel size 15) and pooling layers tokenized the input into 512 sequences at 4-bp resolution. This sequence of 512 tokens was then passed through four transformer blocks. Mirroring the behavior seen in current large-language models (Jiang et al. 2023; Touvron et al. 2023), we observed better performance when using rotary positional encodings (RoPE) (Su et al. 2024) compared to conventional sinusoidal encodings proposed in Vaswani et al. (2017). A dense layer aggregated the (512, C) output across channels, followed by a softplus activation.
ConvNeXt-xLSTM
Lastly, we built an extension of the above methods by using xLSTM in the core of the model. xLSTM is a newly built model that extends LSTM by retaining memory across different states, thereby capturing both local and global contexts (Beck et al. 2024). The input DNA sequence was passed through five ConvNeXt blocks followed by an xLSTM block consisting of three mLSTM and one sLSTM blocks with a kernel size of 4 and 4 heads (Supplemental Fig. S3). The output was then passed through a dense layer with 128 hidden units and softplus activation function.
Hyperparameter tuning
To optimize model performance for ATAC-seq signal prediction, we tuned hyperparameters for each method by maximizing Pearson’s correlation on the validation set of fold 0, comprising held-out chromosomes. For all models, we tested batch sizes of 64, 128, and 256, initial learning rates of 0.005, 0.001, 0.0005, and 0.0001, and a cosine annealing scheduler with warmup ratios of 0.1, 0.25, or 0.5 of total epochs (Loshchilov and Hutter 2017).
For CNN-based methods (CNN and ConvNeXtCNN), we tuned the number of convolutional layers in the core block (2, 3, or 4), the kernel size in the core block (3, 7, or 15), and the number of dense layers in the output block. All combinations of these parameters were tested. Increasing the dense layers from 2 to 3 did not improve performance, so we retained two layers.
For LSTM-based methods (LSTM and ConvNeXtLSTM), we tuned the kernel size of the initial convolutional stem (3, 7, 15, or 26, inspired by DanQ Quang and Xie 2016), the number of bidirectional LSTM layers (1 or 2), the number of dense layers (2 or 3), and their hidden sizes (256, 512, or 1024).
For dilated-CNN-based methods (dCNN and ConvNeXtDCNN), we explored architectures with 7–15 convolutional residual blocks, inspired by Enformer (Avsec et al. 2021a) and ChromBPNet (Pampari et al. 2024). We varied the dilation rate (1–2, in steps of 0.1) and tested three convolutional block configurations within each residual block. These configurations used kernel counts of 128–512 (step size 64), 64–256 (step size 64), or 256–4096 (step size 256), with corresponding kernel sizes of 5–20, 2–5, or 1–4, respectively.
For the ConvNeXtTransformer, we tuned the ConvNeXt stem with 1–4 layers, kernel sizes of 7, 11, 15, or 26, and filter counts of 64, 128, or 256. The core ConvNeXt blocks were tested with 2–6 layers, kernel sizes of 3–11, and inverted bottleneck scales of 0.5, 1, 2, or 4, which adjust channel expansion in ConvNeXt’s depthwise convolutions (Woo et al. 2023). The transformer core was optimized with 2–6 layers and feedforward dimension scales of 0.5, 1, 2, or 4.
Adding mappability information to models
Experimental ATAC-seq signals are influenced by the mappability of the region of interest. Although deep learning models can implicitly learn mappability from DNA sequences, explicitly providing this information can enhance certain models. To achieve this, we predicted mappability values from the initial layers of each model alongside the 4-bp resolution ATAC-seq signal (Supplemental Fig. S7).
The mappability values were calculated as a binary vector based on overlap with Basenji’s unmappable regions (Kelley et al. 2018). Zeros were assigned to the genomic segments overlapping with unmappable regions, and ones were assigned otherwise.
Adding this mappability information stabilized CNN and LSTM models but had no significant effect on dCNN or ConvNeXt-based models (Supplemental Fig. S7). This suggests that architectures capturing long-range contextual information, such as ConvNeXt, may implicitly encode mappability due to their larger receptive fields.
Peak calling
Predicted ATAC-seq signals from each model were saved as bigWig files using the pyBigWig Python package (https://doi.org/10.5281/zenodo.7809144). These were then converted to bedGraphs using bigWigToBedGraph from UCSC Genome Browser utilities (Kent et al. 2010) and sorted by chromosome and start position. Finally, peaks were called from the sorted bedGraphs using MACS2 bdgpeakcall command (version 2.2.9.1) (Zhang et al. 2008) with a minimum score threshold of 1 to filter low-confidence regions.
Variation score
To calculate the robustness of each model to input shifts, we used the variation score as described in Toneyan et al. (2022). For each 512 bp region in our test data set, we constructed 17 distinct 2048 bp long input sequences such that the output
corresponding to each input contains the predictions for the selected 512-bp region. Using these 17 predictions, the coefficient
of variation was calculated for each 4-bp bin; that is, for each bin we calculated the mean and standard deviation of the
predictions. The CoV for a given bin was then defined as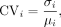 where σi and μi denote the standard deviation and mean, respectively, of the 17 predictions for bin i.
where σi and μi denote the standard deviation and mean, respectively, of the 17 predictions for bin i.
