
Birth of protein-coding exons by ancient domestication of LINE-1 retrotransposon
- 1Laboratory of Genome and Epigenome Dynamics, Department of Animal Sciences, Graduate School of Bioagricultural Sciences, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8601, Japan;
- 2Department of Molecular Life Science, Tokai University School of Medicine, Isehara, Kanagawa 259-1193, Japan;
- 3Division of Omics Sciences, Institute of Medical Sciences, Tokai University, Isehara, Kanagawa 259-1193, Japan;
- 4Division of Interdisciplinary Merging of Health Research, Micro/Nano Technology Center, Tokai University, Hiratsuka, Kanagawa 259-1292, Japan
Abstract
Transposons, occasionally domesticated as novel host protein-coding genes, are responsible for the lineage-specific functions in vertebrates. LINE-1 (L1) is one of the most active transposons in the vertebrate genomes. Despite its abundance, few examples of L1 co-option for vertebrate proteins have been reported. Here, we describe protein isoforms, in which the L1 retrotransposons are incorporated into host genes as protein-coding exons by alternative splicing. L1 ORF1 protein (ORF1p) is an RNA-binding protein that binds to L1 RNA and is required for retrotransposition by acting as an RNA chaperone. We identified a splicing variant of myosin light chain 4 (MYL4) containing an L1 ORF1–derived exon and encoding a transposon fusion protein of L1 ORF1p and MYL4, which we call “Lyosin” in this study. Molecular evolutionary analysis revealed that the Lyosin isoform was acquired before the divergence of Sauropsida (reptiles and birds) during the Paleozoic era. The amino acid sequence of Lyosin had undergone purifying selection although it was lost in some lineages, including the Neognathae birds and snakes. The Lyosin transcript was expressed in the testes of four reptilian species, suggesting that its function is different from that of the canonical MYL4 transcript expressed in the heart. Furthermore, comprehensive sequence searches revealed other splicing isoforms fused to the L1 ORF1 in three genes in vertebrates. Our findings suggest the involvement of L1 for the birth of lineage-specific proteins and implicate the previously unrecognized adaptive functions of L1 ORF1p.
The emergence of lineage-specific proteins is important for the diversity of vertebrates. Vertebrate genomes contain large numbers of transposons, accounting for several percent to tens of percent of host genomes (Sotero-Caio et al. 2017). In humans, transposon-derived sequences are estimated to account for >60% of the genomic sequence (de Koning et al. 2011). Occasionally, transposon-encoded proteins have been “domesticated” as novel host proteins (Volff 2006).
Transposons are typically classified into two classes. Class I comprises retrotransposons transposed by copy-and-paste via RNA intermediates, whereas Class II comprises DNA transposons generally transposed by a cut-and-paste mechanism (Wells and Feschotte 2020). Co-option of transposon-encoded proteins has been reported in both groups (Volff 2006; Johnson 2019). Retrotransposons are further classified into LTR retrotransposons and non-LTR retrotransposons. In mammals, various lineage-specific proteins have been acquired by LTR retrotransposons. They have core gag and pol genes. The gag genes encode capsid proteins, and the gag gene co-option has occurred multiple times in mammals (Pang et al. 2018; Wang et al. 2019; Boso et al. 2021; Henriques et al. 2024). The gag-derived ARC is involved in synaptic plasticity, packaging its mRNA for cell–cell transfer in mammals and flies (Ashley et al. 2018; Pastuzyn et al. 2018). The SIRH families are gag-derived gene families in mammals (Kaneko-Ishino and Ishino 2023), and the SIRH family genes, Peg10 and Rtl1, are involved in placental development (Ono et al. 2006; Sekita et al. 2008). Studies on knockout mice suggest that other SIRH genes contribute to innate immunity in the brain (Irie et al. 2022; Ishino et al. 2023). PNMA2 is expressed in neurons, and its capsid structure is implicated in autoimmune diseases (Xu et al. 2024).
