
Meta-analysis of activated neurons reveals dynamic regulation of diverse classes of alternative splicing
- Keegan S. Krick1,2,3,4,
- Marissa Maroni4,5,
- Erica Korb4,
- Kristen W. Lynch2 and
- Elizabeth A. Heller3,4
- 1Cell and Molecular Biology Graduate Group, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA;
- 2Department of Biochemistry and Biophysics, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA;
- 3Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA;
- 4Epigenetics Institute, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA;
- 5Neuroscience Graduate Group, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA
Abstract
Activity-dependent gene expression in neurons is well established, yet few studies have examined activity-dependent alternative splicing. Alternative splicing regulates >95% of genes and is essential to diverse neuronal functions, including synapse development and calcium channel diversity. Alternative splicing is regulated by the expression and activity of RNA-binding proteins and through changes in the local chromatin environment. To date, most analyses of activity-dependent alternative splicing are focus primarily on microexons, a small subclass of neuron-specific exons. To broaden knowledge of activity-dependent alternative splicing in neurons, we analyzed five independent RNA-seq studies to identify splicing events that consistently respond to potassium chloride (KCl) depolarization. We found that the majority of activity-dependent exons become less included upon activation, are basally constitutive, are not microexons, and reside in genes that are not differentially expressed after KCl treatment. Functionally, alternative splicing of RNA processing machinery and regulators precedes splicing of genes related to neuronal function. Given recent advances in elucidating chromatin-mediated alternative splicing in the brain, we explored the coincident regulation of histone modifications over activity-dependent exons. We found KCl-dependent changes in H3K36me3 and H4K20me1, both enriched in active gene bodies, over a subset of KCl-dependent exons, suggesting coordination of activity-dependent histone modification and alternative splicing. Together, these findings identify a diverse class of activity-dependent alternative splicing and describes the temporality and features of its regulation in cultured neurons.
Alternative splicing regulates the vast majority of mammalian genes, with >95% of multiexon genes undergoing some form of alternative splicing (Pan et al. 2008; Wang et al. 2008). Alternative splicing is involved in a myriad of biological processes, including differentiation, immune cell activation, and cancer (Martinez et al. 2012; Li et al. 2014; Dvinge et al. 2016). The function of alternative splicing to diversify the proteome is vital to producing distinct cellular states from an otherwise homogeneous genome. The mammalian brain consists of thousands of distinct neuronal subtypes that respond to neuronal activity through regulation of the transcriptome and proteome (Lake et al. 2016; Van Oostrum et al. 2023). Consequently, disruption of alternative splicing has been linked to neurodegenerative, neurodevelopmental, and neuropsychiatric diseases (Dredge et al. 2001; Lopez Soto et al. 2019). Neuronal alternative splicing is downstream from calcium-calmodulin-dependent protein kinase (CaMK) signaling, which regulates RNA-binding proteins both post-transcriptionally and post-translationally (Xie and Black 2001; Fredj et al. 2004; Xie et al. 2005; An and Grabowski 2007; Lee et al. 2009). Depolarization has been shown to regulate microexon (<28 nt) splicing networks, including a microexon in neuronal splicing regulator nSR100, which regulates its expression (Quesnel-Vallières et al. 2015, 2016). Heterozygous mutations that force skipping of this nSR100 microexon phenocopy autism spectrum disorder (ASD)–linked splicing and behavioral phenotypes (Quesnel-Vallières et al. 2016).
More recently, chromatin state has been linked to splicing regulation. Global analysis of alternative splicing patterns and histone post-translational modifications (hPTMs) across mouse brain development finds that enrichment of chromatin modifications correlates with, and predicts, alternative exon inclusion (Hu et al. 2017, 2020; Xu et al. 2017, 2018). Alternative splicing of neurexin I (Nrxn1) exon 22 responds to activity through both SAM68, an RBP splicing regulator, and deposition of the hPTM H3K9me3 in a region- and stimuli-specific manner, and underlies learning and memory (Iijima et al. 2011; Ding et al. 2017). H3K9ac is also associated with alternative splicing outcomes of neuronal cell adhesion molecule 1 (Ncam1) exon 18, with neuron depolarization inducing H3K9 hyperacetylation and inhibiting differentiation of N2a cells to mature neurons (Schor et al. 2009, 2013). Finally, cocaine self-administration alters intragenic H3K36me3 levels and alternative splicing of genes in mouse neurons (Xu et al. 2021). CRISPR epigenetic editing of H3K36 methylation targeted to a cocaine-responsive exon in the gene Srsf11 is sufficient to drive exon inclusion and cocaine reward behavior (Xu et al. 2021). These studies demonstrate the putative role of histone modifications, especially H3K36me3, in regulating alternative splicing in the brain. However, the underlying molecular mechanisms through which chromatin regulates signal-induced alternative splicing remain elusive.
Direct membrane depolarization of primary neurons using potassium chloride (KCl) serves as a widely used model of neuronal activity, and it functions by initiating conserved calcium-dependent signaling cascades to activate activity-dependent gene networks (West et al. 2001; Rienecker et al. 2020, 2022). Despite this, the paradigm cannot recapitulate the temporal kinetics of variable stimuli or uncouple localized depolarization, for example, at the synapse, from that at other neuronal features (Rienecker et al. 2020, 2022). As such, these experiments are unlikely to perfectly reflect in vivo dynamics yet offer a robust means to untangle activity-dependent genomic events in homogenous cell populations. In this study, we established the identity, features, and functions of exons that respond to KCl stimulation to define the global coordination of activity-dependent alternative splicing in mouse primary neurons.
