
Continuous infiltration and evolutionary trajectory of nuclear organelle DNA in Oryza
- Chenbo Gong1,
- Yicheng Huang1,
- Mengmeng Liu1,
- Yong Zhou2,
- Yinwei Xu1,
- Nahed Mohammed2,
- Xintong Qiao1,
- Andrea Zuccolo2,
- Weibo Xie1,
- Rod A. Wing2,3,
- Jianwei Zhang1,
- Fei Zhou1 and
- Yongjun Lin1
- 1National Key Laboratory of Crop Genetic Improvement, Hubei Hongshan Laboratory, Huazhong Agricultural University, Wuhan 430070, China;
- 2Center for Desert Agriculture, Biological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955‐6900, Saudi Arabia;
- 3Arizona Genomics Institute, School of Plant Sciences, University of Arizona, Tucson, Arizona 85721, USA
Abstract
Transfer of chloroplast or mitochondrial DNA into the nuclear genome is a common phenomenon in many species. However, little is known about the evolutionary fate and mechanism of transfer of organellar DNA sequences in higher plants. We observe abundant insertions of organelle DNA into the nuclear genomes of 22 genome assemblies across seven Oryza species and further categorize nuclear organelle DNA (NORG) into 3406 orthologous groups. Analysis of the whole-genome resequencing data from 3458 O. sativa, O. glaberrima, and O. barthii accessions indicate that NORGs have intra- and inter-population variability owing to sequence loss and transposon insertion during evolution. Our results also suggest that NORGs have been continuously produced during the evolution of Oryza, and both double-strand break repair pathways and replication-based mechanisms play important roles in integrating organelle DNA into the nuclear genome. Further investigation indicates that complex NORGs are formed through single mutational events before or during the insertion process via ligation of multiple plastid and/or mitochondrial DNA with each other. In summary, this work provides novel insights into the process of endosymbiotic DNA transfer and its role in reshaping genome variation and plant genome evolution.
The observation of “promiscuous DNA,” which is defined as a nucleotide sequence occurring in more than one of the three membrane-bound organellar genetic systems of eukaryotic cells (Ellis 1982), provides evidence for DNA transfer between organelles and the nucleus. Such transfers of mitochondrial DNA (mtDNA) or plastid DNA (ptDNA) to the nucleus result in formation of nuclear mitochondrial DNA (NUMT) and nuclear plastid DNA (NUPT), which are collectively known as nuclear organelle DNA (NORG) (Leister 2005). The consistent and frequent occurrence of organelle-to-nucleus transfer of DNA has been demonstrated by many experimental studies (Thorsness and Fox 1990; Huang et al. 2003; Stegemann et al. 2003).
To date, NUMT has been ubiquitously found in eukaryotic nuclear genomes (Richly 2004; Hazkani-Covo et al. 2010; Dayama et al. 2020), representing an important source of genetic variation in the nuclear genome, and plays an important role in cellular evolution (Liang et al. 2018; Puertas and González-Sánchez 2020). In humans, de novo formation of NUMT occurs approximately once in every 4000 births (Wei et al. 2022), with a higher rate in cancer cells (Ju et al. 2015; Wei et al. 2022), leading to high NUMT diversity across the human population. The insertion of mtDNA into the nuclear genome can cause genomic instability via mutations or disruption of normal gene function, which may result in several human diseases such as carcinogenesis and genetic disorders (Puertas and González-Sánchez 2020; Harutyunyan 2024).
Transfer of mtDNA and ptDNA to the nucleus occurs frequently in plants (Huang et al. 2004; Richly and Leister 2004; Noutsos et al. 2005; Portugez et al. 2018; Zhang et al. 2020). Moreover, the large size, complex structure, and frequent rearrangement events of mtDNA (Gualberto and Newton 2017; Morley and Nielsen 2017; Wu et al. 2022) contribute to the diversity of NORGs in plants. Plant NORG diversity is further increased by the mosaic sequences formed from rearranged DNA originating from both mtDNA and ptDNA (Noutsos et al. 2005; Portugez et al. 2018). Triticeae species tend to have more nuclear organellar genes than other Poaceae species (Chen et al. 2023). In Asian cultivated rice, several NUPT and NUMT events occur specifically in one subspecies (Huang et al. 2005; Guo et al. 2008; Wang and Timmis 2013), and giant NORGs (>10 kb) vary greatly in Asian and African rice (Ma et al. 2020). Currently, little is known about the overall profile and dynamics of NORGs in different Oryza species or across diverse populations.
Several potential mechanisms for the formation of NORGs have been reported. Capture of mtDNA and ptDNA during double-strand break repair (DSBR) has been confirmed in yeast (Ricchetti et al. 1999; Yu and Gabriel 1999; Decottignies 2005) and tobacco (Wang et al. 2012, 2018a). In primates, NUMT exhibits the characteristics of non-homologous end joining (NHEJ) (Hazkani-Covo and Covo 2008; Wei et al. 2022). However, nuclear integration of mtDNA in somatic cells frequently occurs together with other complex rearrangement events, suggesting that formation of NUMTs may involve mechanisms in addition to NHEJ (Ju et al. 2015). In plants, the specific mechanism underlying NORG formation remains elusive.
The Oryza genus is a grass model system for studying molecular evolution. With an evolutionary history of ∼15 million years, Oryza includes two cultivated rice species, Asian cultivated rice Oryza sativa and African cultivated rice Oryza glaberrima, along with 25 wild species. It comprises six diploid genome types (AA, BB, CC, EE, FF, and GG) and five allopolyploid genome types (BBCC, CCDD, HHJJ, HHKK, and KKLL) (Wing et al. 2018). Here, to obtain the landscape of NORGs and understand its variations during the evolution of Oryza, we identified and analyzed NORGs at the pangenome level focusing on evolutionary patterns, population dynamics, formation, and fragmentation mechanisms of NORGs. The results provide a resource for interpreting NORG variation and formation across diverse rice populations and improve our understanding of genome evolution.
