
Multi-species analysis of inflammatory response elements reveals ancient and lineage-specific contributions of transposable elements to NF-kB binding
- Liangxi Wang1,2,3,
- Tiegh Taylor4,
- Kumaragurubaran Rathnakumar2,5,6,
- Nadiya Khyzha6,7,8,
- Minggao Liang1,2,9,
- Azad Alizada2,16,
- Laura F. Campitelli1,10,
- Sara E. Pour1,10,
- Zain M. Patel1,10,
- Lina Antounians1,11,12,
- Ian C. Tobias4,17,
- Huayun Hou2,
- Timothy R. Hughes1,10,
- Sushmita Roy13,14,
- Jennifer A. Mitchell4,
- Jason E. Fish6,7,15 and
- Michael D. Wilson1,2
- 1Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5S 3K3, Canada;
- 2Genetics and Genome Biology, SickKids Research Institute, Toronto, Ontario M5G 0A4, Canada;
- 3Institut de Pharmacologie Moléculaire et Cellulaire, Université Côte d'Azur and Centre National pour la Recherche Scientifique (CNRS), Valbonne 06560, France;
- 4Department of Cell and Systems Biology, University of Toronto, Toronto, Ontario M5S 3G5, Canada;
- 5Division of Developmental Biology, Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio 45229, USA;
- 6Toronto General Hospital Research Institute, University Health Network, Toronto, Ontario M5G 2C4, Canada;
- 7Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario M5S 3K3, Canada;
- 8Basic Sciences Division, Fred Hutchinson Cancer Center, Seattle, Washington 98109, USA;
- 9Department of Comprehensive Pathology, Institute of Science Tokyo, Tokyo 108-0071, Japan;
- 10Donnelly Center for Cellular and Biomolecular Research, Toronto, Ontario M5S 3E1, Canada;
- 11Developmental and Stem Cell Biology, SickKids Research Institute, Toronto, Ontario M5G 0A4, Canada;
- 12Division of General and Thoracic Surgery, The Hospital for Sick Children, Toronto, Ontario M5G 1X8, Canada;
- 13Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Madison, Wisconsin 53726, USA;
- 14Wisconsin Institute for Discovery, Madison, Wisconsin 53715, USA;
- 15Peter Munk Cardiac Centre, University Health Network, Toronto, Ontario M5G 2N2, Canada
Abstract
Transposable elements (TEs) provide a source of transcription factor (TF) binding sites that can rewire gene regulatory networks. NF-kB is an evolutionarily conserved TF complex primarily involved in innate immunity and inflammation. The extent to which TEs have contributed to NF-kB responses during mammalian evolution is not well established. Here, we perform a multi-species analysis of TEs bound by the NF-kB subunit RELA in response to the proinflammatory cytokine TNF. By comparing RELA ChIP-seq data from TNF-stimulated primary aortic endothelial cells isolated from human, mouse, and cow, we find that 55 TE subfamilies are associated with RELA-bound regions, many of which reside near TNF-responsive genes. A prominent example of lineage-specific contribution of transposons comes from the bovine SINE subfamilies Bov-tA1/2/3 which collectively contributed over 14,000 RELA-bound regions in cow. By comparing RELA binding data across species, we also find several examples of RELA motif-bearing TEs that colonized the genome prior to the divergence of the three species and contributed to species-specific RELA binding. For example, we find human RELA-bound MER81 instances are enriched for the interferon gamma pathway and demonstrate that one RELA-bound MER81 element can control the TNF-induced expression of interferon gamma receptor 2 (IFNGR2). Using ancestral reconstructions, we find that RELA containing MER81 instances rapidly decayed during early primate evolution (>50 million years ago [MYA]) before stabilizing since the separation of Old World monkeys (<50 MYA). Taken together, our results suggest ancient and lineage-specific transposon subfamilies contributed to mammalian NF-kB regulatory networks.
Transposable elements (TEs) play a role in shaping gene regulation by serving as a source of novel cis-regulatory elements (CREs) (Chuong et al. 2017; Pontis et al. 2019; Sundaram and Wysocka 2020; Hermant and Torres‐Padilla 2021; Senft and Macfarlan 2021; Fueyo et al. 2022).
Identifying which TFs interact with TEs and under what conditions is an ongoing challenge that requires a range of experimental and computational approaches (Bourque et al. 2008; Macfarlan et al. 2011; Emera and Wagner 2012; Jacques et al. 2013; Ward et al. 2013; Sundaram et al. 2014; Ito et al. 2017; Fuentes et al. 2018). Chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) of an ever growing number of TFs in different cell types, species, and conditions are revealing repeat-associated binding sites (RABS) whereby reproducible TF-DNA interactions fall within subtypes of major repeat families (Bourque et al. 2008). Coincident with their discovery, RABS have been shown to contribute to gene regulatory networks underlying a myriad of processes including early developmental gene regulation, pregnancy, circadian rhythm, and innate immunity (Kunarso et al. 2010; Xie et al. 2010; Lynch et al. 2011; Notwell et al. 2015; Chuong et al. 2016; Trizzino et al. 2017; Fuentes et al. 2018; Pontis et al. 2019; Todd et al. 2019; Bogdan et al. 2020; Judd et al. 2021; Kelly et al. 2022). Evidence of the regulatory potential of TEs comes from high-throughput experiments that perturb the activity of TE families using CRISPR-Cas9 (e.g., Fuentes et al. 2018; Pontis et al. 2019), as well as massively parallel reporter assays (e.g., Du et al. 2022).
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) is an essential mediator of innate immune/inflammatory responses (Zhang et al. 2017), whose activity has been conserved across metazoans (Sen and Baltimore 1986; Satriano and Schlondorff 1994; Correa et al. 2004; Hetru and Hoffmann 2009). In vertebrates, NF-kB is composed of hetero-dimers of five proteins: NFKB1 (also known as p50), NFKB2 (also known as p52), RELA (also known as p65), RELB, and REL, with the most predominant combinations being NFKB1:RELA which can be found in most cell types (Bhatt and Ghosh 2014). The NFKB1:RELA complex mediates canonical NF-kB signaling pathways. This process relies on NFKB1:RELA being sequestered in its inactive form in the cytoplasm through its association with the inhibitory IKB proteins under unstimulated conditions. In response to upstream stimuli, such as the pro-inflammatory cytokine tumor necrosis factor (TNF), the IKB inhibitors are degraded and NF-kB rapidly translocates into the nucleus where it binds promoter-proximal and promoter-distal CREs that drive the expression of multiple innate immunity/inflammation related genes (Zhang et al. 2017).
Although the core protein components of NF-kB are often shared across cell types, the binding of NF-kB to the genome is constrained by cell type–specific chromatin states (Natoli et al. 2005; Ostuni et al. 2013). For example, abundant NF-kB binding occurs at the pre-existing SPI1 (also known as PU.1) transcription factor-bound regions in macrophages (Kaikkonen et al. 2013), whereas NF-kB binding in aortic endothelial cells occurs at ERG- and JUN-bound regions (Hogan et al. 2017). Inter-species comparisons of NF-kB binding in primary aortic endothelial cells stimulated with TNF for 45 min demonstrates that conserved orthologous NF-kB-bound regions were enriched near NF-kB target genes and coincided with increased RELA binding, chromatin accessibility, active post-translational histone modifications (H3K27ac and H3K4me2), and RNA polymerase II activity (Alizada et al. 2021). The functional relevance of conserved orthologous NF-kB-bound regions also corresponded to noncoding genetic variation associated with both inflammatory and cardiovascular phenotypes. However, the extent to which TEs are involved in pan-cell type and conserved orthologous NF-kB gene regulatory networks remains to be seen.
Previous work cloning and sequencing DNA from RELA ChIP led to the conclusion that ∼11% of NF-kB-bound regions contain an Alu-derived NF-kB motif (Antonaki et al. 2011). However, the extent to which specific subfamilies of repetitive elements contribute to NF-kB binding in human and other mammalian species remains to be characterized. Here, we address the global contribution of TEs to mammalian NF-kB binding by performing a comparative genomic analysis of TNF-induced NF-kB binding in primary human, mouse, and cow aortic endothelial cells.