Results
KCl regulates alternative splicing, but not expression, of thousands of exons in mouse primary neurons
Many studies have established robust KCl-dependent transcript abundance changes in primary neurons, yet few have globally examined alternative splicing (West et al. 2001; Kim et al. 2010). We analyzed alternative splicing in five public RNA sequencing data sets of mouse primary neurons following KCl treatment (Fig. 1A; Maze et al. 2015; Quesnel-Vallières et al. 2016; Calderon et al. 2022; Ibarra et al. 2022; Xu et al. 2022). We confirmed sustained increases in immediate early gene (IEG) expression after initial exposure to KCl in all data sets but found no changes in the splicing of canonical IEGs (Fig. 1B,D; Supplemental Fig. S1; Supplemental Table S1), consistent with previous literature (Greenberg et al. 1992; West and Greenberg 2011). Across all five data sets, we found that alternative splicing was globally altered by KCl stimulation, changing inclusion of thousands of events after >1 h of KCl treatment (Supplemental Table S2).
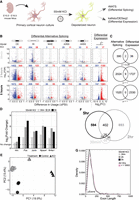
KCl induces widespread changes in alternative splicing in mouse primary cortical neurons. (A) General experiment workflow for depolarization of mouse primary neurons using KCl. (B) Volcano plots of global differential splicing and expression changes following KCl treatment in primary cortical neurons from Maze et al. (2015). Differential alternative splicing is presented as change in percentage spliced in (ΔPSI) between KCl-treated and untreated samples for each treatment duration. Expression changes are reported as log2 of the fold change (FC) between equivalent comparisons. Alternative splicing event types analyzed include the following: (CE) cassette exon, (RI) retained intron, (MXE) mutually exclusive exons, (A3SS) alternative 3′ splice site, and (A5SS) alternative 5′ splice site. Splicing events were considered significant with an FDR < 0.05 and |ΔPSI| > 10%. Differentially expressed genes were considered significant with an FDR < 0.05 and |log2FC| > 1.5. Significant differential splicing events and differentially expressed genes are highlighted in red (more included or upregulated) or blue (less included or downregulated). (C) Venn diagrams of overlap between differentially expressed genes and genes containing a differentially spliced event (all event types). Significance of overlap was determined using a hypergeometric test. (D) Differential expression of immediate early genes (IEGs) upon KCl treatment in Maze et al. (2015). (E) Splicing PCA of KCl-treated and untreated primary neurons from Maze et al. (2015). Individual samples are plotted with treatment groups denoted by shape and time point by color. Principal components are calculated using percentage spliced in (PSI) of 21,618 alternative exons (0.1 < PSI < 0.9) quantified at all time points and treatments. (F) Euler plot of genes containing differentially spliced CEs in response to each duration of KCl treatment from Maze et al. (2015). (G) Smoothed density distribution of exon lengths across differentially spliced alternative exons for each duration of KCl treatment in Maze et al. (2015). Pink line denotes CEs (0.1 < PSI < 0.9; n = 9860) regardless of KCl dependence quantified in all samples. Dashed line denotes the upper size limit of μexons (28 nt).
To highlight details observed for KCl-induced splicing, we focused on data from Maze et al. (2015) that exemplified the trends observed broadly across all five data sets (Fig. 1B; Supplemental Table S2). We identified thousands of alternative splicing events that exhibited different levels of percentage spliced in (PSI) upon KCl treatment (ΔPSI > 10% and FDR < 0.05) (Fig. 1B). KCl-induced splicing events fell across all major classes of alternative splicing, and we noted that skipping of alternative cassette exons (CEs) accounted for >70% of differential events at all time points (Fig. 1B, decreased inclusion of CEs). Furthermore, >95% of KCl-regulated events were changed after 2 or 5 h of KCl stimulation, with only a small subset of events altered after an acute, 30 min treatment. Finally, at all time points, genes in which we observed KCl-dependent alternative splicing events were distinct from those which exhibited KCl-regulated expression (Fig. 1C; Supplemental Table S2). From these findings, we concluded that alternative splicing is regulated independently of transcript abundance in KCl-treated neurons. Such independent regulation of splicing and transcript abundance is consistent with other studies of alternative splicing in cellular development and other stimulations (Martinez et al. 2012; Li et al. 2014; Karlebach et al. 2024). Different durations of KCl treatment regulate distinct classes of gene expression in neurons (Tyssowski et al. 2018). We next determined if such temporal regulation was also observed in alternative splicing. We first performed principal component analysis (PCA) to stratify samples based on global splicing profiles, using the PSI of exons quantified in all samples. We found that alternatively spliced exons differed in the PCA between KCl and control splicing profiles after 2 and 5 h, but not 30 min, of KCl treatment (Fig. 1E). We then assessed which facet of the data sets drove the greatest variance between samples by plotting all samples from all data sets in the same principal component space for transcript abundance and splicing (Supplemental Fig. S2). When analyzing splicing, we observed that KCl-stimulated samples across data sets shifted toward the same PCA space, whereas samples clustered much more strongly by data set when considering transcript abundance. This analysis highlighted a general convergence at the level of exon inclusion, with KCl-stimulated samples looking more similar to one another despite origin, whereas sample heterogeneity was driven largely by data set with transcript abundance (Supplemental Fig. S2). Moreover, genes undergoing differential splicing demonstrated moderate overlap between time points, with >50% of genes being unique at the 2 and 5 h time points (Fig. 1F). The distinct genes that undergo alternative splicing at each time point suggest unique regulatory programs, rather than the accumulation of splicing changes over time.
At least one previous study of KCl-regulated splicing focused on microexons, a subclass of exons that are shorter than most exons (<28 nt) and are highly neuron specific (Quesnel-Vallières et al. 2015, 2016). We identified differential splicing of >70% of the KCl-dependent microexons identified in the original analysis (Quesnel-Vallières et al. 2016), with a Pearson's correlation of 0.92 between ΔPSIs called by each algorithm (Supplemental Fig. S3). However, microexons accounted for only ∼3% of alternative exons altered by KCl across all time points and data sets, as highlighted by the length distribution of KCl-dependent exons in the Maze et al. (2015) data set (Fig. 1G). The size distribution of KCl-regulated exons reflects that of murine exons genome-wide, suggesting there is no size or class bias in the exons regulated by KCl in primary neurons.