Results
Abundant organelle DNA insertions in the nuclear genomes of Oryza species
To obtain the landscape of NORGs and understand its continuous variation during the evolution of Oryza genus, we identified and analyzed NORGs at the pangenome level using 22 high-quality genomes of representative Asian cultivated rice (O. sativa) (Kawahara et al. 2013; Zhou et al. 2020; Song et al. 2021) and six other Oryza species (Table 1; Supplemental Fig. S1; Stein et al. 2018; Ntakirutimana et al. 2023; Thathapalli Prakash et al. 2023). In total, we identified 21,971 NORGs in all 22 genomes. Among these NORGs, 20,085 NORGs in the AA genome were classified into 3406 orthologous NORG groups (ONGs), whereas only 948 ONGs were identified in the IRGSP-1.0 O. sativa ssp. japonica (AA genome) reference genome (Supplemental Table S1). This discrepancy indicates that simply relying on a single reference genome fails to comprehensively represent the diversity of NORGs within the Oryza genus. A total of 138 to 841 kb NUPTs and 180 to 932 kb NUMTs were identified (Table 1; Supplemental Table S2). The total lengths of NORGs across the 22 genomes range from 301 kb to 1574 kb, accounting for 0.12% to 0.42% of the nuclear genomes, with an average of 0.26% in the AA genome (Table 1; Supplemental Fig. S1). All NUPTs in each genome cover the complete plastid genome region, indicating that the ptDNA sequences are ubiquitously transferred to and retained in the nuclear genome (Supplemental Fig. S2), whereas NUMTs do not cover the entire mitochondrial genome (covering 43% to 79% of the mtDNA) (Supplemental Fig. S3). The origins of NUPTs and NUMTs from organellar genomes were not random, with the RPS8 gene region of ptDNA and the RRN5–RRN18 gene region of mtDNA appearing the most frequently (Supplemental Figs. S2, S3).
Summary of NORG content in assemblies of Oryza genus
The number of ONGs in each genome is similar among different accessions and has a positive correlation with the chromosome length (R2 = 0.759) (Fig. 1A,B), but the total length of NORGs demonstrates a relatively weaker association with chromosome length (R2 = 0.0303) (Fig. 1C,D). The majority of ONGs (83%) are <1000 bp, indicating that NORG insertion usually involves short fragments (Fig. 1E; Supplemental Fig. S4). ONGs appear to be uniformly distributed across chromosomes with no definite tendency for the insertion into centromeric regions (Kolmogorov–Smirnov test P-value > 0.01 in 10,000 uniformly distributed simulations) (Fig. 1F; Supplemental Fig. S5). Still, ONGs tend to be located in noncoding regions of the nuclear genome, with only a small number of ONGs overlapping with exons in each genome (chi-square test P-value < 0.01 in 1000 NORG random shuffle simulations; see Methods) (Supplemental Table S3). In plants, some species accumulate longer NUMT than NUPT with genome size as a key factor determining accumulated length of relatively older NUPT and NUMT (Yoshida et al. 2017). The presence of giant NORGs (>10 kb), such as those in Chr 10 and Chr 12 of Nipponbare and CHAO MEO, respectively, might be the reason for the variation in the total length of NORGs.
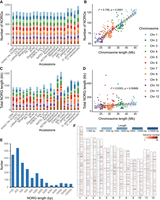
NORG length varies, but number and distribution are relatively constant across Oryza genomes. (A) NORG number per chromosome in each accession. (B) Correlation of NORG number with the chromosome size. (C) Total length of NORGs per chromosome in each accession. The total length of all NORGs from each chromosome was summed. (D) Sum of NORG lengths per chromosome in each accession correlated to chromosome size. (E) ONG length distribution. The x-axis indicates the length range of ONGs, with 200 indicating a length range of 100–200 bp, 300 indicating a length range of 200–300 bp, and so on. (F) Chromosomal distribution of ONGs in 16 O. sativa accessions. Chromosome heatmaps in the left and right represent the length and frequency of ONGs, respectively. A total of 1456 ONGs in 16 O. sativa assemblies were mapped to the O. sativa ssp. japonica cv. Nipponbare genome IRGSP-1.0.
In total, we classified five types of NORGs according to the origin of organelle DNA (Fig. 2A–E). The proportions of different types of NORGs were similar in all genomes (Fig. 2F). Among the NORGs with lengths <500 bp, simple NUMTs and simple NUPTs predominated. In contrast, for NORGs >1000 bp in length, the majority of them were complex NORGs, with sequences matching multiple distinct regions from plastid, mitochondria, or both organelles (Fig. 2G). The three types of complex NORGs also had different numbers of fragments, with nuclear mosaic insertions (NUMINs) (Fig. 2E) having more fragments (Fig. 2H).
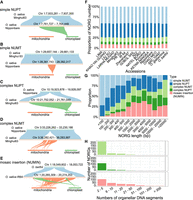
NORG types vary depending on length but are consistent across Oryza genomes. (A–E) Structure of five types of NORGs. (A) A simple NUPT consisting of one segment from plastid. Synteny between Minghui63 Chr 1: 7,933,281–7,937,300, ONG0100220 in Nipponbare, and the plastid of rice. (B) A simple NUMT consisting of one segment from mitochondria. Synteny between Minghui63 Chr 1: 29,657,144–29,661,133, ONG0100760 in Nipponbare, and the mitochondria of rice. (C) A complex NUPT consisting of two segments from the plastid. Synteny between Nipponbare Chr 10: 19,923,878–19,928,097, ONG1000620 in Minghui63, and the plastid of rice. (D) A complex NUMT consisting of eight segments from the rice mitochondria. Synteny between Nipponbare Chr 3: 33,226,262–33,230,186, ONG0301080 in Minghui63, and the mitochondria of rice. (E) A nuclear complex insertion (NUMIN) consisting of two segments from plastid and 26 segments from the mitochondria. Synteny comparison between O. barthii Chr 1: 18,049,602–18,053,722, ONG0100500 in O. sativa IR64, and the mitochondria and the plastid of rice. (F) Relative proportion of five types of NORGs in each accession. (G) Relative proportion of each type of NORGs with different lengths. The length of NORGs was calculated using the median size. (H) Number of organelle DNA segments in complex NORGs.
Analysis of near-intact organellar genes (NIOGs; ≥95% coverage of an organellar gene) within the nuclear genome revealed that 435 (13%) ONGs contain NIOGs (Supplemental Table S6). The NIOG number ranges from 308 to 969, covering 123 (58%) to 189 (89%) organellar genes (Supplemental Table S7). Among these, 34% to 59% of the protein-coding genes lack complete CDS (Supplemental Table S6). The number of nuclear mitochondrial genes was smaller than that of nuclear plastid genes, probably because mitochondrial genomes have a lower gene density than plastids (Supplemental Table S7).
Recent transfer and nonconservation of NORGs
Abundant presence/absence variations (PAVs) of ONGs were identified in 20 AA genome accessions (Fig. 3A). Among the 1728 ONGs in O. sativa genomes, 34% (590/1728) of them are present in all the 16 O. sativa accessions, suggesting that these ONGs that were already present in the common ancestor of O. sativa are termed the O. sativa “common ONGs” (Fig. 3B). Among these “common O. sativa ONGs,” ∼80% (470) of them are shared with both O. glaberrima and Oryza barthii (Fig. 3C). The remaining 66% of ONGs in O. sativa have PAVs (Fig. 3B). Abundant species-specific ONGs were found, suggesting that the insertion of organelle DNA into nuclear genomes is ongoing (Fig. 3D).