Results
Transposable elements contribute to mammalian NF-kB-bound regions
To identify the contribution of transposable elements to NF-kB-bound regions relative to other TFs, we first interrogated genome-wide binding data for 435 human and 266 mouse TFs found in the Gene Transcription Regulation Database (GTRD) (Kolmykov et al. 2021). We calculated the fraction of RABS by overlapping ChIP-seq peak summits with RepeatMasker-annotated TEs (Bao et al. 2015). TE-derived RELA-bound regions were above the median of all transcription factors (84th percentile and 77th percentile of all investigated TFs for human and mouse, respectively), with a median overlap of 33% and 21% for human and mouse, respectively (Supplemental Fig. S1A,B).
To study the evolution of TE-derived RELA binding, we analyzed published work that mapped RELA binding by ChIP-seq in human, mouse, and cow (Alizada et al. 2021). This study was performed in primary human, bovine, and mouse aortic endothelial cells (HAEC, BAEC, and MAEC, respectively) treated acutely (45 min) with a species-matched pro-inflammatory cytokine tumor necrosis factor (Fig. 1; Alizada et al. 2021). To facilitate direct comparisons between this study and Alizada et al. (2021), we aligned RELA ChIP-seq data to human (hg19), mouse (mm10), and bovine (bosTau6) genome builds found in the multiple genome alignment (Ensembl 70 release) used by Alizada et al. (2021). This data set gave a significant number of RELA-bound regions for each species after 45 min of TNF treatment (human: 59,735; mouse: 23,623; and cow: 93,511). There were only 383 RELA peaks unique to hg19 and 420 peaks unique to the hg38 genome build, suggesting that the choice of genome would not significantly affect our results. Although the use of 100-bp single-end reads will be insufficient for mapping the youngest transposon families (Sexton and Han 2019), we reason that this unique comparative epigenomic data set, which includes ATAC-seq (all three species) and H3K4me2 and H3K27ac (human), is well-suited for comparing mammalian repeat associated NF-kB binding between species. We observed 19%, 12%, and 31% overlapping fractions of TEs within RELA-bound regions in human, mouse, and bovine aortic endothelial cells, respectively (Supplemental Fig. S1C). For all species, the observed TE class proportions were significantly different from background expectations (χ2 test, corrected P-values < 1 × 10−7 for all tests) (Supplemental Fig. S1C,D). The most striking example of this difference was for SINE elements in cow, which overlapped 20,188 RELA peaks, at a frequency of 22% (genome background of 17%; χ2 test, 1.3-fold enriched, corrected P-value = 2.6 × 10−267).
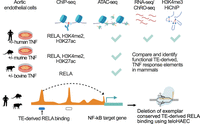
Experimental overview. Comparative epigenomic analysis of the contribution of TEs to NF-kB gene regulatory networks. Primary aortic endothelial cells from human, mouse, and cow were treated with TNF and subjected to ChIP-seq (RELA, H3K4me2, and H3K27ac), ATAC-seq, and RNA-seq. H3K4me3 HiChIP, ChRO-seq, and CRISPR-Cas9 deletions of an exemplar TE-derived region were performed in TeloHAEC. Created in BioRender (https://www.biorender.com).
Next we compared TE-derived RELA-bound regions from each species using whole-genome multiple sequence alignments which allowed us to identify orthologous and species-specific NF-kB binding events (Alizada et al. 2021). As expected, we found most TE-derived RELA-bound regions to be species-specific (χ2 test; human: 90%, mouse: 92%, cow: 95%; corrected P-values < 3.6 × 10−207 for all three species) (Supplemental Fig. S2A). We then took a closer look into the instances where TE-derived RELA-bound regions occurred in the context of conserved orthologous RELA binding. Most conserved orthologous TE-derived RELA-bound regions were classified as being TE-derived in only one species (human: 810/1026; mouse:127/255; and cow: 920/1410). Two explanations for this observation are that: (1) there is an orthologous TE that was not annotated by RepeatMasker in the second species; or (2) a conserved orthologous RELA binding site turned over into an adjacent TE-derived DNA sequence. To explore these possibilities, we split our human conserved RELA binding events into three categories: Category 1 – conserved orthologous RELA binding is not related to TEs (n = 16,522); Category 2 – conserved orthologous RELA binding involving TEs in humans and at least one other species (n = 221); and Category 3 – conserved orthologous RELA binding was TE-derived only in human (n = 899) (Supplemental Fig. S2B).
Both Category 1 and Category 2 regions showed relatively high DNA constraint. In contrast, Category 3 showed lower DNA constraint that was comparable to the DNA constraint observed for human-specific RELA binding. To investigate this further, we extracted and realigned all human RELA-bound sequences (±500 bp from the summit) that had conserved orthologous RELA binding in at least one other species. We then compared the base pair distance between human RELA peak summits and the “lifted-over” summit position from mouse or cow. Consistent with an enrichment of micro-turnover events in Category 3, we found that the lifted-over RELA peak summits in Category 3 were further apart than what was observed for Category 1 or 2 (Supplemental Fig. S2C). We then measured the base pair distance of lifted-over mouse or cow RELA peak summits to the nearest human TE. As would be expected for conserved orthologous TE-derived RELA binding events, we observed that ∼75% of the mouse/cow RELA peak summits in Category 2 were within 25 bp of a human TE. In contrast, ∼50% of the RELA peak summits in Category 3 were within 25 bp of a human TE (Supplemental Fig. S2D). This indicates that Category 3 events likely represent a combination of missing TE annotations in addition to micro-turnovers into adjacent TEs, several of which are primate lineage-specific TEs (Supplemental Fig. S2E,F).
To investigate the TE features that contributed to TE-derived RELA-bound regions, we annotated each RELA peak summit with its corresponding TE subfamily using RepeatMasker (Fig. 2A). We identified 55 significantly enriched TE subfamilies among all three species, seven of which were shared in at least one additional species (Fig. 2A,B). These enriched subfamilies collectively make up 9% (1020/11,365), 13% (369/2903), and 52% (15,475/29,576) of TE-derived RELA-bound regions in human, mouse, and cow, respectively. Notably, 22 subfamilies contained canonical RELA motifs in their consensus sequences (Fig. 2B). LTRs are the largest contributor of RELA-bound regions in human and mouse (human: 68%, mouse: 65%, and cow: 2%). The only LTR that was enriched for RELA binding in all three species was MER21C. In contrast to human and mouse, the SINE class of TEs makes up the majority (68%) of TE-derived RELA-bound regions in the cow genome. Four SINE subfamilies—Bov-tA1, Bov-tA2, Bov-tA3, and SINE2-1_BT—account for 14,914 (74%) of these RELA-bound SINEs. The presence of a RELA motif in the consensus sequence of Bov-tA3, which overlaps 2252 RELA binding events, provides a plausible explanation for these SINE-derived RELA-bound regions (Fig. 2B).

Transposable elements contribute to mammalian NF-kB-bound regions. (A) Ranked overlap values for each TE subfamily with RELA peak summits from human, mouse, and cow. Significantly enriched TE subfamilies found in panel B are colored red. (B) TE subfamilies significantly enriched within RELA-bound regions in human, mouse, and cow were calculated by comparing observed and expected/simulated overlap events at RELA peak summits. The cutoffs to define enriched TE subfamilies: FDR < 0.05, log2 fold change >1 and the number of observed overlap events ≥10. Observed overlap number/total number of each TE subfamily is displayed for each bar. Bars marked with an asterisk indicate the presence of canonical RELA motifs in the consensus sequence. (C) Estimated evolutionary age of all the enriched TE subfamilies. The top panel depicts the median of estimated age of these elements, and the bottom panel presents enrichment values. Dashed gray lines represent estimated human-mouse and mouse-cow divergence time. TE subfamilies shared by more than one species are highlighted in blue. (D) An example (AmnSINE1) of a three-species-conserved TE-derived RELA-bound region. Light blue shades show orthologous regions of the three species.