Endogenous retroviruses (ERVs), derived from retroviruses and classified as LTR retrotransposons, contain an env gene in addition to gag and pol genes. The syncytin gene is derived from the env gene and is involved in placental development, exhibiting fusogenic activities in trophoblast cell–cell fusion (Blond et al. 2000; Mi et al. 2000). Env-derived fusogenic genes have been independently acquired in multiple mammalian lineages (Dupressoir et al. 2005; Heidmann et al. 2009; Cornelis et al. 2013, 2014, 2015, 2017; Esnault et al. 2013; Nakaya et al. 2013; Kitao et al. 2023). Some Env proteins have immunosuppressive activity, suggesting their involvement in maternal–fetal immunity (Mangeney et al. 2007). Other co-option events of retroviral genes include ASPRV1(also known as SASPase), derived from a retroviral protease involved in the protease activity for mammalian skin formation (Matsui et al. 2011); NYNRIN, derived from integrase and contributing to placentation (Plianchaisuk et al. 2022); and RTOM family, derived from reverse transcriptase in monotremes (Kitao et al. 2022). Most of the co-option events have been studied in mammals; however, retroviral proteins have also been reported in birds, reptiles, fish, and eukaryotes (Carré-Eusèbe et al. 2009; Henzy et al. 2017; Aiewsakun et al. 2019; Wang and Han 2021). The recent expansion of available vertebrate genomic data has enabled further computational detection of co-option, revealing more than 100 co-option events of gag and env genes (Wang and Han 2020). A comprehensive genome analysis has revealed that lineage-specific transcription factors have arisen recurrently in vertebrates through the co-option of DNA transposons (Cordaux et al. 2006; Cosby et al. 2021). Co-option of transposon proteins has been reported for DNA transposons and LTR retrotransposons; however, the co-option of non-LTR retrotransposons is still unelucidated.
Long interspersed elements (LINEs) are non-LTR retrotransposons that are found in various vertebrates (Sotero-Caio et al. 2017). LINE-1 (L1) is the only active LINE in the human genome and has open reading frames (ORFs). The ORF2 protein (ORF2p) is an enzymatic protein containing endonuclease and reverse transcriptase domains. The ORF1 protein (ORF1p) is a nonenzymatic RNA-binding protein involved in the other retrotransposition processes, although some LINEs lack such a nonenzymatic protein (Metcalfe and Casane 2014). These LINE proteins interact with host proteins to mediate the complex processes of retrotransposition (Luqman-Fatah and Miyoshi 2023). The domestication (co-option) of LINE protein-coding genes has been reported. L1TD1 consists of two coding exons derived from L1 ORF1 and is conserved in eutherian mammals. This gene was first identified in a screen for reprogramming-related proteins in mouse ES cells, but knockout of L1td1 showed no phenotypic effects on cell reprogramming or mouse development (Iwabuchi et al. 2011). In contrast, other studies suggest that human L1TD1 is involved in cell reprogramming (Närvä et al. 2012). Mechanistically, L1TD1 is involved in post-transcriptional regulation by controlling intracellular protein condensation (Jin et al. 2024). The other example is a ruminant-specific gene, BCNTP97, derived from the tandem duplication of BCNT gene followed by insertion of retrotransposable element-1 (RTE-1) (Iwashita et al. 2006). RTE-1 is classified as a LINE superfamily (Metcalfe and Casane 2014). The BCNTP97 gene encodes an intact endonuclease domain derived from RTE-1; however, its molecular function remains unknown (Iwashita et al. 2006). Although these few LINE-derived genes have been identified in mammals, their evolution and diversity are poorly understood.
In the present study, we report the co-option of L1 ORF1 as alternative exons to encoding transposon fusion protein in vertebrates. Although the contribution of L1 insertions to the gene structures is well documented, this study proposes a previously unrecognized role of L1 in generating evolutionarily conserved protein isoforms.