Abundant presence/absence variations of ONGs in Oryza genomes. (A) Presence and absence information of ONGs in five AA genome species. The presence of a NORG is indicated by blue, and the absence of a NORG is indicated by white. (B) Distribution of ONGs among 16 O. sativa accessions. The “common ONGs” are those present in all accessions; “variable ONGs” are present in more than one but fewer than 16 accessions; “private ONGs” are present in only one accession. (C) Distribution of O. sativa “common ONGs” in O. glaberrima and O. barthii. (D) Position of ONGs on the phylogenetic tree of AA genome species in Oryza. The total number of ONGs in genomes is shown above branches. The number of branch-specific ONGs is shown below branches. The four lines under each node from top to bottom represent O. meridionalis, O. glumaepatula, O. glaberrima and O. barthii, and O. sativa.
Interestingly, the more ancient the evolutionary branch, the fewer shared ONGs were identified. Five hundred ninety ONGs were conserved in all 16 O. sativa accessions; 180 ONGs were present in all 20 accessions of the AA genome, of which only 28 ONGs were present in Oryza punctata (BB genome) (Fig. 3D).This indicates that ONGs were not conserved during evolution, and most of ONGs identified in this study occurred as new insertions after the speciation between ancestors of AA and BB genome species.
To understand the characteristics of conserved ONGs, the ONGs in Nipponbare were divided into three groups, including those fixed in AA species, those fixed in O. sativa, and those not fixed in O. sativa. The distribution of ONG positions relative to genes was then compared. Conserved ONGs are more probably located within genic regions than nonconserved ONGs (Fisher's exact test P-value = 5 × 10−4) (Supplemental Fig. S6), indicating a potential evolutionary constraint on the retention of ONGs during evolution.
Population dynamics of NORG insertions in Asian and African rice
To further investigate the distribution of ONGs in Asian and African rice at the population level, 828 ONGs exhibiting PAVs in O. sativa, O. glaberrima, and O. barthii, and with available relative insertion positions were selected. A total of 74 ONGs that could not be assessed in the whole-genome resequencing (WGS) data were filtered out, leaving 754 ONGs for further analysis. Then, the analysis was conducted using sequencing data from 3458 accessions (Supplemental Table S4).
Multidimensional scaling (MDS) analysis of ONGs distinguishes well the Asian and African populations (Fig. 4A), O. glaberrima and O. barthii (Fig. 4B), four major groups of O. sativa (Fig. 4C), and three subpopulations of the japonica group (Fig. 4D) except for indica groups (Fig. 4E). PAV analysis of ONGs showed that there is considerable variation among and within the subpopulations of O. sativa (Fig. 4F; Supplemental Table S5). Among these ONGs, 422 and 432 ONGs showed different distributions among the four major O. sativa groups and the 15 subpopulations, respectively (Fisher's exact test P-value < 0.01). As shown in Figure 4F, 92 subpopulation-predominant, 183 major group-predominant, 29 O. sativa–predominant, 83 O. glaberrima–predominant, 42 O. barthii–predominant, and 142 O. glaberrima/O. barthii–predominant ONGs were observed. These results indicate that the distribution of ONGs is population and species specific. On this basis, we set out to find the representative ONGs in each major group and obtained three Xian-indica (XI) representative ONGs (ONG0900650, ONG0301080, and ONG1000640), one Geng-japonica (GJ) representative ONG (ONG0300140), one circum-Aus (cA) representative ONG (ONG0500890), and one circum-Basmati (cB) representative ONG (ONG0500930).
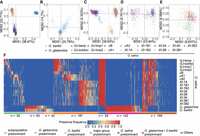
PAVs of ONGs reviews population structure and subpopulation differences. (cA) circum-Aus; (cB) circum-Basmati; (GJ) Geng-japonica, for which (trop) tropical, (subtrp) subtropical, and (temp) temperate; and (XI) Xian-indica. (A,E) Multidimensional scaling plots for all accessions of O. sativa, O. glaberrima, and O. barthii (A); O. sativa (B); O. glaberrima and O. barthii (C); GJ groups of O. sativa (D); XI groups of O. sativa (E). (F) Presence frequencies of ONGs (n = 754) plotted by heatmap.
DNA-mediated organellar gene transfer
Experimental evidence has indicated that mtDNA escape is independent of RNA intermediates in yeast (Shafer et al. 1999), and plastid-to-nucleus transfer of DNA in plants can occur without the elimination of introns (Fuentes et al. 2012) or RNA editing sites (Sheppard et al. 2011). Further, an RNA-mediated model posits that mitochondrial genes could undergo retroprocessing in the mitogenome before being physically transferred as DNA (Wu et al. 2017). We examined the transfer of 15 organellar intron-containing genes and five trans-splicing genes and found no postsplicing forms among NORGs. Only 11 ONGs contained one to four RNA-editing sites that had already changed to T bases, with no NIOGs showing C-to-T change at all RNA editing sites (Supplemental Table S6). These findings support that NORGs are transferred predominantly via DNA rather than via RNA or cDNA intermediates.
Signatures of junction sites of de novo NORG insertions
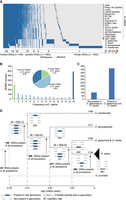
To better understand the mechanisms contributing to NORG formation, we examined the junction sequences of NORG insertions in 828 polymorphic ONGs from O. sativa, O. glaberrima, and O. barthii, for which we could obtain exact organelle DNA insertion sites. Microhomologies, blunt ends, and fillers were observed in the breakpoints (Fig. 5A; Supplemental Table S8), similar to the characteristics of T-DNA insertion (Kleinboelting et al. 2015). Microhomology (≥1 bp) was found in 48% of the NORG junctions, with the length of microhomology significantly exceeding the length expected by chance (chi-square test P-value < 2.2 × 10−16) (Fig. 5A; Supplemental Fig. S7A). Moreover, target site duplication (TSD) and DNA loss were also found in NORG insertion sites (Fig. 5B). Among the 110 ONGs (13%) with TSD lengths >5 bp, 36 had TSD >20 bp, which is significantly longer than the length expected by chance (chi-square test P-value < 2.2 × 10−16) (Fig. 5A,D–E; Supplemental Table S8; Supplemental Fig. S7B). The majority of ONGs (73%, 607/828) appear to have been formed through DSBR, with the primary mechanism being classical non-homologous end joining (c-NHEJ; 59%, 975/1656) (Supplemental Table S8). Among these, 29% (179/607) of the ONGs exhibit distinct formation mechanisms at their two junction sites, with one being c-NHEJ and the other being alternative end joining (alt-EJ) or microhomology-mediated template switching (Fig. 5C; Supplemental Table S8). Fork stalling and template switching/microhomology-mediated break-induced replication (FoSTeS/MMBIR) contributed to the formation of the remaining ONGs (27%, 221/828), resulting in both sequence loss and flanking sequence duplication that could not be explained by DSBR mechanisms (Supplemental Table S8). Collectively, these results indicate that both DSBR and replication-based mechanisms (RBMs) can generate NORGs.
