
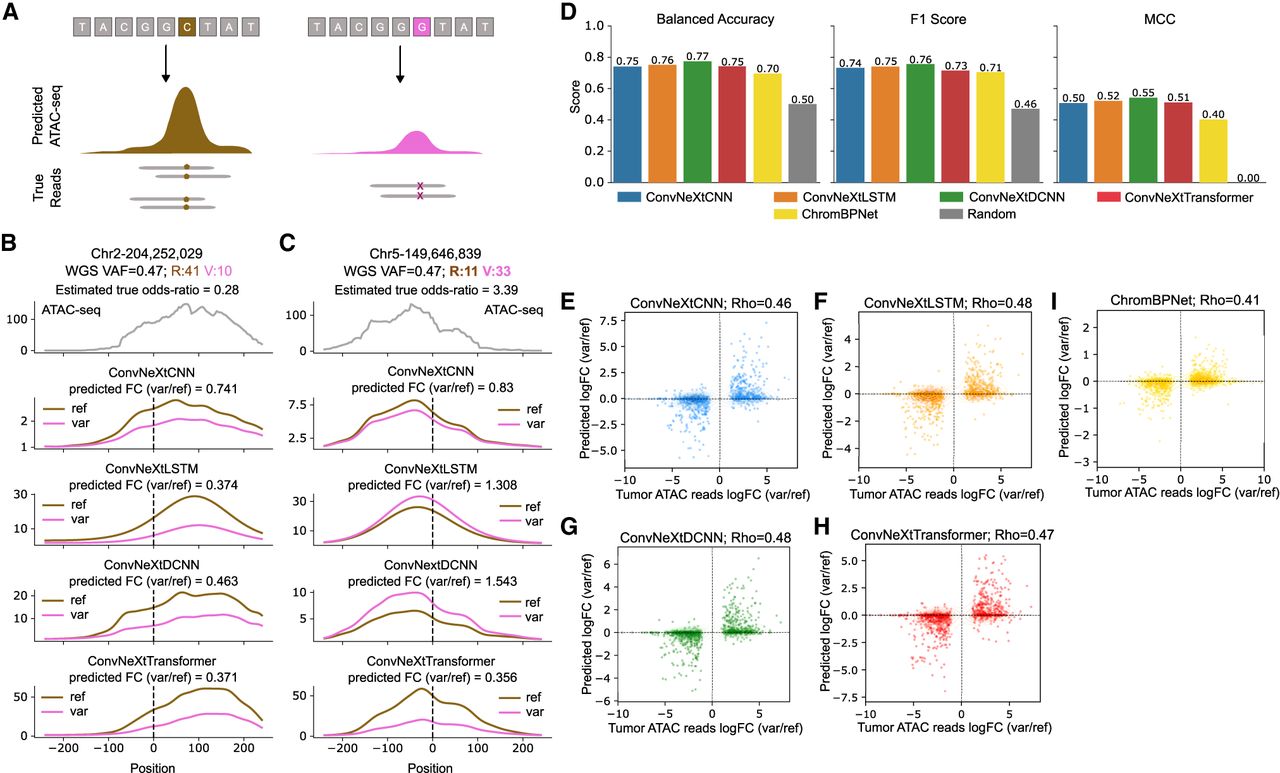
Evaluating model ability to accurately predict effects of single-nucleotide variants on chromatin accessibility. (A) Experimental setup to test each model’s ability to predict the change in chromatin accessibility between the reference DNA sequence (left) and a genomic variant (right). (B,C) Examples of allele-specific chromatin accessibility predictions for the reference allele and a genomic variant in Chromosomes 2 and 5, respectively. WGS VAF: Whole genome sequencing variant allele frequency; R, V: ATAC-seq total reference reads and variant reads, respectively. The model predictions are smoothed using a 1D Gaussian kernel with σ = 8. (D) ConvNeXt-based models are compared against ChromBPNet based on their ability to predict the correct directionality of change in accessibility between reference and variant. A total of 1025 variants are considered across all cancer patients included in this study. Balanced accuracy, F1 score, and Matthews correlation coefficient (MCC) are used as metrics. A random baseline is included to demonstrate the improvements in metrics across all models proposed in this work. (E–H) Scatter plots showing the quantitative change in accessibility by each of the ConvNeXt-based models. (I) Performance of ChromBPNet is also shown for reference. Log odds ratio based on ATAC-seq read counts for reference and variants are compared against log fold change (FC) between model predictions with reference and variant as model inputs and Spearman’s correlation (Rho) is calculated.