Results
Identification of Lyosin: an L1 ORF1p and MYL4 fusion isoform
We conducted MMseqs2 searches using amino acid sequences of L1 families obtained from the Dfam database against RefSeq proteins of American alligator (Alligator mississippiensis) and found that myosin light chain 4 (MYL4) has homology with L1 ORF1p (L1-32_DR, E-value: 9.0 × 10−20). According to RefSeq annotation, the American alligator's MYL4 gene has a noncanonical exon (hereafter called “exon L”) (Fig. 1A). The amino acid sequences encoded by exon L show 30% identity and 51% similarity to L1 ORF1p (Fig. 1B,C). Exon L was predicted to be connected to exon 3 instead of the canonical exon 1 and 2. Therefore, this splicing variant was inferred to encode a transposon fusion protein of L1 ORF1p and the MYL4 protein. We call this putative protein Lyosin (L1-MYL4 fusion protein) in this study. The L1 ORF1p consists of four domains: the disordered N-terminal domain (NTD), coiled-coil (CC), RNA recognition motif (RRM), and the C-terminal domain (CTD) (Naufer et al. 2019). Structural prediction revealed that the Lyosin protein retained domains corresponding to the NTD, CC, and RRM (Fig. 1D). The predicted RRM structure of the Lyosin protein showed a characteristic structure composed of α-helixes and β-sheets (βαββαβ) observed in the L1 ORF1p RRM domain (Fig. 1E). Moreover, superposition of the predicted Lyosin RRM domain with the human L1 ORF1p RRM domain determined by X-ray diffraction of the crystal structure (Khazina and Weichenrieder 2009) showed a high degree of similarity (root mean square deviation [RMSD] = 1.619 Å) (Fig. 1F). These structural analyses suggest that the protein encoded by exon L is homologous to L1 ORF1p. The MYL4 protein and other myosin light chain proteins belong to the EF-hand calcium-binding protein family (Grabarek 2006), although the MYL4 protein itself has lost its calcium-binding capacity (Sitbon et al. 2020). The protein sequence encoded by exon 3 and the later exons included in the Lyosin protein corresponds to the EF-hand calcium-binding motifs (Fig. 1B). Taken together, these structural analyses indicate that the Lyosin protein is a transposon fusion protein composed of the NTD, CC, and RRM domains of L1 ORF1p and the EF-hand domain of the MYL4 protein.

Identification of Lyosin in the alligator genome assembly. (A) UCSC Genome Browser of the MYL4 gene of mouse and American alligator. The noncanonical exon L was annotated in the American alligator's MYL4 gene. (B) Representation of the MYL4 and Lyosin protein structure in American alligator. The amino acid sequences of exons 1 to 2 and exons 3 to 6 correspond to the actin-binding site and the EF-hand calcium-binding domain, respectively, in the MYL4 protein. The Lyosin protein consists of amino acids encoded in exon L and exons 3 to 6. Exon L encodes a 246 amino acid protein similar to the L1 ORF1 protein (ORF1p), which is an RNA-binding protein. The ORF2 protein (ORF2p) of L1 contains an enzymatic protein with endonuclease and reverse transcriptase activity. (C) The amino acid sequence alignment of L1-32_DR ORF1p and exon L of the American alligator's Lyosin protein. (D) Protein structures of human L1 ORF1p (PDB: AF_AFQ9UN81F1) and the Lyosin protein predicted by AlphaFold2 (Jumper et al. 2021) implemented in ColabFold (Mirdita et al. 2022). ORF1p consists of the disordered N-terminal domain (NTD), coiled-coil (CC), RNA recognition motif (RRM), and C-terminal domain (CTD). Lyosin contains NTD, CC, and partial RRM. (E) The βαββαβ structure of the predicted Lyosin RRM domain. (F) Structural comparison of the predicted Lyosin RRM domain and the L1 ORF1p RRM domain determined by X-ray diffraction (Protein Data Bank [PDB; https://www.rcsb.org] 2W7A).
Identification of the Lyosin protein in reptiles and birds
Next, we investigated whether Lyosin is present in any species other than the American alligator. We performed a BLASTP search against the NCBI nonredundant (nr) protein database using the amino acid sequence of the alligator Lyosin protein as a query. The search identified the Lyosin-like proteins in three species of Testudines (turtles), four species of Squamata (lizards and snakes), and five species of Aves (Supplemental Table S1). Alignment of these proteins revealed that sequences corresponding to L1 ORF1p were divergent, whereas those of the MYL4 protein were highly conserved. In particular, deletions in the region corresponding to the RRM domain were observed in birds, except Okarito kiwi (Apteryx rowi) (Supplemental Fig. S1). We then constructed a molecular phylogenetic tree of L1 ORF1p and Lyosin-like exon L to gain insights into their evolutionary relationships. The maximum-likelihood based tree showed that the exon L of Lyosin-like proteins was distinct from L1 ORF1p and further reflected the host evolution (Fig. 2A).