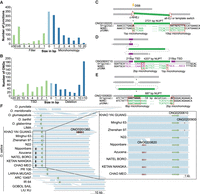
Characteristics of organelle DNA insertions reflecting its formation mechanisms. (A) Size distribution of microhomology and filler DNA at insertion sites of NORGs. (B) Size distribution of target-site duplications and deletions at insertion sites of NORGs. (C–E) Nucleotide-resolution breakpoint sequence of organelle DNA nuclear insertions. Red rectangles highlight sequence microhomology. (F) A region on Chr 2 has four independent NORGs among AA genome accessions.
Formation of complex NORGs
It remains to be determined whether complex NORGs evolved from simple NORG insertions or from more complex/aberrant recombination mechanisms. To address this, we propose three potential formation pathways: (1) single mutational events involving ligation of organelle DNA fragments before or during nuclear insertion (Supplemental Fig. S8A), (2) multiple insertion events at the same site (Supplemental Fig. S8B), and (3) postinsertion rearrangements after organelle DNA insertion (Supplemental Fig. S8C). First of all, methodologically, multiple insertions occurring in nearby regions were treated as independent ONGs, which ensures that complex NORGs formed by multiple insertion events are not categorized as single ONG. In the 828 ONGs mentioned above, 33% (272/828) of the ONGs are complex NORGs (120 NUMTs, 51 NUPTs, and 101 NUMINs). Collinearity analysis of these ONGs and their flanking sequences indicated that these ONGs exhibit PAVs across different accessions, but there was no rearrangement after insertion. The insertion sites of simple and complex NORGs both suggested their formation through RBM and DSBR. Six conserved complex NORGs in O. sativa accessions were further examined. Collinearity analysis revealed these NORGs originated in and fixed in O. sativa without no sequential insertion or structural rearrangements postinsertion, strongly supporting their origin from a single event (Supplemental Fig. S9). In addition, 10 out of the 828 ONGs are found to be located within 5 kb regions around existing ONGs (Fig. 5F; Supplemental Fig. S10). Although rare cases of hypotheses 3 cannot be entirely excluded, systematic collinearity classification and structural analyses indicate that most complex NORGs arise via single mutational events.
Moreover, newly inserted NORGs can also be classified as complex NORGs (Supplemental Fig. S10A,C,G,H). For example, the simple NUPT ONG0200610 was formed in the common ancestor of Oryza glumaepatula and O. sativa (Fig. 5F). Subsequently, in the O. sativa clade, both new complex NORGs and simple NORGs inserted into its flanking region, with the most recently formed NORG (ONG020136) exhibiting the most complex form (Fig. 5F). These results suggest that both simple and complex NORGs can form regardless of the insertion time or rearrangement after insertion.
In addition, among the 828 ONGs mentioned above, 11% (88/828) contained long DNA fillers >50 bp in the insertion sites, suggesting that NORG insertion can occur in conjunction with the insertion of nonorganelle DNA during single transfer events, leading to complex structural variations (Supplemental Table S8). In conclusion, complex NORGs could be formed by single mutational events, and the newly inserted NORGs near existing NORGs may also increase the complexity of NORGs.
NORG fragmentation triggered by transposable elements
Collinearity analysis revealed sequence insertions in 71 ONGs in one or more accessions, which resulted in the splitting of single NORGs into multiple fragments with very large distances between each other. The insertions were mainly composed of transposable elements (TEs), mostly the Ty3/Gypsy LTR retrotransposons (Supplemental Fig. S11). Among the transposon insertion polymorphisms in rice, Ty3/Gypsy elements are most abundant (Huang et al. 2008). It has also been observed that the content of Ty3/Gypsy is particularly high in centromeric regions compared with in other regions (Song et al. 2021). Once a NORG is integrated into nuclear genome, it becomes part of the nuclear DNA and, like other sequences, is subjected to the influence of TEs. Thus, the presence of Ty3/Gypsy insertions within NORGs may reflect their general abundance and activity within the genome, rather than a NORG-specific association.
Based on their original and derived forms, we further interpreted the dynamic change process of NORGs. For example, ONG0400870 appears to have been a 66 kb insertion of mtDNA along with >10 kb sequences from chromosomal DNA in the ancestor of the AA genome, which underwent independent changes (i.e., sequence loss and TE insertion) in different clades (Fig. 6A,B). After speciation, seven insertions occurred in Oryza meridionalis, whereas an independent insertion and large segment loss occurred in O. glumaepatula. In the common ancestor of O. sativa, O. glaberrima, and O. barthii, a segment loss occurred in the middle of the NUMT. In O. sativa, several independent insertions occurred within NORGs, resulting in their dispersion and fragmentation (Fig. 6A,B). Similarly, ONG0400650 formed in the common ancestor of O. sativa, which contains two separate NUPTs. The second NUPT then underwent several independent TE insertions in different accessions (Fig. 6C,D). Multiple changes from transposons were likewise found in ONG0100920, ONG0900660, and others (Supplemental Fig. S12).
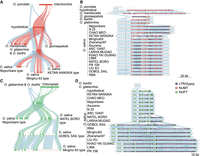
Transposons result in the fragmentation of NORGs. (A) Dynamic process of ONG0400870. The “hypothetical” indicates the insertion form of ONG0400870 in the presumed ancestor of the AA genome, which is inferred from the form of ONG0400870 in the AA genome. The orange color indicates the mtDNA sequence. (B) Synteny of the ONG0400870 region in the O. punctata (BB genome) and AA genomes. The NORG is not present in O. punctata. Orange indicates the NUMT. (C) Dynamics of ONG0400650. The “hypothetical” indicates the insertion form of ONG0400650 in the presumed ancestor of the O. sativa genomes, which is inferred from the form of ONG0400650 in O. sativa. Green color indicates ptDNA sequence. (D) Synteny of the ONG0400650 region in O. barthii, O. glaberrima, and O. sativa. The NORG is not present in O. barthii and O. glaberrima. Green color indicates NUPTs.