KCl consistently alters splicing of high-confidence alternative exons
We next sought to expand the collection of activity-dependent exons beyond microexons by defining a set of high-confidence alternative exons that reproducibly responded to KCl. Exons were considered high confidence if they were significantly differentially spliced in the same direction upon KCl stimulation in at least two independent data sets. We further stratified the data into “moderate” (1–3 h) and “long” (5–6 h) durations of KCl treatment to identify exons that were involved in different temporal windows of splicing (Fig. 2A). Such categories were based on grouping time points in independent experiments that displayed similarly enriched GO terms (Supplemental Fig. S4). Each data set we analyzed had between 25% and 60% of exons that were identified in at least one other data set (Supplemental Table S3; Supplemental Fig. S5). This allowed us to define a set of high-confidence exons as exons that exhibited significant changes in the same direction in at least two data sets at the same time point. We observed a strong positive correlation between the ΔPSIs of the high-confidence exons in each pairwise comparison (moderate KCl, R = 0.61; long, R = 0.73) (Fig. 2B). In all, we defined approximately 1200 high-confidence alternative exons in each time window, the majority of which were found in two or three data sets and mirrored the proportions seen for transcript abundance (Fig. 2C,D). The inclusion of >80% of these exons decreased after KCl (moderate KCl, 87%; long KCl, 81%), similar to the direction of regulation we observed in individual data sets (Fig. 2D).
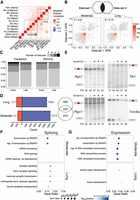
Integrative analysis defines activity-dependent exons of high confidence. (A) Heatmap of weighted Pearson's correlations between all data sets used in analysis (for description of calculation, see Methods). Data sets categorized as “moderate” and “long” after KCl stimulation are designated. Q-V is equivalent to Quesnel-Vallières et al. (2016). (B) Tile plot of ΔPSIs for shared events between pairwise data sets in the moderate (left) and long (right) classifications. Correlation is calculated using Pearson's R with significance considered P < 0.05. Plot space is organized into a 50 × 50 grid with greater color denoting a higher number of events mapping to a given tile. (C) Number of data sets in which a high-confidence event is significant for expression (left) and splicing (right). (D) Summary of all high-confidence events in the moderate and long alternative exon sets. Euler plot of gene-level overlaps between the two sets is shown at right. (E) 32P-labeled RT-PCR of four candidate high-confidence CEs in primary mouse cortical neurons stimulated with 55 mM KCl for 2 h. PSI values for each sample are quantified beneath the gel. (F,G) Enriched Gene Ontology terms for differential splicing (F) and differential expression (G) high-confidence events in the moderate and long sets. Terms are derived only from the “Biological Process” domain and are filtered to contain fewer than 2500 genes in each term. The top five terms by descending −log10(FDR) are shown for each condition, with the size of the shape corresponding to the number of genes annotated in a given GO term. Gene ratio is calculated as the proportion of genes identified in experimental samples over the total number of annotated genes in the GO term. Analysis of long high-confidence-set differential expression (G, lower) yielded only two significantly enriched GO terms.
We next biochemically validated a subset of exons identified in the meta-analysis. We selected five candidate exons from the high-confidence set that defined features highlighted in our analysis. Importantly, given the bias toward exon skipping in this set, we selected four exons whose inclusion decreased after KCl treatment and one whose inclusion increased. We treated primary mouse cortical neuronal cultures with 55 mM KCl for 2 h to reflect “moderate” treatment. We assayed splicing using 32P-radiolabeled RT-PCR (Fig. 2E) and validated the predicted changes in PSI, in both direction and magnitude, for four of the candidate exons (mean predicted ΔPSI: argonaute 1 [Ago1], −0.125; transducin-like enhancer of split 1 [Tle1], −0.236; protein tyrosine phosphatase, nonreceptor type 1 [Ptpn1], −0.120; trinucleotide repeat containing 6a [Tnrc6a], +0.302) (Fig. 2E). KCl altered splicing of distinct genes in the moderate and long treatments (Fig. 2D), suggesting distinct functions of KCl-dependent alternative splicing over time. Gene Ontology analysis identified unique functional terms enriched among high-confidence spliced exons at moderate versus long KCl treatment (Fig. 2F). Moderate treatment altered splicing of RNA processing and histone modification genes, whereas long treatment altered splicing of synaptic function and cell-to-cell communication genes (Fig. 2F). Consistent with previous work (Tyssowski et al. 2018; Rienecker et al. 2022), we also found differences in Gene Ontology of differentially expressed genes at moderate versus long KCl treatment (Fig. 2G; Supplemental Tables S4, S5; Supplemental Fig. S4). The function of genes regulated by splicing was similarly distinct from that regulated by transcription (Fig. 2F,G). Genes altered first by KCl were directly related to gene expression (transcription and RNA processing). Conversely, genes with altered splicing at later time points were more associated with neuronal activity (synaptic trafficking/structure), whereas differentially expressed genes showed no functional enrichment. Importantly, neither splicing nor transcript abundance reflected the activation of stress pathways (Supplemental Table S5). In all, this analysis defined exons whose inclusion strongly associated with KCl treatment across multiple data sets and demonstrated a tuned temporal response to stimulation at the levels of expression and splicing.
KCl-regulated exons show distinct genomic features that reflect direction of inclusion
Having defined a set of high-confidence alternative exons, we next sought to identify features that reflect mechanisms of KCl-dependent splicing regulation. As an appropriate control, we identified a set of alternative exons in expression-matched genes whose splicing did not change with KCl treatment (0.1 < PSI < 0.9, FDR > 0.1). We then compared KCl-dependent exons at both the moderate and long time points to control exons. Exons whose inclusion decreased after KCl had higher GC content at both time points but had no difference in length at the moderate time point and were generally shorter at the long time point (Fig. 3A,B). Conversely, exons whose inclusion increased after KCl stimulation at both time points had lower GC content and greater exon length (Fig. 3A,B). We also examined if GC content and exon length were linked; however, we saw equivalent changes in GC content between KCl-dependent and control exons when binned across exon lengths (10 equivalent bins) (Supplemental Fig. S4A). Finally, introns flanking more included exons had lower GC content as opposed to introns adjacent to less-included exons, which were shorter and more GC-rich (Fig. 3C,D). Taken together, our results suggest that the genomic context of these exons contributes to their regulation by neuronal activity, with less-included exons occupying coding dense and highly GC-rich regions of genes.