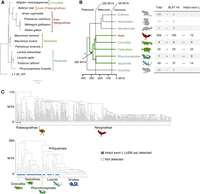
Identification of Lyosin in the genome assemblies of Tetrapoda. (A) Maximum likelihood-based phylogenetic tree of the Lyosin proteins obtained by NCBI BLASTP search. Open circles in internal nodes indicate >95% ultrafast bootstrap support (1000 replicates). (B) Schematic summary of the detection of the Lyosin sequence in the genome assemblies of Tetrapoda. The numbers of the analyzed genome assemblies (total), Lyosin-detected genome assemblies by BLAT searching (BLAT hit), and intact Lyosin’s exon L-detected genome assemblies are listed on the right. The co-option for the Lyosoin protein occurred before the divergence of reptiles and birds, 280 million years ago (MYA) in the Paleozoic era. (C) The evolutionary history of Lyosin in reptile and bird lineages. Phylogenetic trees of species with divergence times were based on the TimeTree (Kumar et al. 2022). The species predicted to have an intact exon L with more than 200 amino acid coding sequence were reflected on the tree nodes (Supplemental Table S3).
Lyosin in genome assemblies of Tetrapoda
The RefSeq annotations include predicted gene models. In later analyses, we evaluated whether the Lyosin transcript is an existing isoform. The Lyosin protein is encoded by a noncanonical splicing variant, and it is probable that the Lyosin protein has not been annotated in the most genome assemblies, resulting in its absence of protein databases. Therefore, we conducted a comprehensive database-wide search for the Lyosin sequences. First, we performed an intron-considered BLAT search on the genome assemblies of Tetrapoda using the full-length amino acid sequences of the Lyosin protein as queries. Approximately 40% of the Lyosin instances correspond to the canonical MYL4 protein. Therefore, we set the threshold for the detection to a query coverage of ≥50%. As a result of this search, the Lyosin sequences were detected in the genome assemblies of Aves (185 out of 556 assemblies), Crocodilia (four out of four assemblies), Testudines (25 out of 25 assemblies), Rhynchocephalia (one out of one assembly), and Squamata (37 out of 49 assemblies), but not in those of Mammalia and Amphibia (Fig. 2B). This suggests that the Lyosin isoform arose in the ancestor of the Sauropsida clade (reptiles and birds) during the Paleozoic era, at least 280 million years ago. We could not determine whether exon L was captured at an earlier time but lost in the other clades (e.g., Mammalia) because no clear traces of L1 insertion could be confirmed owing to its old insertion date. We also performed a BLAT search for the canonical MYL4 protein to confirm that the identified Lyosin sequence was the isoform of the MYL4 gene. Among the 252 genome assemblies with significant hits for the Lyosin protein, 249 genome assemblies showed an overlap of the best hits for the canonical MYL4 and Lyosin proteins. This suggests that our BLAT search identified the actual Lyosin locus rather than the L1 ORF1 unrelated to the MYL4 gene. Furthermore, in 211 out of 252 genome assemblies, the Lyosin locus was located within 100 kb of the BLAT hit for the protein of CDC27 gene, a neighboring gene of MYL4. In the three genome assemblies, the CDC27 gene was >100 kb apart, and in 38 assemblies, it was located on a different scaffold (Supplemental Table S2). Note that in cases of truncated assembly or loss of synteny in the genomes, proximity to the CDC27 gene could not be confirmed.
It is also possible that coding capacities or splicing sites of the exon L were lost even in species in which Lyosin sequences were detected by BLAT search. Therefore, we considered those that retained canonical 5′ splice sites (5′-GT-3′) and encoded more than 200 amino acids (sufficient length to include the NTD, CC, and RRM domains) as the intact exon L. Among the genome assemblies with the Lyosin isoform confirmed by BLAT search, the intact exon L was present in the genome assemblies of Aves (15 assemblies), Crocodilia (four assemblies), Testudines (23 assemblies), and Squamata (14 assemblies) (Fig. 2B). Crocodilia was the only group in which all species retained the intact exon L.