To broadly classify TEs into ancient and lineage-specific categories, we approximated the evolutionary age of the 55 enriched TE subfamilies using the sequence divergence of subfamily instances relative to their consensus sequence. Seven of the enriched TE subfamilies were considered ancient (Tigger7, MER21C, MER81, AmnSINE1, UCON26, Plat_L3, MamSINE1) and likely to have colonized the mammalian genome near or prior to the divergence of the three species (∼90 MYA [Kumar et al. 2017]) (Fig. 2C). Of these ancient subfamilies, AmnSINE1, MER81, and UCON26 were enriched for NF-kB binding in at least two of the three species. For example, AmnSINE1 colonized the mammalian genome before the radiation of amniote animals (Nishihara et al. 2006) and has been shown to contribute CREs to conserved gene regulatory networks active during brain development (Sasaki et al. 2008; Hirakawa et al. 2009). AmnSINE1 elements were significantly enriched for human and cow RELA binding. Although this TE was not considered to be significantly enriched in mouse, we found nine RELA-bound instances (4.6-fold enrichment; one example is shown in Fig. 2D). A clear example of an ancient RELA-associated TE was MER81, a 114-bp non-autonomous DNA transposon (Bao et al. 2015). MER81 was enriched for RELA binding in all three species (10.9-fold change in human, 10.6-fold change in mouse, and 6.2-fold change in cow). Thus, both ancient and lineage-specific TE subfamilies have contributed to the NF-kB binding repertoire.
RELA-bound TEs possess active enhancer features for target gene regulation
To explore the regulatory potential of the TE-derived RELA-bound regions, we assessed epigenomic proxies for active CREs in TNF-treated and untreated ECs in each species. Starting with the 55 TE subfamilies in human, we investigated the dynamic changes in RELA binding, H3K27ac (active enhancers/promoters), H3K4me2 (active or latent enhancers/promoters), and chromatin accessibility (ATAC-seq) before and 45 min after TNF treatment (Alizada et al. 2021). We first subcategorized these elements into “old-TEs” and “young-TEs” where “old” TE subfamilies were designated based on their estimated colonization of mammalian genomes being prior to the divergence of the selected mammals (90 MYA).
We found that the old and young TE-derived RELA-bound regions were associated with high RELA binding signals after TNF treatment and were comparable to that of conserved RELA-bound regions (Supplemental Figs. S3, S4A). Increased chromatin accessibility was also observed for both TE-derived groups post-TNF treatment. Although a clear signature of active CREs, characterized by H3K4me2 and H3K27ac, was associated with TE-derived groups, this TE-derived epigenomic signal was far lower than what was observed for the conserved RELA-bound regions and slightly lower than the human-specific group. TE-derived RELA-bound regions being associated with active CRE characteristics was also observed for mouse and cow TE-derived RELA- bound regions (Supplemental Fig. S4B,C).
To further assess the enhancer features of TE-derived subsets in human, we analyzed data from a genome-wide global RNA polymerase run-on assay, which we previously generated from a karyotypically normal, immortalized human aortic endothelial cell line (TeloHAEC) (Fish et al. 2017; Alizada et al. 2021). This profiling was achieved using ChRO-seq (Chromatin Run-On sequencing [Chu et al. 2018]) which reveals bi-directional transcription of enhancer RNAs (eRNAs), a hallmark of active enhancers (Kaikkonen et al. 2013). Conserved RELA-bound regions showed the highest eRNA signal, followed by old TE-derived RELA-bound regions. The majority of the two TE categories reside within the distal genomic regions (more than 3 kb away from gene TSS). We found a clear pattern of bi-directional transcription of eRNAs in all four RELA groups at both distal (>3 kb) and proximal (≤3 kb) regions relative to the gene TSS (one TSS was considered per gene) (Fig. 3A). Overall, this analysis supports the possibility that transposons contribute active enhancers which are involved in NF-kB-mediated gene regulation.
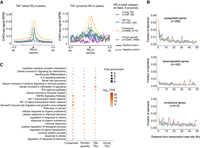
RELA-bound TEs possess active enhancer features for target gene regulation. (A) eRNA signal from ChRO-seq of TeloHAEC around RELA-bound TEs that are distal or proximal to gene TSS with TNF stimulation for 45 min. The cutoff for distal/ proximal elements: 3 kb relative to gene TSS. (B) The distribution of RELA-bound TEs near upregulated, downregulated, and constitutive genes. The three groups of genes were defined using matched RNA-seq data (see Methods). “Background” represents the distribution of all the genomic TEs near the three gene groups, shown in gray. (C) Functional enrichment of RELA-bound TEs by GREAT (McLean et al. 2010).
We assessed the likelihood that RELA-bound TEs influenced NF-kB target gene expression by comparing the distribution of the four categories of RELA-bound regions around upregulated, downregulated, and constitutive genes (Fig. 3B; see Methods). We observed a significant enrichment of RELA-bound regions near the promoters of upregulated genes compared to constitutive genes, with the old TE-derived RELA group showing the highest enrichments, followed by the conserved RELA group (binomial test for occurrences within 10 kb relative to upregulated vs. constitutive gene TSS; Old TEs: 3.6-fold, P-value 6.9 × 10−9; Conserved: 1.8-fold, P-value < 2.2 × 10−16; Human-specific: 1.6-fold, P-value < 2.2 × 10−16; and Young TEs: 0.9-fold, P-value = 0.85). Accordingly, TE-derived RELA-bound regions were enriched for annotated immune/inflammatory functions (Fig. 3C). Although there was no enrichment of the young TE-derived RELA group (n = 561), we found comparable enrichment signals in functionally related terms for the other three groups. We noted several terms particularly enriched in the old TE-derived RELA group, including “IFN-γ pathway” (binomial FDR of 2.2 × 10−7) and “IL6 signaling pathway” (binomial FDR of 1.06 × 10−6). These observations suggest the possibility that ancient TE-derived RELA-bound elements have preferentially contributed to IFNG/IL6 signaling during human evolution.
Whereas linear proximity between a cis-regulatory element and a promoter is associated with potential function, three-dimensional chromatin interactions provide additional direct evidence of a physical connection between distal CREs and gene promoters (Chepelev et al. 2012; Mifsud et al. 2015; Fulco et al. 2019). To profile the interaction of TE-derived RELA-bound regions with gene promoters, we performed HiChIP (Fang et al. 2016; Mumbach et al. 2016) in TeloHAECs before and 45 min after TNF stimulation using an antibody against H3K4me3, a post-translational histone modification enriched at active gene promoters. We found a union of 19,372 significant chromatin interactions before and 45 min after TNF stimulation, 1605 of which involved NF-kB upregulated genes. Considering the portion of distal elements (at least 5 kb away from any gene TSS) from the four categories, conserved RELA-bound regions have the highest fraction of overlap with the distal anchor from the identified loop sets (28%), followed by old TE-derived (15%), human-specific (15%), and young TE-derived (11%) RELA-bound regions. We next focused on the subset of chromatin interactions (n = 1605) that connect 1180 distal RELA- bound regions and 224 NF-kB upregulated genes. A subset of these elements (186 out of 1180) were derived from TEs, 39 of which belong to the significantly enriched transposon families enriched for RELA binding (Supplemental Table S1).
TE-derived RELA-bound regions coincide with cell type–specific TF motifs
The NF-kB signaling pathway is active across diverse cell types and involves common and cell type–specific target gene expression (Tabruyn and Griffioen 2007; Gerondakis and Siebenlist 2010; Liu et al. 2017). Although cell type–specific NF-kB binding is known to occur in collaboration with cell type–specific TFs (Kaikkonen et al. 2013; Hogan et al. 2017; Alizada et al. 2021), it was not known if specific cell types would be enriched for distinct TE subfamilies at NF-kB-bound regions. We looked for TE-associated RELA binding after TNF stimulation in: human umbilical vein endothelial cells (HUVECs; n = 12,442 [Brown et al. 2014]), lymphoblasts (LCLs; n = 52,563 [Kasowski et al. 2010]), and adipocytes (n = 58,043 [Schmidt et al. 2015]) (Supplemental Fig. S5). Whereas HUVECs showed a subset of the TE-derived RELA binding that we observed in HAECs, RELA binding in both adipocytes and LCLs enriched for additional TE families (Supplemental Fig. S5). Notably, MER81 showed the high enrichment values in all four cell types (10.6-, 9.2-, 18.4-, and 8.6-fold enrichment for HAECs, HUVECs, adipocytes, and LCLs, respectively).