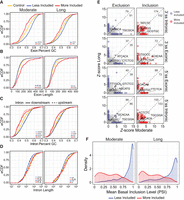
High-confidence activity-dependent exons are enriched in genomic features. (A) Cumulative distribution function of exon GC content for high-confidence KCl-dependent exons and control, expression-matched alternative exons that are not responsive to KCl at each duration of treatment. Blue and red designate KCl-dependent exons that become less or more, respectively, included upon KCl treatment. A Kolgomorov–Smirnov test (KS) was performed to assess the similarity of the KCl and control distributions, and statistical significance was considered P < 0.05. (n.s.) P > 0.05, (*) P < 0.05, (**) P < 10−3, (***) P < 10−6, (****) P < 10−9. (B) As above, looking at exon length. (C) Cumulative distribution function of intronic GC content for high-confidence KCl-dependent exons and control, expression-matched alternative exons that are not responsive to KCl at each duration of treatment. Dashed line represents upstream (5′) introns; solid line, downstream (3′) introns. Blue and red designate introns adjacent to KCl-dependent exons, which become less or more, respectively, included upon KCl treatment. A KS test was performed to assess the similarity of the KCl and control distributions between upstream and downstream introns with statistical significance considered P < 0.05. (n.s.) P > 0.05, (*) P < 0.05, (**) P < 10−3, (***) P < 10−6, (****) P < 10−9 (D) As above, looking at intron length. (E) Enrichment of 6-mers in less included (left) and more included (right) KCl-dependent alternative exon sets relative to control alternative exons unresponsive to KCl treatment. Enrichments are performed using sequence of shallow (100 bp) upstream and downstream introns, as well as exons. Number of significantly enriched k-mers (Fisher's exact P-value > 0.05) for moderate and long KCl exon sets are noted in top right corner. The top-ranking k-mers that contain a motif for RNA-binding proteins previously implicated in KCl-regulated splicing in neurons are indicated on plot: UGC (SRRM4), CA-rich sequences (HNRNPL/LL), UC-rich sequences (PTBP1), and GCAUG (RBFOX2). A full table of significantly enriched k-mers and their P-value is given in Supplemental Table S6. (F) Density plot of mean basal inclusion level (PSI) across all data sets of high-confidence KCl-dependent exons prior to KCl stimulation. Blue and red designate exons that become less or more, respectively, included upon KCl treatment.
We also discovered hexanucleotide sequences associated with both more and less exon inclusion after KCl stimulation, although motif enrichment analysis did not return a consensus motif (Fig. 3E; Supplemental Table S6). We observed enrichment near our high-confidence exons, compared with the expression-matched control set, for binding motifs of RBPs previously implicated in KCl-regulated splicing in neurons, including SRRM4 (also known as nSR100) (UGC), HNRNPL/LL (CA-rich sequences), PTBP1 (UC-rich sequences), RBFOX2 (GCAUG), and several SR proteins, which are denoted in Figure 3E (Quesnel-Vallières et al. 2016; Raihan et al. 2019; Mazille et al. 2022). Ptbp1, Rbfox2, and Srsf11 were themselves alternatively spliced after stimulation, leading to the regulation of characterized premature termination codons and regulatory protein domains (Supplemental Table S7). Therefore, although all of these RBPs may contribute to a subset of KCl-induced alternative splicing, our analysis suggests that none of these factors explain the entirety of such regulation.
Finally, we investigated the resting inclusion levels of the KCl-responsive exons by calculating the mean basal inclusion level in unstimulated samples across all data sets and both time points. Exons that increased inclusion upon KCl treatment had broadly distributed PSI levels prior to stimulation (Fig. 3F), whereas exons that decreased inclusion were predominantly constitutive at baseline (Fig. 3F, blue curve). This bias toward high inclusion at basal levels confirms that the KCl-induced loss of inclusion for these exons is not an artifact of limitations in detection and quantification of lowly included exons. Moreover, >85% of KCl exons reside in the open reading frame (ORF) and ∼50% preserve the ORF, pointing to the functional diversity produced by these splicing events at the protein level (Supplemental Fig. S6D). For these exons, we intersected Pfam protein domains and found a broad range of domains impacted by their splicing. Protein kinase domains were the most frequently altered, but this represents a small percentage of the total (moderate, 1.6%; long, 1.8%) (Supplemental Fig. S7; Supplemental Table S9). We find that 50% of exons induce either a +1 or +2 codon frameshift, which may trigger nonsense-mediated decay (NMD) and result in depletion of the transcript (Supplemental Fig. S6E). Taken together, we found that KCl treatment led to loss of dominant, coding exons that likely impact the function of resulting protein derived from target genes.
H3K36me3 redistributes over KCl-dependent alternative exons in response to KCl
Previous work by ourselves and others demonstrate the impact of chromatin modifications on alternative splicing (Schor et al. 2009, 2013; Luco et al. 2010; Ding et al. 2017; Hu et al. 2017, 2020). We found that histone modification was a highly enriched functional term for genes whose splicing changed with moderate KCl treatment, including enzymes involved in writing and erasing a diverse set of modifications and target residues (Supplemental Fig. S8A). In particular, the modification H3K36me3 is implicated in alternative splicing (Luco et al. 2010; Hu et al. 2017, 2020). We found KCl-dependent alternative splicing of Setd5 and Ash1l, which encode enzymes that catalyze H3K36me3 and H3K36me1/2, respectively (Sessa et al. 2019). Therefore, in parallel to our analysis of RBP regulation and RNA, we also assessed the chromatin landscape surrounding high-confidence KCl-regulated exons.