Discussion
In this study, a high-throughput identification pipeline for NORGs was established and used to reveal the landscape of NORGs in Oryza using 16 O. sativa genomes representative of all 15 subpopulations and six other wild relatives of Oryza. Our methods based on collinearity between different accessions were used to identify and analyze ONGs, thereby providing a more accurate detection process of organelle DNA insertion and change, which is similar to the approach and advantages described by Uvizl et al. (2024). NORGs can be fragmented by insertions into multiple segments separated at distances of several to tens of kilobases (Fig. 6). At the same time, there are also cases in which new NORGs are inserted within 5 kb of pre-existing NORGs (Fig. 5F). Such changes cannot be identified by determining whether the adjacent sequences from organelle DNA are the same NORGs according to a threshold (such as 5 kb intervals).
Importantly, we found that the predominant forms of NORGs in Oryza were mostly <1000 bp in length (Fig. 1E), and longer NORGs were mainly complex NORGs (Fig. 1G). Few intact and contiguous long organelle DNA fragments were observed. Similarly, the majority of NUMTs in humans (Wei et al. 2022), as well as the NUMTs and NUPTs in Triticum/Aegilop complex species (Zhang et al. 2024b) and Toxoplasma (Namasivayam et al. 2023), are short insertions, indicating that the short-fragmented nature of NORGs is widespread in eukaryotes. Small fragments of NUMT can be inserted into the site of DSBR in tobacco, which may result from extensive fragmentation of mtDNA (Wang et al. 2012). Previous studies have found that microinjected plasmid DNA in HeLa and COS cells disappeared within 50–90 min owing to the rapid plasmid degradation caused by cytosolic nuclease(s) (Lechardeur et al. 1999). In addition, the rate of DNA diffusion through the cytoplasm is heavily dependent on the DNA size (Lukacs et al. 2000), and therefore, small fragments of free organelle DNA are more likely to enter the nucleus. In summary, we suspect that damage or fragmentation of free organelle DNA in the nucleus may be the primary reason for the presence of more short-fragment NORGs than long-fragment NORGs.
The escape of DNA from chloroplast to nucleus is estimated to occur once per 16,000 pollen grains (Huang et al. 2003) and once per 5 million somatic cells (Stegemann et al. 2003). Such a high frequency has also been observed in mitochondria, with a rate of approximately 2 × 10−5 per cell per generation in yeast (Thorsness and Fox 1990) and once in every 4000 births in humans (Wei et al. 2022). This work estimates the rate of organelle-to-nucleus DNA transfer (and retention) at an evolutionary scale in plants. In this study, it was estimated that the insertion rate of ONGs in AA genome was 631.12–943.81 insertions per million years (Fig. 3D), suggesting that there has been a significant amount of de novo insertions of organelle DNA in the nucleus genome during the evolution of Oryza and that this process is ongoing and frequent.
In addition, the evolutionary branches with greater distances tended to have fewer conserved ONGs, indicating that some NORGs may be gradually lost owing to nuclear genomic variations during evolution (Fig. 3D). Once formed, NORGs will be subjected to persistent mutations in the nuclear genome, such as base mutations and insertions and deletions (indels) (Huang et al. 2005), and may be lost in progeny owing to their instability (Sheppard and Timmis 2009). Here, we found that NORGs, especially nonconserved ones, are primarily located in intergenic regions (Supplemental Fig. S6; Supplemental Table S3), suggesting selection against insertions that may disrupt genes. In Triticum/Aegilops complex species, NUPTs and NUMTs also tend to be located in intergenic regions, as accompanied by high levels of DNA methylation and low levels of active histone modification (Zhang et al. 2024b). These findings collectively suggest that NORGs primarily constitute nonfunctional sequences, which may be the reason for the low conservation of NORGs observed in this study. Moreover, we suspect that TE activity is an important cause of NORG fragmentation and diverse variations in different accessions (Fig. 6; Supplemental Fig. S12). Under the action of mutations, sequence loss, and TE activity, NORGs may be lost or become no longer detectable, which may also lead to the nonconservation nature of NORGs. These factors together contribute to the rich polymorphisms of NORGs together with continuous and frequent NORG insertions, which have continuously reshaped the Oryza nuclear genome.
Unlike conservation of mitochondrial gene content in metazoa (Castellana et al. 2011; Shtolz and Mishmar 2023), the loss of mitochondrial and plastid genes in angiosperms is ongoing (Mohanta et al. 2020; Mower 2020). Several organellar genes, such as the mitochondrial ribosomal protein genes (Adams et al. 2000, 2002) and the plastid acetyl-CoA carboxylase subunit D (accD) gene (Harris et al. 2013; Rousseau-Gueutin et al. 2013), have been lost and transferred to the nucleus multiple times during angiosperm evolution. There are abundant copies of organellar genes in the nuclear genome of each accession (Supplemental Tables S6, S7). Among the 180 shared ONGs in AA genomes, 14 contain NIOGs. Only two of these ONGs (ONG0600060 and ONG0700470), which include ORF241 (OrsajM_p39) and OrsajCt151, were also found in O. punctata (BB genome) (Supplemental Table S6). These observations suggest that NIOGs are continuously formed as organellar DNA is inserted into the nuclear genome, and they lack conservation during evolution, which is similar to the findings of Chen et al. (2023). The expression patterns of plant organelle genes are different from those of nuclear genes (Liere et al. 2011; Zhang et al. 2023). To become functional, NIOGs typically require acquisition of a nuclear promoter, a polyadenylation signal for expression, and, if necessary, signal peptides or alternative mechanisms to target the protein back to the mitochondria or plastid (Stegemann and Bock 2006; Liu et al. 2009). Unlike parasitic genetic elements, DNA from organelles might insert into the regulation regions, such as promoters, untranslated regions, and introns, or land in the nuclear genome with intact reading frames or protein domains, which help contribute the raw genetic material needed for gene expression regulation and de novo gene birth.
Our data demonstrated that inter- and intra-population polymorphism of ONGs and PAVs of ONGs can be utilized for reliable clustering of rice populations (Fig. 4A–E). Most of these ONGs are shared by at least two subpopulations (K = 15) in O. sativa (Fig. 4F), indicating some admixture among subpopulations during the domestication and breeding of O. sativa. As NORGs are homoplasy-free and easily identified (Hazkani-Covo 2009; Liang et al. 2018), they will be valuable tools for phylogenetic studies.
Direct transfer of organellar DNA into the nuclear genome and transfer via mRNA/cDNA intermediates have been considered as the two main mechanisms for endosymbiotic gene transfer. We did not observe gene transfer that involves the removal of introns in NORGs (Supplemental Table S6). Because of the presence of mutations and U encoding at the DNA level, the few RNA-edited sites in NIOGs (Supplemental Table S6) may not result from RNA- or cDNA-mediated gene transfer. These findings collectively suggest that the organellar gene transfer to the nuclear genome predominantly occurs via DNA rather than via RNA or cDNA intermediates, although the possibility of RNA-mediated transfer cannot be entirely excluded.