Finally, the variation score was obtained by taking the average CoV across all the bins. For the whole-genome evaluation, this was repeated for all the 512-bp regions of the test chromosomes whereas for peak-centered evaluation, only those 512-bp regions were selected that contained an ATAC-seq peak.
Motif discovery
To interpret sequence features underlying model predictions, we computed base-resolution feature attributions using integrated
gradients (Sundararajan et al. 2017). Attribution scores were calculated with respect to the input DNA sequence for each prediction. Following the approach described
in Pampari et al. (2024), the attribution of each input feature was defined as its effect on the weighted sum of the predicted profile. Specifically,
for the predicted profile 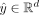 , the attribution of each input nucleotide was calculated for the target
, the attribution of each input nucleotide was calculated for the target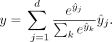 For computational efficiency, attributions were calculated on 30,000 randomly sampled peaks per cell line. Integrated gradients
were implemented with the PyTorch Captum library (Kokhlikyan et al. 2020), using 50 interpolation steps and 20 dinucleotide-shuffled sequences as the baseline.
For computational efficiency, attributions were calculated on 30,000 randomly sampled peaks per cell line. Integrated gradients
were implemented with the PyTorch Captum library (Kokhlikyan et al. 2020), using 50 interpolation steps and 20 dinucleotide-shuffled sequences as the baseline.
The resulting attribution or importance maps were processed with TF-MoDISCo Lite (Shrikumar et al. 2018) using “-n 100,000 -w 500” parameters to cluster recurring 100,000 high-attribution sequence patterns into representative motifs. Consensus motifs were generated with the tfmodisco-lite report workflow, using representative motifs with Tn5 variants obtained from the ChromBPNet repository (https://github.com/kundajelab/chrombpnet/blob/master/chrombpnet/data/motifs.meme.txt). The discovered motifs and their associated transcription factors were then reported to highlight the biological relevance of the features captured by each model.
SNV filtering
We define a single-nucleotide variant (SNV) as either a somatic mutation or a SNP. For the four primary tumor samples, we obtained somatic mutation calls from the study of pan-cancer analysis of whole genome (PCAWG) (Goldman et al. 2020). The SNP calls were obtained from GDC data portal (Grossman et al. 2016).
For each patient, the SNVs were filtered to retain only those SNVs that significantly changed the chromatin accessibility profiles. First, somatic mutations that did not pass the quality control measures of PCAWG were removed. Similarly, we only selected those SNPs that had a heterozygous genotype and a high confidence score (uncertainty value <0.1). The filtered files were then combined, and total read counts from the whole genome sequencing (WGS) and ATAC-seq experiments were calculated for the reference and variant alleles for each SNV. Using these values, the variant allele frequency (VAF) for each SNV was calculated, giving us WGS VAF and ATAC VAF. As we estimated the expected change in ATAC-seq signal for each SNV using the ATAC-seq read counts, we expected the SNVs to be unbiased towards either allele. Therefore, we selected SNVs with WGS VAF in the range of [0.4, 0.6]. Finally, to obtain SNVs that have a significant change in ATAC-seq, we calculated the significance of change using the Fisher’s exact test and the odds ratio. Each of these quantities was calculated using the reference and alternate allele read counts in WGS and ATAC-seq experiments. SNVs that had a P-value < 0.01 and odds-ratio ∈ [0.67, 1.33] were considered to have a significant change in ATAC-seq signal and therefore, were selected for SNV analysis.
This filtering led to a total SNV count of 1025, with 80 somatic mutations and 945 SNPs across the four cancer patients.
Code availability
ASAP is available as a Python package at https://pypi.org/project/atac-asap/, released under the MIT license. Detailed documentation and installation instructions are provided at GitHub (https://github.com/BoevaLab/ASAP/wiki). All results presented in this paper can be reproduced using the source code and scripts, which are deposited at Zenodo (https://doi.org/10.5281/zenodo.15377297) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This project is partially funded by the Swiss Data Science Center (SDSC) collaborative projects grant (C22-09); Swiss Government Excellence Scholarship (ESKAS-Nr: 2021.0468) to A.G.
Author contributions: V.B. and A.G. conceived the study. V.B. and E.K. supervised the project. A.G. and L.K. processed the data sets. T.M. and L.K. developed the different neural network architectures with the assistance of L.Z. and A.G. The experiments were devised and interpreted collectively by all authors. L.K., T.M., and A.G. implemented the experiments and analyzed the results. A.G. composed the figures. A.G., T.M., and L.K. wrote the manuscript while all other authors provided feedback.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.281042.125.
-
Freely available online through the Genome Research Open Access option.
- Received June 11, 2025.
- Accepted December 18, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