We measured H3K36me3 over KCl-dependent exons in primary neurons using a publicly available ChIP-seq data set of four histone methylation marks (Wang et al. 2015). We first performed peak calling using the SICER algorithm to identify hPTM peaks across the genome and intersected these with the KCl-dependent and control exon sets. We found that ∼50% of KCl-dependent exons intersected a peak for H3K36me3 (moderate KCl, 53%; long KCl, 46%) called in the KCl-stimulated condition (Fig. 4A, more saturated bars), whereas only 41% and 42% of control exons intersected an H3K36me3 peak (Fig. 4A, less saturated bars). We analyzed three additional modifications included in the data set, H4K20me1, H3K4me1, and H3K4me2, all of which showed a minimal difference in the number of peaks over KCl-dependent exons versus control exons (Fig. 4A). Although H4K20me1 had similar coverage over KCl-dependent exons as H3K36me3, these modifications were altered over different sets of KCl-dependent exons, suggesting that they do not act synergistically (Supplemental Fig. S8B). As expected given their established enrichment at promoters and enhancers, we observed few intragenic peaks for H3K4me1/2, with a minimal difference between KCl-dependent and control exons. These overall trends in histone modifications are exemplified by a KCl-dependent exon in Ago1, which gained an H3K36me3 peak after stimulation but showed minimal change in other histone modifications (Fig. 4B).
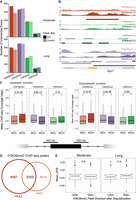
H3K36me3 dynamically marks a subset of alternative exons in response to KCl. (A) Number of KCl-dependent (saturated bar) and control (unsaturated bar) exons that overlap an intragenic H3K36me3 peak (Wang et al. 2015) across moderate and long KCl treatments. Exons and H3K36me3 peaks were considered overlapping with a >90% overlap. Equivalent analysis was performed for H4K20me1, H3K4me1, and H3K4me2. (B) Genome tracks of ChIP-seq coverage and peaks for H3K36me3, H3K4me1, H3K4me2, and H4K20me1 over a candidate KCl-dependent CE in Ago1. (C) Mean ChIP-seq coverage of H3K36me3, H4K20me1, and H3K4me1 at upstream and downstream exon–intron junctions (100 nt intron + 100 nt exon) of KCl-dependent alternative exons. A Mann–Whitney U test was used to test significance of mean ChIP coverage between treatments, and significance was considered at P < 0.05. (D) Euler plot of H3K36me3 peaks called between KCl and untreated primary cortical neurons. Peaks were considered overlapping with reciprocal overlap of >10%. (E) Mean ΔPSI of KCl-dependent exons under a changing H3K36me3 peak. Gain and loss were characterized as being unique to the +KCl (D, nonoverlapping left circle) or −KCl (D, nonoverlapping right circle) sets, respectively. An unpaired t-test was performed to assess difference in mean ΔPSI between exons under a KCl-dependent H3K36me3 peak that disappears (decreased) or appears (increased) after KCl stimulation.
We also found that KCl-treatment increased H3K36me3 enrichment at the splice junctions of KCl-dependent alternative exons (Fig. 4C). Because few of the genes containing the alternative exons also displayed differential expression upon KCl treatment, the increase in H3K36me3 does not appear to regulate transcription levels. This is further supported by the lack of change in the permissive hPTM, H3K4me1, at these KCl-dependent splice junctions (Fig. 4C). The majority of H3K36me3 peaks were not shared between untreated and treated cells, with 72% (8087 peaks) being unique to the KCl+ condition and 61% (4910 peaks) to the KCl− condition (Fig. 4D). This lack of overlap suggests that intragenic H3K36me3 patterns are dynamically altered by KCl. Finally, we sought to understand if there was a connection between gain or loss of H3K36me3 and the direction of inclusion change. The mean ΔPSI values for KCl-dependent exons that either gained or lost an H3K36me3 peak showed no bias toward either inclusion or exclusion after KCl (Fig. 4E). This lack of directionality suggests that H3K36me3 may require further local sequence context, such as RBP binding sites, to regulate KCl-dependent exons.
Together, these results suggest that histone modifications may be repositioned in response to KCl, possibly through alternative splicing of histone-modifying enzymes. Such redistribution of H3K36me3 appears to be concomitant with KCl-dependent changes in alternative splicing at a subset of exons, in support of a model for histone modifications contributing to activity-dependent splicing in neurons.
Discussion
Here, we integrate RNA sequencing data sets of mouse primary cortical neurons treated with KCl from five studies to identify activity-dependent splicing events of high confidence. By incorporating multiple data sets, we alleviated some of the inherent variability between culture conditions and stimulation parameters and focused only on alternative exons that were independently reproduced. We found more than 1000 alternative exons that consistently respond to KCl across different mouse strains and conditions, the majority of which become less included upon stimulation. These KCl-repressed exons generally exhibit higher GC content than do expression-matched controls and are enriched in genes related to neuronal function. Although the set of high-confidence splicing exons we define include previously described microexons, these are a minor subset of a larger program of alternative splicing that encompasses a normal distribution of largely constitutive exons.
Critically, >95% of all genes containing KCl-dependent splicing events are not differentially expressed after KCl treatment. The lack of apparent change in gene expression for most of the genes that have frame-shifting changes in splicing (Supplemental Fig. S9) initially seemed inconsistent with the prediction of NMD. However, NMD efficiency is dictated by several features beyond premature termination codon, such as distance from the final splice junction and length of downstream 3′ UTRs, which may not all be satisfied by these events (Nagy and Maquat 1998; Sato and Singer 2021). Indeed, future study of the proteome in response to KCl stimulation may uncover proteoforms of these frameshifting exons that escape NMD and are successfully translated. Moreover, loss of transcripts through NMD has been observed in some instances to be compensated for by an increase in transcription owing to autoregulatory feedback to compensate for loss of protein (Mühlemann et al. 2001; Iborra et al. 2004; Zhao et al. 2020). In sum, these results demonstrate the importance of considering splicing as well as transcript abundance when interpreting the impact of neuronal activity on the transcriptome.