To gain further insight into the nature of the shared and cell-specific TE-derived RELA binding, we investigated TF motif usage within TE-derived RELA-bound regions from each species (Fig. 4). Supporting a direct role of DNA sequence features in NF-kB recruitment, motifs from RELA and other NF-kB subunits (RELB, REL, NFKB1, and NFKB2) were consistently detected in most TE subfamilies from all the species and cell types (Supplemental Fig. S6). For example, 16 out of 22 subfamilies in HAEC showed strong NF-kB motif enrichments. We also found multiple transposable elements with high AP-1 motif enrichment that lacked NF-kB motif enrichment (such as Tigger7 and MER44B/C/D in HAECs). Additional evidence for the collaboration of ETS-related factors in TE-derived RELA-bound regions was observed in aortic endothelial cells from human and cow (7/22 and 11/18 enriched TE subfamilies in human and cow, respectively) (Fig. 4A,B). We found enrichments of nine subfamilies (e.g., MER20 and L1MB2) for the adipocyte-specific master regulator CEBPA in human adipocyte RELA ChIP-seq data (Freytag et al. 1994; Darlington et al. 1998). These results indicate that further cell type–specific ChIP-seq experiments are needed to capture the full repertoire of TE subfamilies associated with NF-kB binding.
Bovine SINEs mediate RELA binding via a restricted repertoire of RELA motifs
We then investigated the sequence features within the ∼14,000 RELA-bound regions observed in the bovine SINE subfamilies Bov-tA1, Bov-tA2, and Bov-tA3. These three bovine SINE subfamilies make up ∼15% of all RELA binding observed in bovine aortic endothelial cells. Bov-tA elements originated through recombination between tRNA-Glu-derived SINEs and a fragment of BovB element before the radiation of Pecoran ruminants (∼25–30 MYA) (Nijman et al. 2002; Nilsson et al. 2012). Collectively, Bov-tA1/2/3 gave rise to more than 750,000 copies in the modern cow genome with a consensus sequence length of ∼210 bp. Given the sequence divergence of transposon instances relative to the subfamily consensus, Bov-tA1 is estimated to be the oldest subfamily, followed by Bov-tA2 and Bov-tA3 (Fig. 2C). Unlike the Bov-tA1 and Bov-tA2 consensus sequences, the Bov-tA3 consensus sequence contains a RELA motif. We found that a one nucleotide difference at the 130- to 140-bp position of the aligned consensus sequences can explain the presence of the RELA motif in Bov-tA3 (Supplemental Fig. S7A).
We observed a significant increase in motif density at the 130- to 140-bp position in RELA-bound regions (χ2 test; 4.4-fold, 3.8-fold, 2.0-fold enrichment for Bov-tA1/2/3, respectively; P-value < 2.2 × 10−16 for all three subfamilies) (Fig. 4C). Given that a single mutation at the 130- to 140-bp position of Bov-tA1 or Bov-tA2 can generate a RELA motif, we next asked if the RELA-bound instances were biased towards specific motif words. The most abundant RELA motif matching word (“RELA-word1”) corresponded to the consensus sequence of Bov-tA3 and was present in 18%, 12%, and 54% of RELA-bound Bov-tA1, Bov-tA2, and Bov-tA3 elements, respectively (Supplemental Fig. S7). The second most frequent RELA motif-containing word (“RELA-word2”) was found in all three subfamilies (present in 22%, 35%, and 14% of RELA-bound Bov-tA1, Bov-tA2, and Bov-tA3 elements, respectively).
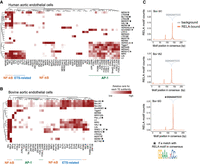
Multiple TF motifs contribute to TE-derived RELA-bound regions across species and cell types. Heat maps of significantly enriched TF motifs from (A) human aortic endothelial cells and (B) bovine aortic endothelial cells. Rows and columns of the heat maps indicate TE subfamilies and TF motifs, respectively. The intensity of heatmaps represent the rank of significantly enriched TF motifs for each TE. (C) RELA motif distribution relative to the consensuses of Bov-tA1, Bov-tA2, and Bov-tA3. Gray and orange represent all instances and RELA-bound instances, respectively.
DNA transposon MER81 carries two RELA motifs responsible for enhancer activity
In human aortic endothelial cells, we also observed an enrichment of RELA motifs at specific positions in MER41B and MER81. MER41B is a primate LTR subfamily bound by interferon gamma (IFNG)-inducible TFs STAT1 and IRF1 (Chuong et al. 2016). We find that the RELA motif occurs at positions 378 to 387 bp of the consensus sequence in MER41B (Supplemental Fig. S7D). MER81 was the only TE subfamily enriched in RELA-bound regions from all tested species and cell types. Using RepeatMasker annotations from a representative set of 24 mammals (Chuong et al. 2016), we found MER81 to be present in all 23 placental mammals and absent in the non-placental mammal representative, opossum (Supplemental Table S2). In placental mammals, a median of ∼2800 copies were identified in each species. There is a notable lineage-specific reduction of annotated MER81 elements in Eulipotyphla (hedgehog) and Rodentia (mouse, rat, and prairie vole) species (∼500 copies in each species). In human, 3639 MER81 elements were annotated in the genome, among which 734 had RELA motifs. From our HAEC ChIP-seq data, we identified 88 extant MER81 copies bound by RELA in response to TNF stimulation. Consistent with the general features of RELA-bound TEs, RELA-bound MER81s also exhibit active CRE characteristics measured by H3K4me2, H3K27ac, and ATAC-seq signal before and after TNF stimulation (Fig. 5A; Supplemental Fig. S8A). Furthermore, RELA-bound MER81 elements were enriched in the vicinity of genes that are annotated with immune-related functions (Supplemental Fig. S8B), such as “increased hematopoietic cell number” (binomial P-value = 4.4 × 10−6) and “IFN-γ pathway” (binomial P-value = 5.5 × 10−5). For example, the proximal promoter region (0.3 kb upstream of the TSS) of the gene CXCR5, which plays an important role in B cell maturation and migration (Pereira et al. 2010), has one instance of the MER81 element with RELA binding that is shared across all four human cell types. We also found several interferon-signaling-related genes in the vicinity of RELA-bound MER81 instances, including IFNAR1 (surface receptor of IFN1A signaling; 58 kb downstream from the TSS), TYK2 (downstream kinase required for IFN1A signaling; 5 kb upstream of the TSS), IFNGR2 (surface receptor of IFNG signaling; 19 kb upstream of the TSS), JAK2 (downstream kinase required for IFNG signaling; 47 kb upstream of the TSS), and IRF1 (known transcription factor activated by IFNG signaling; 15 kb downstream from the TSS) (Supplemental Table S3). Distal RELA-bound MER81 elements showed characteristic bi-directional ChRO-seq signal, indicative of active enhancers (Supplemental Fig. S8C).
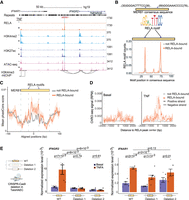
DNA transposon MER81 carries two RELA motifs responsible for enhancer activity. (A) Genome browser view of an instance of a MER81-derived RELA-bound region. “−” and “+” denote basal and TNF stimulation, respectively. (B) Location of RELA motifs within the MER81 consensus sequence. Orange and gray represent RELA-bound and unbound instances determined by ChIP-seq, respectively. (C) Mean phastCons score for RELA-bound and unbound MER81 sequences. (D) Chromatin run-on sequencing signal (TeloHAEC ChRO-seq) at MER81-derived RELA sites before and after TNF stimulation. (E) Impact of CRISPR-Cas9 deletions of the RELA-bound MER81 element at the IFNGR2 and IFNAR1 locus. The plots show the qPCR-measured expression levels of IFNGR2 and IFNAR1 in wild-type and two MER81 knockout TeloHAEC lines (with/without TNF simulation for 45 min). One-sided t-test for comparisons that involve the same genotype (e.g., stimulated vs. unstimulated mutants) and unpaired one-sided t-test for ones that involve different genotypes. Error bars indicate standard error of the mean. Each biological replicate is shown with a unique shape in the figure.
To understand how MER81 could recruit RELA for binding, we examined its sequence features. We found two canonical RELA motifs in its 114-bp consensus sequence, which could explain RELA binding (Fig. 5B). To expand the repertoire of RELA-bound MER81 elements, we additionally searched the pool of annotated RELA-bound regions from the GTRD database, which contains data from several human cell lines (Supplemental Table S4). Together with our HAEC data, we obtained 327 MER81 instances that overlapped a RELA peak summit in at least one human cell type. We observed a clear enrichment of normalized RELA motif counts in RELA-bound MER81 elements compared to unbound ones (0.16 vs. 0.03 for MER81 RELA-motif 1, and 0.19 vs. 0.03 for MER81 RELA-motif 2) (Fig. 5B). In other words, the loss of RELA binding in extant MER81 copies could be largely explained by the loss of RELA motifs. In addition, RELA-bound MER81 sequences show increased sequence constraints relative to unbound ones (34% increase in averaged phastCons score; one-sided t-test; P-value = 0.008), further indicating their potential functional importance (Fig. 5C).