Previous studies have revealed that organelle DNA can be captured during DSBR (Ricchetti et al. 1999; Yu and Gabriel 1999; Decottignies 2005; Wang et al. 2012, 2018a), but the molecular mechanism of organelle DNA insertion into the nucleus has not been fully elucidated. DNA double-strand break (DSB) can be repaired by c-NHEJ (involving blunt end and short microhomologies from 1 to 4 bp), alt-EJ (involving microhomologies <20 bp), homologous recombination (HR; typically requires >50 bp homology) and single-strand annealing (SSA; typically requires >20 bp homology) (Chang et al. 2017). In Toxoplasma gondii, the capture of ptDNA (from apicoplast, a type of plastid) and mtDNA at DSB sites is dependent on Ku80 protein, which plays a crucial role in the c-NHEJ pathway of DNA repair, suggesting that DSBR via the c-NHEJ pathway is the key mechanism for the formation of NORGs in T. gondii (Namasivayam et al. 2023). In this study, only microhomologies <20 bp were found (Fig. 5A), indicating that the insertion of NORGs does not depend on HR or SSA throughout Oryza evolution. In addition, we found that alt-EJ or microhomology-mediated template switching could also contribute to integration of organelle DNA. Consistent with this, a previous study revealed that DNA polymerase theta–mediated DNA end joining (TMEJ; the predominant mediator of alt-EJ in most eukaryotes) promotes random integration of exogenous DNA in mouse embryonic stem cells (Zelensky et al. 2017). Moreover, long TSDs (>20 bp) and deletions (>10 bp) were observed in the insertion site of some NORGs (Fig. 5D,E; Supplemental Table S8), which are difficult to explain by c-NHEJ- or alt-EJ-mediated mechanisms. In addition to DSBR pathways, RBMs such as FoSTeS/MMBIR could explain nonrecurrent structural variants such as complex genomic rearrangements containing multiple breakpoints, insertions of DNA segments, and microhomology at the breakpoint junctions (Lee et al. 2007; Burssed et al. 2022). These mechanisms can result in complex rearrangements in genomic disorders (Lee et al. 2007; Zhang et al. 2009), template insertions in cancer cells (Osia et al. 2021), and structural variants in rice (Qin et al. 2021). In human cancer cells, somatic mtDNA integration events frequently occur together with other rearrangements of the nuclear genome (Ju et al. 2015), suggesting that mtDNA may be used as a “filler material” for DNA replication through MMBIR (Ju et al. 2015; Ju 2016). Similarly, ptDNA and mtDNA may also act as DNA fillers and be captured during DNA repair or DNA replication. Therefore, we hypothesize that NORGs result from DSBR and replication errors without requiring long homologies.
Based on this study, we propose that the formation of complex NORGs occurs before or during the process of insertion via ligation of ptDNA and/or mtDNA rather than by subsequent insertion or rearrangement after insertion. In addition, insertion of NORGs can be accompanied by filling with nonorganelle DNA, resembling the insertion pattern of mtDNA in human somatic cells (Ju et al. 2015). Similarly, particle bombardment in transgenic papaya SunUp resulted in insertion of a 1.6 Mb foreign DNA sequence containing several newly formed NUPTs, NUMTs, and sequences from the nuclear genome (Yue et al. 2022). DSBR could also result in complex insertions originating from various regions on different chromosomes via multiple sequential invasion and annealing events (Min et al. 2023). Because of possible damage or fragmentation of organelle DNA entering the nucleus, template switching may be further triggered during integration of organelle DNA, forming complex NORGs containing several to hundreds of fragments through mechanisms such as FoSTeS/MMBIR. Additionally, new organelle DNA insertions can accidentally occur near the existing NORGs owing to continuous formation of NORGs, which further increases the complexity of NORG structure.
Our study relies on a single mitochondrial reference genome (O. sativa) for NUMT identification, necessitated by the lack of published mtDNA assemblies for other Oryza species. Although mitochondrial sequence composition is highly conserved across the genus, structural variations such as recombination or lineage-specific rearrangements may result in undetected NUMTs. Alignment-based methods like BLAST are particularly susceptible to missing NUMTs derived from divergent mtDNA regions, as they prioritize sequence continuity over fragmented homology. Recent advances in alignment-free approaches (Li et al. 2019; for review, see Xue et al. 2023) offer promising solutions, which can detect NUMTs even when structural divergence obscures sequence collinearity. Future applications of such k-mer-based analyses may further resolve species-specific NUMTs and provide a more comprehensive understanding of NORG diversity in Oryza.
In summary, we classified NORGs into ONGs and analyzed their characteristics and polymorphisms in Oryza at the pangenome level. Our findings reveal continuous insertion and nonconservation of NORGs during evolution, resulting in intra- and inter-species variations. Our results indicate that organelle DNA might be integrated into the nucleus through DSBR and replication-based template switch, and complex NORGs may arise from single insertion events. The ongoing insertions and losses of NORGs, coupled with TE-induced fragmentation, collectively contribute to the polymorphism of NORGs throughout Oryza evolution. All these findings provide new insights into the ongoing and dynamic evolutionary processes of intracellular DNA promiscuity in plants.
Methods
Genome resources
A total of 22 genomes covering AA, BB, and FF genome species in the Oryza genus were collected, including 16 high-quality genome assemblies covering 15 subpopulations of Asian cultivated rice O. sativa (Kawahara et al. 2013; Zhou et al. 2020; Song et al. 2021) and six genome assemblies of other species (O. glaberrima, O. barthii, O. glumaepatula, O. meridionalis, O. punctata, and Oryza brachyantha) (Stein et al. 2018; Ntakirutimana et al. 2023; Thathapalli Prakash et al. 2023). The genome sequences in FASTA format were obtained for each assembly. The genome sequences and gene annotations of O. sativa Nipponbare were downloaded from Ensembl Plants (http://plants.ensembl.org/). The genome sequences and gene annotations of other O. sativa accessions were downloaded from RGI (Yu et al. 2023; https://riceome.hzau.edu.cn), and the assembly data of CHAO MEO::IRGC 802731 (GCA_009831315.1), Azucena (GCA_009830595.1), KETAN NANGKA::IRGC 19961-2 (GCA_009831275.1), ARC 10497::IRGC 12485-1 (GCA_009831255.1), PR 106::IRGC 53418-1 (GCA_009831045.1), Minghui 63 (GCA_001623365.2), IR 64 (GCA_009914875.1), Zhenshan 97 (GCA_001623345.2), LIMA::IRGC 81487-1 (GCA_009829395.1), KHAO YAI GUANG::IRGC 65972-1 (GCA_009831295.1), GOBOL SAIL (BALAM)::IRGC 26624-2 (GCA_009831025.1), LIU XU::IRGC 109232-1 (GCA_009829375.1), LARHA MUGAD::IRGC 52339-1 (GCA_009831355.1), N22 (N 22::IRGC 19379-1; GCA_001952365.2), and NATEL BORO::IRGC 34749-1 (GCA_009831335.1), plus the remaining assemblies of O. glaberrima::IRGC 96717 (GCA_000147395.3), O. barthii::IRGC 105608 (GCA_000182155.4), O. glumaepatula (GCA_000576495.2), O. meridionalis::OR44 (W2112; GCA_000338895.3), O. punctata:: IRGC 105690 (GCA_000573905.2), and O. brachyantha::IRGC 101232 (GCA_000231095.3) were downloaded from NCBI's Genome database (https://www.ncbi.nlm.nih.gov/datasets/genome/).