Possible truncation of the exon L coding sequence in some lineages
To trace the evolution of the Lyosin isoform, we mapped the presence or absence of the exon L sequence onto a species tree obtained from the TimeTree database (Kumar et al. 2022). In Aves, we found that the intact exon L was detected only in the species of Palaeognathae (Fig. 2C). Similarly, the intact exon L was not detected in snakes (Serpentes, Squamata) (Fig. 2C). Although some analyzed species were not shown in the tree of Figure 2B owing to the lack of those species in the TimeTree database (Kumar et al. 2022), we confirmed that no genome assemblies of Neognathae or Serpentes contained intact exon L. Even in the genome assemblies of Palaeognathae, Testudines, Rhynchocephalia, and the nonsnake Squamata (lizards), the intact exon L was not detected in several species, suggesting that the coding sequences of exon L have been truncated independently by mutations. For example, a shared nonsense mutation was observed in three species of the genus Nothopracta (Supplemental Fig. S2). One nonsense mutation and two indels causing frameshift were identified in Varanus komodoensis. Although the possibility of sequencing errors cannot be excluded in all cases, these shared and multiple mutations strongly suggest that the coding sequence of exon L was truncated by actual mutations.
Molecular evolution of amino acid sequences in the Lyosin protein
Frequent loss of the Lyosin isoform raises the suspicion that these sequences have remained by chance and are not evolutionarily conserved as functional protein-coding exons. Therefore, we next investigated the conservation of the amino acid sequences of exon L. Alignment of the amino acid sequences revealed that the conservation level differed in protein domains. In lizards, in which the sequences were relatively diverse, the amino acids were conserved at the N terminus of the CC domain and at both termini of the RRM domain (Fig. 3A). The same trend was also observed for Palaeognathae and Testudines. As for the RRM domain, the conservation levels were high at the structured α-helix and β-sheet, whereas those were relatively low at the loop region (Fig. 3B). These data suggest that the functional constraints of the Lyosin protein structure dictated their amino acid sequence conservation.
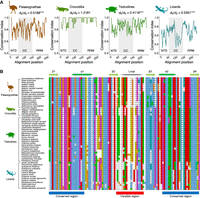
Evolutionary conservation of each amino acid site in the Lyosin protein. (A) Amino acid sequences encoded in exon L were aligned. The conservation index was calculated by summing of the squares of the amino acid rates at each site. Sites with gaps were excluded. Plots were obtained along a sliding window of five sites for the conservation index. The dN/dS values were analyzed by the likelihood ratio test; (***) P < 0.001. (B) An alignment of the RRM domains of Lyosin. The top part of the alignment shows the structure of the RRM domain (see Fig. 1E).
We next conducted molecular evolutionary analyses on the amino acid sequence of exon L. We calculated the ratio of nonsynonymous and synonymous codon substitution (dN/dS) in species that retained the intact exon L. A purifying selection on the amino acid sequence of exon L was observed in Palaeognathae (dN/dS = 0.5188, P = 3.679 × 10−9), Testudines (dN/dS = 0.4116, P = 3.479 × 10−23), and lizards (dN/dS = 0.5351, P = 1.295 × 10−13), although the dN/dS value was found to be greater than one in Crocodilia (dN/dS = 1.2181, P = 0.602) (Fig. 3A). We also calculated the dN/dS values of the MYL4 exon3: Palaeognathae (dN/dS = 0.0393, P = 1.321 × 10−19), Testudines (dN/dS = 0.0258, P = 1.211 × 10−32), lizards (dN/dS = 0.0063, P = 8.086 × 10−49), and Crocodilia (dN/dS = 0.0001, P = 1.799 × 10−4). The higher dN/dS ratios of the exon L suggest that the exon L amino acid sequence is under weaker negative selection compared with the canonical MYL4 amino acid sequence. We also evaluated positively selected amino acid sites in the exon L and found that only one site in Palaeognathae was statistically supported as being under positive selection, suggesting that positive selection was not a prevalent feature of the Lyosin protein (Supplemental Fig. S3). Together, these site-specific conservation and purifying selections on the amino acids suggest that exon L of the Lyosin protein was maintained as a protein-coding exon.