Further support that these RELA-bound MER81 sequences can serve as enhancers was obtained by ChRO-seq analysis revealing the bi-directional signal associated with active enhancers which are modulated by TNF stimulation. This feature was not seen at MER81 elements that were not bound by RELA (Fig. 5D). To further explore the functionality of these MER81 elements, we performed luciferase reporter assays to assess the enhancer activity of the sequences. We first tested the activity of the MER81 consensus sequence, which serves as an “ancestral” representative of all MER81 instances. We found a modest increase enhancer activity of the MER81 consensus sequence compared to an empty vector control under basal condition (one-sided t-test; 1.6-fold; P-value = 0.03), which was enhanced upon TNF stimulation (one-sided t-test; 3.0-fold; P-value = 0.02). This modest enhancer activity of the putative ancestral MER81 sequence was lost when the RELA motifs within the sequence were disrupted (one-sided t-test, P-values of 0.81, 0.10, and 0.40 for TNF-stimulated mutant1, mutant2, and mutant3, respectively) (Fig. 5E; Supplemental Fig. S9A). Altogether, our results are consistent with our ChIP-seq results showing the MER81 instances containing intact RELA motifs can enhance gene expression after TNF stimulation.
Next, we selected one native RELA-bound MER81 element in human that resides in the vicinity of interferon signaling genes to explore its potential enhancer function using the same luciferase assay (Fig. 5A). This instance is located between two interferon receptor genes, IFNAR1 and IFNGR2 (+58 kb and −19 kb relative to IFNAR1 and IFNGR2 TSS, respectively), both of which we identified as RELA target genes in our HAEC RNA-seq data. Our epigenomic data indicate that this MER81 sequence coincides with the largest RELA peak at the locus and is within an accessible chromatin region with a strong H3K4me2/H3K27ac signal. Using our H3K4me3 HiChIP data from TeloHAECs, we identified a chromatin loop connecting this instance and the downstream target gene IFNGR2. We first attempted to validate the enhancer activity of this sequence via luciferase reporter assay (Supplemental Fig. S9B). This instance only retained the first RELA motif. Consistent with our luciferase results, using the ancestral MER81 sequence with a single RELA motif showed little to no enhancer activity which increased slightly in response to TNF stimulation (one-sided t-test for TNF-stimulated native sequence; 1.5-fold; P-value = 0.07),whereas the motif-shuffled counterpart exhibited loss of enhancer activity (one-sided t-test for TNF-stimulated mutant sequence, 0.91-fold, P-value = 0.73) (Supplemental Fig. S9B).
To test the function of this enhancer in its chromatin context, we deleted the genomic region in TeloHAECs using CRISPR-Cas9 (Supplemental Fig. S9C). We performed two deletion experiments: “Deletion 1” which spans from 160 bp upstream of to 163 bp downstream from the 101-bp MER81 instance; and “Deletion 2” which is restricted to the first ∼80 bp of this MER81 instance and includes the RELA motif. We then checked the effects of these deletions on the neighboring IFNAR1 and IFNGR2 genes (note that IFNGR2 had an order of magnitude higher expression than IFNAR1 before and after TNF). Both IFNAR1 and IFNGR2 were significantly induced by TNF (2.7-fold change of pre-mRNA transcript expression; paired one-sided t-test, P-value of 2 × 10−4 and 4.7-fold change of pre-mRNA transcript expression; paired one-sided t-test, P-value of 7 × 10−3, respectively). We found a small but significant effect on induced expression in IFNAR1 upon the loss of this MER81 instance in Deletion 1 (0.61-fold change; unpaired one-sided t-test, P-value of 2 × 10−3) and no significant change in the more specific Deletion 2 (0.81-fold change; unpaired one-sided t-test, P-value of 0.13) (Fig. 5E). In contrast, both Deletion 1 and Deletion 2 abolished the ability of IFNGR2 to be induced by TNF (paired one-sided t-test, P-value of 0.74 and 0.61 for Deletion 1 and 2, respectively) (Fig. 5E). These results indicate that this MER81 instance functions as a bona fide enhancer, which is required for the regulation of the downstream target gene IFNGR2. Furthermore, the fine-mapping of the functional sequence (Deletion 2) reveals an essential 80-bp fragment in this element which encompasses one intact ancestral RELA motif. Altogether, our results support that MER81 has functional RELA motifs capable of regulating target gene expression in vivo.
Ancestral reconstruction reveals a stabilization of RELA motifs during MER81 evolution
The enrichment of RELA binding in extant MER81 copies appears to be a common feature across cells and mammalian lineages. However, the extant copies of MER81 with RELA binding ability tend to be species-specific. This is underscored by the mouse-specific loss of MER81 copies, with only 500 elements remaining in the mouse genome, compared with ∼3600 in human and ∼2600 in cow. Furthermore, 79% of MER81-derived RELA binding events in human aortic endothelial cells are human-specific (Supplemental Table S5). To delineate the evolutionary trajectories of the MER81 elements over the course of human evolution, we utilized ancestral whole-genome reconstructions of human and 10 other mammalian species which cover an evolutionary time window of ∼100 million years (Campitelli et al. 2022). With this strategy, we were able to recover the genome sequences of each node/common ancestor (Fig. 6A). By pooling all 10 reconstructed genomes together with the modern human genome, we achieved 11 nodes of genome sequences and were able to study mammalian and primate evolution from a human-centric perspective. We found the total copy number of MER81 was highly stable during evolution; in particular, ∼2800 MER81 elements were detected in each node. The stability of MER81 copies suggests no or a low level of transposition activity in the recent ∼100 million years. Secondly, a large proportion (20%–45%) of MER81 elements were identified with RELA motifs, and the proportion containing a RELA motif decreased steadily over time, with >45% of MER81 elements containing a RELA motif in node 10 (or the human–star-nosed-mole common ancestor) to only ∼20% containing a RELA motif in modern human (Fig. 6B).
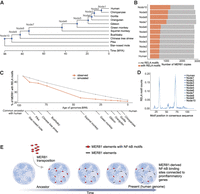
Ancestral reconstruction reveals a stabilization of RELA motifs during MER81 evolution. Modern human together with 10 related mammalian species were selected to reconstruct a series of ancestral genomes based on multiple sequence alignments. (A) Phylogenetic tree of the 11 selected species to reconstruct ancestral genomes. Each node in the tree represents a reconstructed genome of the corresponding common ancestor. (B) The number of MER81 copies in each human ancestor genome. Gray and orange represent total numbers and copies containing RELA motifs, respectively. (C) RELA motif change in MER81 along human evolution. The percentage of MER81 proportion containing RELA motifs is shown as a function of the estimated evolutionary age of each node/common ancestor. A simulation of MER81 sequence change based on nucleotide substitution is shown in gray. (D) The distribution of RELA motifs in MER81 instances in all human ancestors. (E) Schematic of a MER81 co-option model during human evolution. Created in BioRender (https://www.biorender.com).
Relative to a simulated RELA motif decay rate derived using the estimated nucleotide substitution rate (see Methods), we observed a rapid decrease of RELA motifs in the early stages of MER81 colonization followed by a stabilizing trend starting around the last common ancestor of human and squirrel monkey (Fig. 6C). To put this evolutionary trajectory into context, we expanded our analysis to include the additional human TE subfamilies associated with RELA binding. We also queried the motif for the AP-1 family transcription factor JUN, which is well known to bind together with NF-kB and is prominently expressed in endothelial cells (Oeckinghaus et al. 2011; Dorrington and Fraser 2019). We also found JUN motifs to be enriched within distinct subsets of TE-associated RELA binding sites (Fig. 4). Although we observed no signal from scrambled RELA or JUN motifs, distinct deceleration patterns of RELA and JUN motif decay from multiple transposons were observed (e.g., MER57F, MER77B, and MER44D) (Supplemental Fig. S10). The high and stable signals of JUN motifs, and the absence of RELA motif signal, from MER44B/C/D is also consistent with our motif enrichment analysis (Fig. 4A), as well the motif enrichment analyses obtained from accessible chromatin regions induced by the bacterial infection of macrophages (Bogdan et al. 2020). In contrast, the NF-kB binding in MER81 (which was the only significant TF motif we found enriched in this subfamily) (Fig. 4A) showed no signal for JUN motifs.