Given the vast complexity of cell types in the brain, direct stimulation of primary neurons in culture provides a powerful means through which to characterize the mechanisms that may contribute to alternative splicing. The global meta-analysis we present here builds on single-exon studies of calcium-dependent pathways through which stimulation can dynamically regulate gene networks relevant to neuronal function. Targets of these signal-induced splicing programs include important genes related to neuronal function, such as neurexin and NMDA/GABA receptor subunits, as well as genes linked to autism (Lee et al. 2009; Iijima et al. 2011; Quesnel-Vallières et al. 2016). However, how activity-dependent alternative splicing is globally regulated in the brain remains an outstanding question of neurobiology.
One mechanism well described to generally contribute to signal-regulated alternative splicing is alterations in expression or function of RBPs that directly bind pre-mRNAs and direct the recognition or skipping of exons by the splicing machinery (Heyd and Lynch 2011; Fu and Ares 2014). In particular, dynamic alternative splicing has been shown for several neuronal RBPs, including the splicing factor Rbfox1. Splicing of an activity-dependent alternative exon in Rbfox1 regulates its nuclear localization and the splicing patterns of its targets (Lee et al. 2009). We found a homologous alternative exon in Rbfox2, an Rbfox1 paralog, that consistently responded to stimulation and likely alters its localization. More broadly, our analysis identified KCl-responsive alternative exons in many RNA processing factors that may regulate the activity of these factors by changing their localization, stability, or binding partners. These include changes in splicing of other proteins previously implicated in KCl-dependent splicing, such as Srsf11, Rnps1, and Ptbp1 (Supplemental Table S7). We find these exons to be largely spliced after 1–3 h of stimulation, after which they return to baseline inclusion levels (Supplemental Fig. S6). This result points to a global change in the splicing regulatory landscape of the cell following the induction of IEGs. Notably, this splicing of RBPs precedes changes in splicing of neuronal genes, suggesting that neuronal genes respond to the adaptation of splicing networks. Further study will be needed to ascertain how long the splicing of these neuronal genes can persist after activation and under what conditions these splicing events are observed in vivo.
In line with this temporal hypothesis, we find binding motifs for RBPs regulated by KCl enriched near KCl-dependent exons including for RBFOX2 and other RBPs above. However, we are not able to identify a consensus motif, which would suggest highly specific regulation by a small cohort of RBPs. We also do not find specific calcium-responsive elements (CaRRE) sequences that have been described in other studies to regulate activity-dependent exons in neurons (Xie and Black 2001; Xie et al. 2005). However, these CaRRE elements have been described to reside within GC-rich regions and adopt conserved secondary structures. We find that KCl-dependent exons have higher GC content than expression-matched controls, and therefore, perhaps GC-rich structures are more critical to direct splicing regulation than a specific CaRRE primary motif. Exonic GC content has also been associated with splicing outcome through regulating intron–exon definition, other RNA secondary structures, and transcription elongation rates (Kudla et al. 2006; Warf and Berglund 2010; Amit et al. 2012). In short, our data are most consistent with a model in which a diverse cohort of RBPs contributes to the regulation of KCl-responsive exons. These may be RBPs that are themselves alternatively spliced in response to KCl, or via altered binding affinities following from differences in RNA Polymerase II kinetics owing to local GC content.
Alternative splicing occurs predominantly cotranscriptionally and thus takes place in close proximity to the DNA from which the transcript originated (Reimer et al. 2021). Beyond being impacted by transcription elongation rates, we and others have demonstrated that certain chromatin modifications directly regulate alternative splicing in neurons at the single-gene level (Schor et al. 2009; Xu et al. 2021). In the current study, we found that, in addition to splicing regulatory proteins, histone-modifying enzymes were overrepresented among genes undergoing KCl-dependent alternative splicing. Splicing of histone-modifying enzymes has the potential to change their binding partners, subcellular localization, or catalytic efficiency, resulting in dynamic changes in chromatin modifications (Fiszbein et al. 2016; Agosto et al. 2023). One example is the methyltransferase EHMT2 (also known as G9a), which deposits H3K9me3. Specifically, we find that an alternative exon shown to regulate EHMT2 nuclear localization during neurodevelopment and to change global patterns of H3K9me3 also responds to KCl stimulation (Fiszbein et al. 2016). H3K9me3, in turn, has been linked to alternative splicing outcomes in learning and memory, highlighting its importance for neuronal function (Ding et al. 2017).
Given that we observed changes in the splicing of histone-modifying enzymes in response to KCl, we explored changes in histone modifications that have been linked to splicing regulation (Supplemental Fig. S8A). We were only able to find one public data set that contained global hPTM profiling in primary mouse neurons treated with KCl, which did not include replicates. This lack of replicates for each hPTM makes interpretation more difficult; however, we were able to compare between hPTMs as a means of strengthening our analysis. We are also cautious in extrapolating the ChIP data onto our RNA-seq analyses, given the variability we have observed between data set origin, especially at the level of transcript abundance. Despite this, we found KCl treatment altered the enrichment of H3K36me3 across a subset of KCl-responsive exons. H3K36me3 is a histone modification that is typically enriched in active gene bodies and is correlated to splicing outcome across neurodevelopment (Hu et al. 2017, 2020). Similar to GC content, histone modifications, including H3K36me3, have also been linked to changes in transcription rate across the gene body (Pokholok et al. 2005; Li et al. 2007). In turn, manipulation of transcription rate using genetic perturbations of RNA Polymerase II can change splicing patterns involved in neurodifferentiation (Maslon et al. 2019). Therefore, our data support a model in which activity-induced redistribution of H3K36me3 contributes to the regulation of splicing at a subset of activity-dependent exons, possibly via a combination of GC content and histone modification modulating elongation rate over target exons.