Testis-specific expression of the Lyosin transcript
To obtain insights into the physiological function of the Lyosin protein, we investigated its tissue expression. The MYL4 gene is known to be expressed specifically in the heart of adult mammals (Sitbon et al. 2020). We analyzed the transcriptome data set of green anole (Anolis carolinensis), which is predicted to retain the intact exon L of the Lyosin transcript. Similar to mammals, the MYL4 gene was highly expressed in the green anole heart, whereas low expression was observed in other tissues (Fig. 4A). To examine the expression level of the Lyosin splicing variant, we counted the RNA-seq reads spanning the introns. Although canonical splicing between exon 2 and exon 3 was detected dominantly in heart (Fig. 4B), splicing between exon L and exon 3 for the Lyosin transcript was specifically identified in the testis (Fig. 4C). The genome browser view of the RNA-seq reads mapped to the MYL4 locus also confirmed the selective expression of the Lyosin transcript in testis (Fig. 4D,E). To determine whether this expression pattern is similar in other species, we analyzed transcriptome data of the American alligator (A. mississippiensis), Chinese soft-shelled turtle (Pelodiscus sinensis), and emu (Dromaius novaehollandiae). Although the expression of the Lyosin transcript was not confirmed in any emu's tissues, including the testis, the American alligator and Chinese soft-shelled turtle showed the expression of the Lyosin transcript in the testis (Supplemental Fig. S4). In addition, reverse-transcription PCR (RT-PCR) was performed on the heart and testis of Madagascar ground gecko (P. picta) (Noro et al. 2009), which is also predicted to retain the intact exon L of the Lyosin transcript. The canonical MYL4 transcripts were detected in the heart, whereas the Lyosin transcripts were detected in the testis (Fig. 5A).
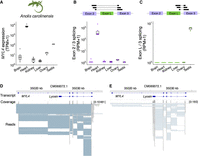
Tissue expression of the Lyosin transcript. (A) Box plot and point representations of the transcripts per million (TPM) of MYL4 expression obtained from RNA-seq data of the green anole tissues. (B,C) Box plot and point representations of splice junction reads spanning exons. The read counts were normalized as reads per million (RPM): reads spanning exons 2 and 3 (B) and reads spanning exons L and 3 (C). (D,E) Genome browser view of the MYL4 locus with RNA-seq reads of green anole. The broad blue bands represent transcripts. Transcripts of canonical MYL4 and Lyosin were constructed by genome-guided de novo assembly in this study. The gray bands below indicate mapped reads, and the blue lines between mapped reads are the gaps corresponding to the introns: heart (D) and testis (E).
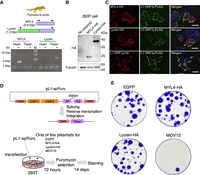
Molecular characterization of the Lyosin protein. (A) RT-PCR targeting the MYL4 and Lyosin transcripts. RNA was extracted from the heart and testis of a sexually matured male Madagascar ground gecko. Samples without reverse transcriptase (RT−) were analyzed as negative controls. (B) Western blotting of MYL4-HA and Lyosin-HA expressed in human 293T cells. (C) Fluorescence immunostaining images in human 293T cells. White arrows indicate L1 ORF1p cytoplasmic foci. The scale bars represent 20 µm. (D) Illustration of the mechanism of L1 retrotransposition assay. (E) Results of L1 retrotransposition assay. The indicated genes and L1 reporter were coexpressed in 293T cells. L1 retrotransposition results in cell colonies resistant to puromycin. MOV10 is used as a positive control that was reported to suppress L1.