We further investigated the distribution of RELA motifs within MER81 elements that contain the motif and confirmed that these motifs were still centered on the two positions from the consensus sequence and that no apparent motif turnover or shift occurred within these elements (Fig. 6D). Together, our analysis supports a model where the DNA transposon MER81 colonized the genome prior to the radiation of placental mammals, followed by a long-term selection for the NF-kB motifs it contained (Fig. 6E). At least a subset of these elements may have been co-opted as active enhancers to regulate target gene expression in an NF-kB-dependent manner.
Discussion
In this study, we explored the contribution of transposable elements to genome-wide binding profiles of NF-kB in three mammalian lineages. Our comparative analyses of RELA binding, together with epigenetic and transcriptional profiles, revealed conserved and lineage-specific TE subfamilies that are likely to participate in mammalian NF-kB regulatory networks. Given the active enhancer features and proximity to multiple NF-kB target genes, it is likely that a subset of these TE-derived RELA-bound elements have been incorporated into the host NF-kB regulatory networks.
The expansion of RELA-bound regions in the bovine genome largely results from bovine SINE subfamilies Bov-tA1/2/3, which contribute over 14,000 RELA-bound regions to the cow genome (Fig. 2A,B). This bovine-specific expansion even exceeds the ∼10,000 SINE-B2 derived CTCF sites observed in the mouse genome (Bourque et al. 2008; Schmidt et al. 2012), making it one of the most striking examples of lineage-specific TF binding expansions described in mammals to date.
Although the null hypothesis is that most all Bov-tA1/2/3-derived NF-kB sites would be non-functional, the large number of these lineage-specific expansion of Bov-tA1/2/3 raises the possibility that a small subset of these elements influences ruminant gene regulatory networks. Kelly et al. (2022) recently discovered a substantial expansion of IFNG-inducible elements by a ruminant-specific SINE, Bov-A2 (∼12,000 instances), for the regulatory evolution of ruminant innate immunity. Bov-A2 and Bov-tA1/2/3 constitute the family BovA (Nijman et al. 2002; Nilsson et al. 2012; Bao et al. 2015), and the related evolutionary trajectory highlights a potentially pervasive function of the BovA family in ruminant innate immunity. For both Bov-tA1 and Bov-tA2, we observed evidence of “maturation” of transposon instances from imperfect “pre-sites” in ancestral sequences to perfectly matched RELA binding motifs consistent with the “proto-motif” model (MacArthur and Brookfield 2004; Bourque et al. 2008; Żemojtel et al. 2011; Sundaram and Wysocka 2020; Judd et al. 2021). It is intriguing to speculate that the Bov-tA (and other TE subfamilies) expanded their foothold in the genome by promoting their own expression through NF-kB binding. Although studies have demonstrated NF-kB activation in relevant cell types and developmental stages in a variety of mammals (e.g., bovine oocytes and preimplantation embryos [Paciolla et al. 2011] as well as rat and mouse testes [Delfino and Walker 1998; Lilienbaum et al. 2000; Rasoulpour and Boekelheide 2005]), no experimental evidence exists to support such a mechanism.
Prior work demonstrated the ability of Alu sequences to bind RELA and suggested that Alu elements contributed substantially to RELA binding in the human genome (Apostolou and Thanos 2008; Antonaki et al. 2011). This work involved cloning of 366 sequences obtained by RELA ChIP in HeLa cells combined with the analysis of ENCODE RELA ChIP-seq (26-bp single-end data) from the lymphoblast cell line GM12878 and led to the conclusion that ∼11% of NF-kB-bound regions contain an Alu-derived NF-kB motif (Antonaki et al. 2011). Although our study in primary endothelial cells did not identify Alu as a major contributor of TE-derived RELA-bound regions, it is important to note that the reported NF-kB motif containing Alu transposons (including AluSx, AluSg, and AluY) are young elements (Bennett et al. 2008), many of which would not be uniquely mappable with the 100-bp single-end reads used in our RELA ChIP-seq experiments (Sexton and Han 2019). Another explanation for this discrepancy with the prior work comes from our stringent definition for TE-derived RELA-bound regions, which required an overlap between a known TE and the summit of a RELA peak. In comparison, Antonaki et al. (2011) allowed for an overlap with any portion of a RELA peak. In the discussion of their study of Alu- associated NF-kB sites, Antonaki et al. (2011) proposed that such sites could serve as transcriptionally inert docking sites of NF-kB which could prevent excessive targeting of promoters. Likewise, one could speculate that the large number of Bov-tA-associated NF-kB binding sites could be tolerated if they somehow collectively provide a fitness advantage by attenuating the bovine inflammatory response.
Growing evidence has indicated that TEs, especially evolutionarily young TEs, act as a source of cis-regulatory elements, which primarily innovate regulatory programs in a species- and tissue-specific manner (Buecker and Wysocka 2012; Chuong et al. 2016; Fuentes et al. 2018; Todd et al. 2019; Du et al. 2022; Kelly et al. 2022; Lee et al. 2022; Choudhary et al. 2023). Although fewer in number, older TEs have been demonstrated to contribute to species-specific (Notwell et al. 2015) or conserved (Lynch et al. 2011) gene regulatory networks. Our comparative epigenomic suggest that the TE subfamilies, AmnSINE1, UCON26, Plat_L3, MamSINE1, MER21C, and MER81 may have played a role in the evolution of NF-kB gene regulatory networks. Indeed, the ∼300-million-year-old AmnSINE1 subfamily has been shown to contribute TF binding sites to mammalian gene regulatory networks (Sasaki et al. 2008; Hirakawa et al. 2009; Chuong et al. 2016). Although it is unlikely that the examples of TE-associated RELA binding shared across species are due to systematic errors in TE annotation, it is important to acknowledge that a recent study estimated that 10%–14% of Alu and L1 subfamilies are discordantly annotated in human and chimpanzee genomes (Carey et al. 2021).
One of the central discoveries in this study is the contribution of the ancient DNA transposon, MER81, to NF-kB-mediated regulatory networks. Most MER81-derived RELA-bound regions are species-specific (79%), indicating that the ancient MER81 elements may play core NF-kB functions in a human-specific manner. In human, the majority (>80%) of MER81-derived RELA-bound regions are shared across cell types, which increases the potential for MER81 to influence core components of NF-kB-mediated transcriptional regulation in different cellular contexts. This observation is supported by a recent study that found that: (1) the chromatin accessibility of MER81 elements increased after bacterial or viral infection of human macrophages; and (2) that these elements showed unique enrichment for NF-kB motifs (Chen et al. 2023).
TF binding sites provided by ancient TEs can be thought of as “natural experiments” that can be followed through speciation events and studied in situ in relevant cell types. We propose that MER81, an eutherian DNA transposon with low replicative efficiency (Muñoz López and García-Pérez 2010; Wells and Feschotte 2020), serves as a clear example of this phenomenon, in which thousands of copies of a short DNA sequence containing two RELA motifs were dispersed throughout the genome prior to the divergence of placental mammals. However, it is important to acknowledge that other chromatin features and transcription factor (TF) motifs, not necessarily originating from transposable elements, likely play a role. For example, it has been well established that regions robustly bound by NF-kB typically contain multiple high-affinity motifs within an accessible chromatin context (e.g., Michida et al. 2020; Alizada et al. 2021), which may explain the modest ability of the MER81 element at the human IFNGR2 locus to increase gene expression in a luciferase reporter assay. Although additional higher throughput experiments focusing on the collaborating TFs and chromatin features are needed, our results demonstrate the ability of TE-derived NF-kB motifs to contribute to RELA binding at active CREs.