In sum, we describe here a conserved set of alternative exons that are tightly regulated in response to depolarization of primary neurons, altering the isoform expression of over a thousand genes. These alternatively spliced genes are separate from those that are regulated at the level of expression in response to depolarization, and together contribute to a changing proteomic landscape in response to neuronal stimulation. We find that activity-dependent splicing predominantly regulates the levels of constitutive, GC-rich exons that are not microexons, causing them to become less included after stimulation. Importantly, it is well documented that precise regulation of alternative splicing and chromatin is necessary for successful neurodevelopment and cognitive functions, such as learning and memory (Zovkic et al. 2013; Salinas et al. 2020). Moreover, chromatin regulators and splicing factors are overrepresented in mutations associated with neurodevelopmental and neurological disorders (Stessman et al. 2017; Faundes et al. 2018). As such, our analysis identifies a subset of activity-dependent exons that also display activity-dependent changes in the histone modification H3K36me3 that may contribute to splicing regulation. Our data complement previous mutational studies to suggest that a first wave of splicing changes in RNA processing factors and histone-modifying enzymes seeds the eventual splicing regulation of neuronal genes after sustained stimulation. We anticipate our findings will be useful in guiding future studies to directly test the mechanistic predictions put forward by this meta-analysis.
Methods
Selection and preparation of KCl-treated RNA sequencing data sets for analysis
Public RNA sequencing data sets of mouse primary neurons were selected for analysis and downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) using the following accession numbers: GSE69807 (Maze et al. 2015), GSE172427 (Calderon et al. 2022), GSE166959 (Ibarra et al. 2022), GSE201636 (Xu et al. 2022), and GSE89984 (Quesnel-Vallières et al. 2016). Sample accession numbers and details are summarized in Supplemental Table S8. Data sets were required to have more than 30 million reads per sample to ensure adequate coverage for differential splicing discovery. Raw reads were downloaded using the SRA-toolkit. Read quality was assessed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were then trimmed as needed with BBTools bbduk (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/) using the included adapters.fa database to remove 3′ sequencing adapters and low-quality bases (Q < 10; ktrim=r k=23 mink=11 hdist=1 qtrim=r trimq=10).
Discovery of differential alternative splicing events
For splicing analysis, cleaned reads were aligned to the mm39 genome (GENCODE vM31 annotations) using the splice-aware aligner STAR v2.7.9a (Dobin et al. 2013) with standard ENCODE parameters (‐‐outFilterType BySJout ‐‐outFilterMultimapNmax 20 ‐‐alignSJoverhangMin 8 ‐‐alignSJDBoverhangMin 1 ‐‐outFilterMismatchNmax 999 ‐‐outFilterMismatchNoverReadLmax 0.04 ‐‐alignIntronMax 1,000,000 ‐‐alignMatesGapMax 100,000). Differential alternative splicing events were called using replicate Multivariate Analysis of Transcript Splicing (rMATS) v4.1.2 (Shen et al. 2014) using the ‐‐variable-read-length flag to include trimmed reads in the analysis. rMATS accounts for differences in both (1) read depth and (2) isoform abundance in modeling changes in splicing. Comparisons were performed between equivalent duration KCl treatment and control sample (e.g., 6 h KCl vs. 6 h control) or between KCl treatment and untreated samples depending on available samples. Differential events were considered significant with an FDR < 0.05 and an absolute change in inclusion (|ΔPSI|) >10%, in line with standards in the field (Shen et al. 2014; Vaquero-Garcia et al. 2016).
Discovery of differentially expressed genes
To identify genes differentially expressed between KCl and control samples, cleaned reads were pseudoaligned to the mm39 genome (GENCODE vM31 annotations) and then quantified using kallisto (Bray et al. 2016) with correction for sequence-specific biases. Transcript quantifications were loaded and collapsed to gene-level identifiers using tximport (Soneson et al. 2015). Differentially expressed genes were called using DESeq2 (Love et al. 2014). Comparisons reflected those used in splicing analysis. Genes were considered significantly differentially expressed with an adjusted P-value < 0.05 and an absolute log2 fold change of >1.5.
PCA of Maze et al.
To assess the global differences in splicing and expression between samples from Maze et al. (2015), we first used PSI quantifications from rMATS to define exons that were quantified in every sample, resulting in a set of 21,618 exons. We also defined a set of 33,610 genes that were expressed at more than one transcript per million (TPM) in all samples. Using these sets, we performed PCA using prcomp and plotted the calculated principal components using ggplot2 (Wickham 2016).
Identification of enriched functional terms using Gene Ontology
Gene Ontology analysis was performed using gprofiler2, an R implementation of the gProfiler web tool (Raudvere et al. 2019). Gene sets were considered from only the “Biological Process” source. The background set was defined as genes expressed at more than one TPM in all samples of a given comparison to prevent spurious enrichment. Terms were considered significantly enriched with an adjusted P-value < 0.05.
Defining KCl-dependent exons of high confidence
Data sets were sorted into “moderate” (1–3 h) and “long” (5–6 h) of KCl treatment. For each data set in a given time interval, significantly changing alternative exons were identified (see the section “Discovery of differential alternative splicing events”), and common exons were identified for each pairwise comparison. Intersections were performed using the alternative exon coordinates, as well as flanking exon coordinates. Common exon sets were further refined by removing exons whose ΔPSI displayed an opposite sign between two data sets. A control set of nonchanging alternative exons was defined for moderate and long durations by filtering all nonsignificant (FDR > 0.1 and ΔPSI < 3%) exons in all data sets. We defined alternative as having a mean PSI between 10% and 90% across replicates. From this set, we selected exons so that the expression distribution matched the KCl-dependent set using the MatchIt package. We verified that distributions were statistically identical in expression using a Kolmogorov–Smirnov (KS) test.