Identification of ONGs
We used the O. sativa chloroplast genome to identify NUPTs owing to the chloroplast genome is highly conserved among the species used in this study. The mitochondrial genome of O. sativa (NCBI GenBank [https://www.ncbi.nlm.nih.gov/genbank/] NC_011033) was used as the reference for NUMT identification. This choice was necessitated by the lack of published mitochondrial assemblies for other Oryza species included in our analysis. Despite the structural diversity of plant mitochondrial genomes, their sequence composition is highly conserved across species. To validate the suitability of the O. sativa mitochondrial genome as a universal reference, we compared it to the mitochondrial assembly of Oryza minuta (BBCC genome, NCBI GenBank: NC_029816.1), a closely related species, using whole-genome alignment (NUCmer). The comparison revealed 97% sequence identity and 83.1% coverage, supporting the use of O. sativa mtDNA for NUMT detection in closely related Oryza species.
We used NUCmer (Marçais et al. 2018) for sequence alignment (version 4.0.0beta2, parameters: -c 50 ‐‐maxmatch). NC_001320 (NCBI GenBank) was used for plastid genome, and NC_011033 (NCBI GenBank) was used for mitochondrial genome as the query sequence, respectively. All anchor matches, regardless of their uniqueness, were used (parameter: ‐‐maxmatch), and the minimum length of a cluster of matches was set to 50 (parameter: -c 50). These parameters allow the inclusion of shorter matches and retain more matches than the default settings. The overlapping hits were merged by the BEDTools (Quinlan and Hall 2010) merge command, and the results were utilized for calculating the total length of the NORGs. Then, the alignments separated by <5 kb of DNA of nonorganelle origin were merged by BEDTools merge command, and the results were temporarily used as NORG clusters.
Each NORG cluster including its flanking sequences of 300 bp was aligned to all genomes using NUCmer with the default parameters, followed by show-coords (parameters: -rclT -O). First, the locations of the NORG clusters in other genomes were determined by the .coords file generated by show-coords. If the genome contained the same NORG cluster without structure variations, the alignment was marked as “CONTAINS” in the .coords file. For the remaining NORG clusters, the best location of each NORG cluster on each genome was determined by show-tiling (parameters: -g 20000). The NORG clusters in the same locations were recognized as the same NORGs by BEDTools intersect.
Because some NORGs were separated by sequence insertions >5 kb, multiple NORG clusters at the same location were merged. In the first step, the alignment of NORGs with flanking sequence of 300 bp from the previous step was used directly to merge partially dispersed NORGs. The dot plots of each NORG cluster produced by mummerplot were used as an aid to manual calibration. In the second step, the flanking sequences of 2000 bp from each NORG cluster in step one were aligned with the genomes without the identification of that NORG. The .delta file from the alignment was then treated with show-tiling (parameters: -g 20000 -v 80) to determine the position of the flanking sequences in each genome. In the third step, several NORGs with multiple large insertions were corrected manually using BLASTN (Camacho et al. 2009) and BLAT (Kent 2002). To assess the accuracy of this workflow, synteny plots of each ONG were generated by genoPlotR (Guy et al. 2010) to visually validate the NORGs.
Identification of NORG insertion position
For each NORG, the flanking 2 kb sequences were merged and aligned with the genomes in which the NORG was not detected using NUCmer, after which the positions of the flanking 2 kb merged sequences in other genomes were obtained using show-tiling (parameters: -g 20000 -v 80), and the NORGs whose flanking sequences could not be obtained in a relative position by show-tiling were ignored. Afterward, the R package genoPlotR was used to plot the collinearity of all NORGs and their flanking sequences in all genomes in which NORGs and insertion positions could be detected, which was used to assist manual correction.
Statistical analysis of NORG overlap with gene and exon regions
The gene annotations of O. sativa Nipponbare were downloaded from Ensembl Plants (http://plants.ensembl.org/). The gene annotations of O. sativa Minghui 63 and Zhenshan 97 were downloaded from Rice Information GateWay (RIGW) (Song et al. 2018; http://rice.hzau.edu.cn/rice_rs3/). The gene annotations of other O. sativa accessions were downloaded from RGI (Yu et al. 2023; https://riceome.hzau.edu.cn). To assess whether the observed overlap between NORGs and gene or exon regions was statistically significant, random permutation was performed using BEDTools. First, the “bedtools shuffle” command was employed to randomly permute the genomic locations of the identified NORGs while maintaining the original number and length of the NORGs. This generated a set of simulated NORGs with randomized positions across the genome. This process was repeated 1000 times to generate a distribution of expected overlaps. Next, the “bedtools intersect” command was used to count the number of simulated NORGs that overlapped with gene regions and exon regions in each permutation, which could provide the expected values for the overlap. Similarly, “bedtools intersect” was used to determine the actual number of NORGs overlapping with gene regions and exon regions.
For each permutation, a chi-square test was performed to compare the observed overlaps with the expected overlaps using the “CHISQ.TEST” function in Excel. The P-value was calculated for each permutation, and then, the average P-value across all 1000 permutations was taken to assess the statistical significance of the observed overlaps.