Methods
ChIP-seq
To annotate mouse TEs, we performed H3K4me2 and H3K27ac ChIP-seq with and without TNF treatment from mouse aortic endothelial cells. One biological replicate of primary mouse aortic endothelial cells (Cell Biologics, cat# C57-6052, lot# B092913T2MP) was selected and cultured following the methods of Alizada et al. (2021). Cells were then treated with 10 ng/mL recombinant mouse TNF (Cell Applications, cat# RP2031-20) for 45 min in basal Endothelial Cell Growth Media MV2 (PromoCell) without supplements (see Supplemental Methods). Cells were crosslinked in 1% formaldehyde for 10 min at room temperature and were then lysed for nuclei isolation as described in Schmidt et al. (2009). Mouse anti-H3K27ac monoclonal (Millipore, # 05-1334) and rabbit anti-H3K4me2 polyclonal (Millipore, # 07-030) antibodies were used. Samples were sequenced on Illumina HiSeq 2500 with a 100-bp single-end run to obtain ∼20–25 million single-end reads per sample.
H3K4me3-based HiChIP
We prepared TeloHAECs with/without TNF stimulation for HiChIP experiments (two replicates per condition). TeloHAECs were cultured following the protocol from Alizada et al. (2021). For TNF and vehicle treatment, the protocol was as described at the beginning of the ChIP-seq method, except here we used 10 ng/mL recombinant human TNF (Cell Applications, cat# RP1111-50). HiChIP was performed from a modified PLAC-seq protocol (Fang et al. 2016; see Supplemental Methods). The clear cell lysate was mixed with 500 µL of antibody-(2.5 μg of H3K4me3; Millipore, #04-745) coated Dynabeads M-280 (Sheep anti-Rabbit, Thermo Scientific). The biotin pull-down procedure was performed according to the Swift Biosciences Accel-NGS 2S Plus DNA Library kit. Libraries were sequenced on a NovaSeq 6000 S4 for PE150.
Luciferase reporter assay
For the investigated sequences, we designed four MER81 consensus-related sequences and two sequences from a native MER81 instance (located at Chr 21: 34,755,496–34,755,596 of hg19). The consensus sequence of MER81 was obtained from Repbase (Bao et al. 2015), and the native MER81 sequence was downloaded from the UCSC Genome Browser for coordinates of Chr 21: 34,755,496–34,755,596 (hg19). The mutant sequences were designed by randomly shuffling the RELA motifs in consensus/native sequences. We included a synthetic sequence with tandem 5 × RELA motifs as a positive control and generally observed >20-fold change with TNF stimulation compared to an empty pGL3 control for all experiments (values of data points are mentioned in the legend). Overall, we prepared six biological replicates with three technical replicates each for each investigated sequence to ensure reproducibility (see Supplemental Fig. S9 for more details).
CRISPR‐Cas9 deletions
Cas9-targeting guide RNAs (gRNAs) were selected flanking the selected MER81 instance (Chr 21: 34,755,496–34,755,596 of hg19) for deletion. On- and off-target specificity of the gRNAs was calculated as described in Doench et al. (2016) and Hsu et al. (2013), respectively, to choose optimal guides. Guide RNA sequences can be found in Supplemental Table S6. Guide RNA plasmids were assembled with gRNA sequences using the protocol described by Mali et al. (2013). TeloHAECs were transfected with 5 µg each of the 5′ gRNA, 3′ gRNA, and pCas9_GFP plasmids (Addgene, ID#44719; [Ding et al. 2013]) using the Neon Transfection System (Invitrogen). Forty-eight hours post-transfection, GFP-positive cells were isolated on a BD FACSAria. After recovery, single cells were seeded in 96-well plates and inspected for colonies after 1–2 weeks. All clones identified from the initial screen were sequenced across the deletion to confirm deletion boundaries. Four homozygously-deleted clones were selected for the following experiments (as shown in Supplemental Fig. S10).
RNA isolation and qPCR
TeloHAECs were treated with/without TNF for 45 min. Total RNA was purified from single wells of >85% confluent six-well plates using the RNeasy Plus Mini kit (Qiagen), and an additional DNase I step was used to remove genomic DNA. RNA was reverse-transcribed with random primers using the high-capacity cDNA synthesis kit (Thermo Fisher Scientific). Primary IFNAR1 and IFNGR2 gene expression was detected by primers targeting the first and fourth intron, respectively. The standard curve method was used to calculate expression levels using TeloHAEC genomic DNA to generate the standard curves. Levels of TUBA1A RNA were used as an internal control to normalize expression values. The primer sequences for IFNAR1, IFNGR2, and TUBA1A can be found in Supplemental Table S6.
Genomic data processing
RELA ChIP-seq data (including all cross-species and cross–cell type RELA ChIP-seq data) were mapped using the Burrows-Wheeler Alignment tool (BWA) (Li and Durbin 2009) as described in Alizada et al. (2021), followed by an additional quality filter (MAPQ ≥ 3) to retain high-quality uniquely mapping reads. We compared this filtering to what we would obtain using the “XT:A:U” flag in the SAM file. For each of the three species, all of our MAPQ filtered reads (MAPQ ≥ 3) were contained within the uniquely mapping reads obtained by the “XT:A:U” flag in the SAM file, suggesting that MAPQ ≥ 3 is a sufficiently stringent means to obtain uniquely mapping reads for our analysis. The histone ChIP-seq and ATAC-seq data were analyzed as described in Alizada et al. (2021) (without MAPQ filtering). H3K4me3 HiChIP was processed using the MAPS pipeline (Juric et al. 2019) with default parameters. Significant loops were called using MAPS with default parameters for each sample. Because chromatin loops are expected to stay largely unchanged within a rapid induction system (in our case, TNF treatment for 45 min), we merged the loops identified from basal and TNF treatment conditions to generate a union set of active chromatin loops (in total 19372) for TeloHAECs. These loops vastly map active enhancer-promoter or active promoter-promoter connections (see Fig. 5A for an example).
Enrichment analysis for TEs overlapping RELA-bound regions
We used the GAT tool (https://gat.readthedocs.io/en/latest/contents.html) to determine the potential overrepresentation of TEs within RELA-bound regions (Heger et al. 2013). To run the enrichment analysis, we used the peak summits of RELA peaks overlapping RepeatMasker-annotated TE subfamilies. We set the workspace (available genomic regions for simulation) as all nonrandom chromosomes. Ten thousand iterations were requested for empirical distribution estimation, and other parameters were set as default. The TE annotation files (matched annotation for hg19, mm10, and btau6) by RepeatMasker were downloaded using the table browser under the UCSC Genome Browser (https://genome.ucsc.edu). Simple repeats were excluded from all the TE annotations. The information of overlapping TEs for each RELA peak summit from human/mouse/cow can be found in Supplemental Table S7.
Estimation of evolutionary ages of TEs
Age was calculated for each TE instance using percent divergence of sequence (obtained from the aforementioned TE annotation files from UCSC Table Browser) divided by estimated nucleotide substitution rates of the corresponding species from Bulmer et al. (1991) and Pace et al. (2008). The median value of all instances from the same subfamily was calculated as the estimation of the subfamily age. Two point-estimations of human-mouse and mouse-cow divergence time (Fig. 2C) were acquired from the TimeTree database (Kumar et al. 2017). Human-mouse divergence time was later used to divide TEs into “young” and “old” categories based on whether the predicted age of TEs was younger or older than when humans and mice diverged (Supplemental Fig. S4).
Analysis of TE overlap with TF-bound regions
We took advantage of all the processed public TF ChIP-seq data from GTRD (Kolmykov et al. 2021) (v2018.3) to investigate TE and TF connections globally. The bound regions of TFs documented in GTRD were generated by merging ChIP-seq peaks from diverse conditions and cell types. This feature represents a comprehensive repertoire of bound regions for each TF, which allows us to study the general relationship between TEs and TFs in an unbiased manner. Note the way GTRD controls for multiple mapping reads in TF ChIP-seq data is slightly different from what we did for the multi-species RELA ChIP-seq data: GTRD uses MAPQ ≥ 10 to filter Bowtie 2–aligned reads (Langmead and Salzberg 2012), whereas our pipeline only enables high-quality uniquely mapping reads for downstream analysis. We downloaded all human/mouse GTRD peak clusters called by MACS2 (Zhang et al. 2008). The peak summits were then extracted to investigate the overlap between TEs and TF-bound regions (Supplemental Fig. S1).