Quantification of GC content between KCl and control alternative exons
The sequences for KCl-dependent and control exons were extracted from mm39, and the number of G and C nucleotides were counted, summed, and divided by the total exon length to calculate GC content.
Enrichment of k-mers near high-confidence KCl-dependent alternative exons
Exons in the KCl-dependent set were randomly subsampled (moderate, n = 696; long, n = 653), and then nonchanging exons (see section “Defining KCl-dependent exons of high confidence”) were selected for GC content and expression to create a GC-matched control set using the MatchIt package. From these sets, sequences 50 nt into a target exon from either the 3′ or 5′ splice site, 300 nt of a upstream intron, and 300 nt of a downstream intron were extracted from the mm39 genome for both the KCl-dependent and control sets using custom Python scripts (Supplemental Code S1). Sequences were then divided into k-mers (4-, 5-, 6-mers) and counted across each sequence of interest. A per k-mer Z-score was then calculated, and a Fisher's exact test was performed to assess statistical enrichment of a given k-mer in the KCl-dependent set compared to the GC-matched control set. We then randomly reassigned each exon to either the test or control set and reperformed this analysis using these shuffled sets. K-mers were considered enriched in KCl-dependent exons if they showed significant enrichment by a Fisher's exact test in the unshuffled analysis but not in the shuffled analysis.
Intersection of high-confidence KCl-dependent alternative exons and histone modification peaks
To assess the presence of histone modifications over KCl-dependent alternative exons, we first retrieved raw sequencing data from Wang et al. (2015) (GEO; GSE63271). We then performed end-to-end alignment of these reads to mm39 using the Bowtie 2 ‐‐dovetail option and filtering duplicate reads and read with an alignment score Q < 10 (Langmead and Salzberg 2012). Using these alignments, we called peaks for each hPTM (H3K36me3, H4K20me1, H3K4me1, H3K4me2) using the SICER2 algorithm (-w 200 -g 600 -fdr 0.001) for stimulated and nonstimulated conditions (Zang et al. 2009). We then filtered for intragenic peaks using gene body coordinates from GENCODE mm39 v31 annotations using BEDTools intersect (Quinlan and Hall 2010). We overlapped peaks to define those shared between both conditions using permissible cutoffs (fraction reciprocal overlap >10%) to decrease the likelihood of false-negative shared peaks. With these peaksets, we intersected each with KCl-dependent and control exons using BEDTools intersect with strict cutoffs, requiring the KCl-dependent exon and peak to overlap by >90%.
Quantification of histone modifications over high-confidence KCl-dependent junctions
To quantify global levels of histone modification, we downloaded bedGraph files summarizing mean read coverage for each histone modification and condition. We intersected these with KCl-dependent exon files using plyranges, and a Mann–Whitney U test was used to determine global differences in coverage.
Ex vivo culture and stimulation of mouse primary neurons
All animal work was performed under approval from University of Pennsylvania School of Medicine IUCAC. Pregnant female C57BL/6J mice were anesthetized using CO2 and then sacrificed by cervical dislocation. Cortices were dissected from E16.5 embryos and cultured in neurobasal medium (Gibco 21,103,049) supplemented with B27 (Gibco 17,504,044), GlutaMAX (Gibco 35,050,061), penicillin–streptomycin (Gibco 15,140,122) in TC-treated 12-well plates coated with 0.05 mg/mL Poly-D-lysine (Sigma-Aldrich A-003-E). At day in vitro (DIV) 3, neurons were treated with 0.5 µM AraC. KCl stimulation was performed at DIV 10. All neurons were pretreated with 100 μM D-AP5 and 1 μM tetrodotoxin 1 h prior to stimulation to quiesce the cells. For KCl-stimulated neurons, one-third of the medium was removed and the following was added to make a 3× KCl solution: 170 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES (pH 7.9). This 3× KCl solution was added back and incubated for a 2 h time point. For control neurons, neuronal media were added at one-third the total volume in the well. Neurons were harvested directly in 500 uL Qiagen P1 resuspension buffer and frozen at −80°C prior to RNA isolation.
32P-radiolabeled RT-PCR
Primers were designed in the flanking exons adjacent to candidate high-confidence CEs at the “moderate” time point. Ideal primer parameters were between 20 and 25 nt in length and a predicted melting temperature >75°C. RNA was harvested from mouse primary cortical neuron cultures (described above) using a Qiagen RNeasy kit (Qiagen 74,104) with the standard protocol. RNA was hybridized with 1 ng of reverse-target primer and reverse-transcribed using MMLV for 1 h. Reverse-target primer was labeled with 32γ ATP using T4 polynucleotide kinase and purified using a phenol–chloroform extraction. Reverse-transcribed RNA was amplified by PCR using Taq polymerase and 32P-labeled reverse-target primer. All radioactive PCRs were performed with an annealing temperature of 70°C and 1 min extension time. The optimal cycle number was determined empirically using cycle courses. Boiled 32P-labeled PCR products were resolved on a prerun 5% TBE-urea-acrylamide gel for 30 min at 1200 V and 30 mA and imaged using an Amersham Typhoon imager. Relative usage (PSI) was quantified using ImageJ and calculated as the normalized signal for included product/(normalized signal for excluded product + normalized signal for excluded product).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Institutes of Health: R35-GM118048 to K.W.L., R01-DA052465 and DP1-DA044250 to E.A.H., and 5T32-GM008216 to K.S.K. We recognize Matthew Gazzara for his important feedback on the analyses included in this work.
Author contributions: K.S.K. conceptualized the project, performed computational analyses and experiments, and wrote the manuscript. K.W.L. and E.A.H. designed experiments and wrote the manuscript. M.M. and E.K. performed mouse primary neuronal culture experiments and gave critical feedback on the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280082.124.
- Received October 1, 2024.
- Accepted April 7, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