NORG insertion rate estimations
Similar to the method of Zhou et al. (2023), to estimate the NORG insertion rate (NIR), we divided the total number of ONGs in pairs of genomes by twice the time to
the most recent common ancestor (TMRCA). The estimate is calculated using the following equation: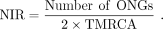 (1)
In detail, the number of ONGs present only in O. sativa was 919. The number of ONGs present only in O. glaberrima and/or O. barthii was 471. The diverged time was 0.81 million years ago (MYA). The estimate was calculated using the following equation:
(1)
In detail, the number of ONGs present only in O. sativa was 919. The number of ONGs present only in O. glaberrima and/or O. barthii was 471. The diverged time was 0.81 million years ago (MYA). The estimate was calculated using the following equation: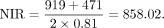 The number of ONGs present in O. sativa, O. glaberrima, and/or O. barthii but not present in other genomes was 1491. The number of ONGs present only in O. glumaepatula was 340. The diverged time was 0.97 MYA. The estimate was calculated using the following equation:
The number of ONGs present in O. sativa, O. glaberrima, and/or O. barthii but not present in other genomes was 1491. The number of ONGs present only in O. glumaepatula was 340. The diverged time was 0.97 MYA. The estimate was calculated using the following equation: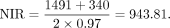 The number of ONGs present in genomes other than O. meridionalis is 2238. The number of ONGs present only in O. meridionalis was 804. The diverged time was 2.41 MYA. The estimate is calculated using the following equation:
The number of ONGs present in genomes other than O. meridionalis is 2238. The number of ONGs present only in O. meridionalis was 804. The diverged time was 2.41 MYA. The estimate is calculated using the following equation: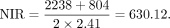
Characterization of NORG junction sites
To avoid errors caused by mutations and for more accurate characterization of the NORG junctions, we selected 18 genomes of O. sativa, O. glaberrima, and O. barthii for analysis. Through the insertion position of NORGs obtained previously, two genotype sequences of the presence and absence of NORGs were obtained. The sequences of the presence genotype of NORGs were the sequences of the NORG with its flanking 2 kb sequences, and the sequences of the absence genotype of NORGs were obtained by the position of the flanking 2 kb sequences of the NORGs in the genome that does not contain the NORGs. For each NORG, the sequences of both genotypes were aligned by BLASTN, and the HSPs flanking the NORG closest in distance to the NORG were retained, respectively. The number of overlapping base pairs of the closest HSP to the NORG was defined as microhomology (≤20 bp) or homology (>20 bp). The number of base pairs of the closest HSP to the NORG that were >0 bp was defined as a sequence insertion. The closest HSP with exactly 0 bp distance from NORG was defined as a blunt end. The formation mechanisms were characterized as follows: classical NHEJ, >5 bp filler or <5 bp microhomology in the junction sites; alt-EJ, 5–20 bp microhomology in the junction sites; SSA, 21–50 bp homology in the junction sites; HR, >50 bp homology in the junction sites; and FoSTeS/MMBIR, >20 bp TSD or >10 bp deletion in the junction sites.
Determination of ONG presence or absence based on WGS data sets
The WGS data set was collected from 3K-RG (Wang et al. 2018b) and the studies of Cubry et al. (2018), Meyer et al. (2016), and Choi et al. (2019) from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJEB6180, PRJEB21312, PRJNA315063, and PRJNA453903, respectively.
To identify the PAVs of ONGs in the rice WGS data set, we developed a pipeline based on presence/absence genotype across assemblies through four major steps: (1) sequences of presence/absence genotype of ONGs were prepared using the identification results described above; (2) to reduce the mapping errors caused by repetitive sequences and organelle genome sequences, the rice TE library “rice6.9.5.liban” from EDTA (Ou et al. 2019) and rice organellar genome NC_001320 (NCBI GenBank) and NC_011033 (NCBI GenBank) were merged to the sequences of the previous step; (3) paired-end reads were mapped to the merged sequences by BWA-MEM2 (Vasimuddin et al. 2019); and (4) the presence/absence genotype was verified by the mapping results.
To verify that this pipeline could accurately identify ONG presence/absence, we chose five WGS accessions (CX140 | IRGSP1.0, CX145 | MH63RS3, CX133 | ZS97RS3, CX145 | AzucenaRS1, IRIS_313-8813 | Os117425RS1) from 3K-RG corresponding to the genome assemblies used in this study. After filtering out the misidentified ONGs, 754 polymorphic NORGs were retained.
The MDS plots were constructed using the presence/absence information of 754 ONGs for each accession. The distance matrices were calculated using the “bray” method in the “vegdist” function from the R package “vegan” and subsequently computed the MDS using the “cmdscale” function in R (version 4.2.3) (R Core Team 2023).
The presence frequency of ONGs was calculated by using R software (version 4.2.3). The heatmap was plotted by TBtools software (Chen et al. 2020).
Identification of population specific ONGs
We defined the ONGs with presence frequency ≥90% in one group and ≤10% in other groups as representative ONGs among the four major groups. Considering that the same criteria could not be applied to the subpopulations, we lowered the subpopulation presence frequency threshold to 80%.
TE annotation
The TEs were identified by RepeatMasker v4.1.0 with parameter “-nolow ‐‐gff -xsmall -pa 4 -species Oryza” using the RepBase-20181026 and Dfam_3.1 library.
Visualization of ONGs
The schematics for five types of NORGs and the NORG change trajectories were plotted using NGenomeSyn (He et al. 2023). The synteny plots of NORGs were generated by genoPlotR (Guy et al. 2010). The R package RIdeogram (Hao et al. 2020) was used to visualize the location of ONGs in the chromosomes. The insertion frequency (depth) plots were generated by ggcoverage (Song and Wang 2023), and the genome maps of mtDNA and ptDNA were generated by OGDRAW (Greiner et al. 2019) and PMGmap (Zhang et al. 2024a), respectively.
Software availability
The source codes used in this study are available as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was supported by the National Key R&D Program of China (2023ZD04073), the Foundation of Hubei Hongshan Laboratory (2022HSZD014), Major Project of Hubei Hongshan Laboratory (2022HSZD031), National Natural Science Foundation of China (31771752), the Fundamental Research Program of Hubei Province (2024AFE001), the Start-up Fund of the Huazhong Agricultural University (HZAU) to J.Z., and the Earmarked Fund for China Agriculture Research System (CARS-01-01). We thank the National Key Laboratory of Crop Genetic Improvement, HZAU for providing the bioinformatics computing platform in this study. We thank Dr. Laura Bartley for constructive criticism of the manuscript.
Author contributions: F.Z., J.Z., and Y.L. designed and conceived the research. C.G., Y.H., M.L., Y.Z., Y.X., N.M., X.Q., and A.Z. performed the research. R.A.W., J.Z., and Y.Z. contributed the materials. Y.H., M.L., Y.X., and X.Q. analyzed data. C.G., W.X., R.A.W., F.Z., J.Z., and Y.L. wrote and edited the paper. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279609.124.
- Received May 27, 2024.
- Accepted March 27, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