TF motif analysis
In this study, we have performed two types of motif analysis: (1) motif enrichment analysis for a group of sequences; (2) motif scan analysis for individual TE instance or consensus sequence. The vertebrate TF motifs documented in JASPAR CORE (746 TFs in total [Castro‐Mondragon et al. 2022]) were selected as the database for both analyses. For motif enrichment (1) analysis, CentriMo from MEME suite (Bailey et al. 2015) (version 4.12.0) was used with the threshold “-ethresh” set to 1. The query sequences—for example, from Figure 4—were from RELA-bound TE instances. The overlapped RELA peak summits were first extracted for all these instances and extended with 250 bp in both directions. CentriMo was then performed for instances of each TE subfamily that were identified to be significant, as shown in Figure 2. RepeatMasker annotation was used to determine the corresponding position in the consensus sequence for each identified motif from instances. The occurrences of motifs in each position were then counted and normalized by the total number of instances.
Motif-word analysis for several example TE subfamilies
Motif-word analysis has been performed for Bov-tA1/2/3 and MER41B. According to the motif distribution plots (e.g., Fig. 4C; Supplemental Fig. S7A), the position of the most prominent “suboptimal” RELA motif (a sequence that is only one or two nucleotides away from a matched RELA motif) in the consensus sequence was determined for each TE subfamily. Next, the RELA motif was scanned over all the instances, and only matches with RELA motifs in instances that corresponded to the position of the “suboptimal” RELA motif in consensus were recorded. The recorded RELA motif-words were then counted for occurrences, and the most abundant motif-words were picked for closer investigation based on the elbow plot (Supplemental Fig. S7B). Finally, the occurrences of these motif-words in the queried position were counted for all the instances (denoted as “background”) and RELA-bound instances (Fig. 5D; Supplemental Fig. S7B).
Visualization of sequence constraints and epigenetic signal profile
We used phastCons scores (Siepel et al. 2005) (downloaded from hg38.phastCons100way.bw) to evaluate the sequence constraints around MER81 instances (Fig. 5C). To achieve the compatibility with phastCons scores from hg38, we lifted sequences from hg19 to hg38 using liftOver (with all default parameters) (Hinrichs et al. 2006). For epigenetic data, we used the UCSC Genome Browser (Tyner et al. 2017) to visualize the signal from ChIP-seq, ATAC-seq, and loops identified from H3K4me3 HiChIP on the whole-genome scale. As to the aggregate plot of ChIP-seq and ATAC-seq signal around RELA-bound TEs (e.g., Fig. 5D; Supplemental Figs. S3, S4), we used computeMatrix and plotProfile functions from the deepTools2 suite (Ramírez et al. 2016) (v3.1.3). To visualize stranded eRNA signals from ChRO-seq, we used HOMER (Heinz et al. 2010) (v4.11). Details and parameter choices were as in Alizada et al. (2021).
Target gene association and functional enrichment analysis for RELA-bound TEs
The significantly upregulated, downregulated, and constitutive human gene sets used in this study were previously defined in Alizada et al. (2021). The distance of the four groups of RELA-bound regions relative to the three gene groups was determined using the TSS annotation of GENCODE (v19) (Fig. 3B). Note that we considered merely one TSS per gene. The occurrences were counted in 10-kb bins and then converted to the corresponding fraction value for the distribution in Figure 3B. For the background reference, all the annotated TE instances were considered and the corresponding fraction was calculated accordingly. The functional enrichment analysis was performed using GREAT (McLean et al. 2010) (v3.0.0).
Cross-species and cross–cell type comparison of RELA-bound regions
We define conserved RELA binding events in one species as the ones that can be detected in the orthologous regions of at least another species with at least 1-bp overlap. The workflow to identify the conservation degree (either conserved or species-specific) of any given RELA-bound region is as in Alizada et al. (2021). Based on this rule, human RELA peaks were divided into conserved and human-specific groups, as shown in Figure 3. Similarly, when compared across human cell types, a RELA-bound region is shared if another site is identified from at least one other cell type with at least 1-bp overlap.
Ancestral genome reconstruction, MER81 annotation, and RELA motif detection
The genomes were reconstructed using Ancestors 1.1 (Diallo et al. 2010) which was applied to an alignment of 58 eutherian genomes extracted from UCSC's 100-way whole-genome alignment. Briefly, the tool employs a maximum likelihood tree-HMM approach with a context-dependent substitution algorithm to infer indels and substitutions within each branch of the phylogenetic tree and derive the ancestral sequence at each node. Its accuracy has been extensively demonstrated, especially for primate ancestors (Blanchette et al. 2004). RepeatMasker (Tempel 2012) with Dfam libraries (Hubley et al. 2016) was run for each reconstructed genome to annotate all human transposable elements. More details were extensively described by Campitelli et al. (2022). To acquire high-quality MER81 annotations, we additionally required MER81 hits to be marked as nonredundant and contain a minimal length of 90 bp with RepeatMasker. For the detection of RELA motifs, we extracted sequences of all the valid MER81 instances from these reconstructed genomes and used FIMO (Grant et al. 2011) to scan RELA motifs. The motif occurrences within identified MER81 instances for each genome were counted and visualized in Figure 6, B and C. The corresponding position of motif matches relative to the consensus sequence was also recorded for each genome (Fig. 6D).
Simulation for neutral decay of MER81 elements during evolution
MER81 elements were extracted from the oldest reconstructed genome (the common ancestor of human and star-nosed mole, or node 10). A primate-specific estimate of nucleotide substitution rate (2.2 × 10−9/ [year × site] Bulmer et al. 1991; Pace et al. 2008) was used to simulate sequence change for the following 10 evolutionary time points. Starting from the collection of the oldest MER81 elements (real observations), all the following time points were estimated solely on nucleotide substitution rate. For each node/time point, all the simulated MER81 elements were scanned for RELA motifs using FIMO, and the percentage of elements with significant motifs was recorded. We iterated 1000 times to obtain the mean values for each node, which were used to plot the gray line in Figure 6C.
We used several published data sets: the human/mouse/cow ChIP-seq and ATAC-seq data were downloaded from ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) under accession numbers E-MTAB-7889 and E-MTAB-7878, respectively (Alizada et al. 2021). HAEC RNA-seq and TeloHAEC ChRO-seq were downloaded from ArrayExpress under accession numbers E-MTAB-7896 and E-MTAB-9425 (Alizada et al. 2021). The RELA ChIP-seq data for HUVEC (Brown et al. 2014), LCL (Kasowski et al. 2010), and adipocytes (Schmidt et al. 2015) were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE54000, GSE19486, and GSE64233, respectively.
Data access
All raw and processed sequencing data generated in this study have been submitted to the ArrayExpress database (https://www.ebi.ac.uk/biostudies/arrayexpress/) under accession numbers E-MTAB-12212 and E-MTAB-12213. Custom scripts and files used for the analysis are available at Zenodo (https://doi.org/10.5281/zenodo.8156730) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Alan Moses, Dustin Sokolowski, and Cadia Chan for helpful discussions, the Centre for Applied Genomics (TCAG) for assistance with DNA sequencing, and the anonymous reviewers for helpful suggestions. Funding for this project was provided by the Canadian Institutes of Health Research (CIHR) (201603PJT-364832) to M.D.W. and J.E.F. and the National Institutes of Health (S.R., M.D.W., and J.A.M.; R01-HG010045-01). J.E.F. and M.D.W. were supported by an Early Researcher Award from the Ontario Ministry of Research and Innovation and Tier 2 Canada Research Chairs from CIHR. T.R.H., L.F.C., S.E.P., and Z.M.P. were supported by a CIHR Foundation Grant (FDN148403) to T.R.H. N.K. received a Canada Graduate Scholarship from the Natural Sciences and Engineering Research Council of Canada (NSERC) and an Ontario Graduate Scholarship. L.A. was supported by a NSERC CGS-M. K.R. was partially supported by a postdoctoral fellowship from the Ted Rogers Centre for Heart Research. L.F.C. was supported by a NSERC PGS-D scholarship. H.H. and M.L. were supported by NSERC grant RGPIN-2019-07014 to M.D.W.
Author contributions: L.W., H.H., and M.L. performed data analysis and wrote the manuscript. A.A., K.R., and L.A. designed and performed experiments; N.K. performed luciferase assays; T.T. and I.C.T. performed CRISPR-Cas9 experiments; L.F.C., Z.M.P., and S.E.P. performed ancestral genome analyses; T.R.H., S.R., J.A.M., and J.E.F. provided funding and supervision. M.D.W. designed experiments, wrote the manuscript, acquired funding, and supervised the project. All authors edited and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280357.124.
- Received December 19, 2024.
- Accepted April 23, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