Molecular characterization of the Lyosin protein
We next attempted the molecular characterization of the Lyosin protein. The coding sequences for the MYL4 and Lyosin proteins of Madagascar ground gecko were cloned into a mammalian expression plasmid with the C-terminal HA tag. Then, the plasmids were transfected into human embryonic kidney 293T cells. Western blotting analysis showed that the bands of expressed proteins were detected at the expected positions, confirming that the proteins were not cleaved (Fig. 5B). We examined their subcellular localization in comparison with human L1 ORF1p, which is known to aggregate and to form the cytoplasmic foci required for retrotransposition (Goodier et al. 2007). Unlike L1 ORF1p-HA, the Lyosin-HA was dispersed in the cytoplasm and did not exhibit the foci formation (Fig. 5C). Furthermore, neither MYL4-HA nor Lyosin-HA was present in foci formed by coexpressed L1 ORF1p-FLAG (Fig. 5C). A mammalian L1 ORF1-derived gene, L1TD1, has been suggested to be a repressor of L1 on the basis of evolutionary analyses (McLaughlin et al. 2014). Given that the Lyosin transcript is expressed in the testis, it may repress the L1 activity to protect germline DNA from transposon insertion. To examine the effect of the Lyosin protein on L1 retrotransposition, we performed a reporter-based L1 retrotransposition assay in 293T cells (Fig. 5D). The results showed that the Lyosin protein did not affect L1 retrotransposition, whereas human MOV10 helicase, which is known to be an inhibitor of L1 mobility (Li et al. 2013), reduced L1 retrotranspositions (Fig. 5E). Although it should be noted that these molecular characterizations were performed in human cells using human L1, we obtained no evidence suggesting that the Lyosin protein affects L1 activity.
Other examples of the origination of coding exons by the ORF1p co-option
The emergence of the Lyosin protein in the Sauropsida clade raises the possibility that the vertebrate genomes may harbor other protein isoforms fused with LINE ORF1p. To assess this possibility, we screened the vertebrate RefSeq proteins for protein-coding genes harboring both ORF1p-like and non-ORF1p-like isoforms (see Methods) (Fig. 6A). We identified four protein-coding genes with ORF1p fusion isoforms, including the MYL4 gene. It should be noted that the L1 ORF1p-derived L1TD1 gene was not detected because it is not a fusion isoform with a host gene. BCNTP97 was also not detected, as it is derived from the fusion with the LINE enzymatic protein (i.e., ORF2p). The identified ORF1p-like exons were classified as either the first or last coding exons (Supplemental Fig. S5). The RFX5 gene, which encodes DNA-binding protein RFX5, had an alternative noncanonical first coding exon similar to L1 ORF1p in catfish (order Siluriformes) (Fig. 6B). The NUP42 gene, encoding nucleoporin 42, and the USP4 gene, encoding ubiquitin-specific peptidase 4, had ORF1p-like final coding exons in Afrotheria and Carnivora, respectively (Fig. 6C). These transposon fusion proteins were termed L1-RFX5, NUP42-L1, and USP4-L1. Structural prediction of the proteins encoded by these ORF1p-like exons confirmed the presence of CC and RRM domains as in L1 ORF1p in all three genes, and an almost complete CTD structure was confirmed in the NUP42-L1 protein (Fig. 6D). Based on the RepeatMasker tracks on the UCSC Genome Browser, the ORF1p-like exon of the NUP42-L1 transcript overlapped with L1M4c in Elephas maximus indicus (Indian elephant), and that of the USP4-L1 transcript overlapped with L1MA9 in Mirounga angustirostris (Northern elephant seal) (Fig. 6E,F). We could not identify any overlap with the RepeatMasker track in the Lyosin or L1-RFX5 isoform. We also confirmed the lack of overlap with tracks of our repeat consensus sequences made by RepeatModeler2 (Supplemental Fig. S6; Flynn et al. 2020). A possible explanation for this is that the copies of their ancestral LINEs were highly fragmented and are not detected as repeat sequences at present.
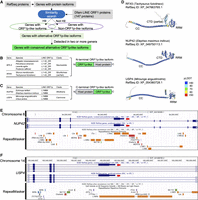
Other ORF1p fusion isoforms in vertebrates. (A) Schematic workflow of the detection of ORF1p fusion isoforms from RefSeq proteins. (B) In the nonmammalian vertebrate RefSeq proteins, two protein-coding genes had alternative ORF1p-like exons. The top hit LINE ORF1p was described in each fusion isoform. In both genes, the amino acid sequences encoded by the first coding exons were similar to ORF1p. (C) In the mammalian vertebrate RefSeq proteins, two protein-coding genes had alternative ORF1p-like exons. The amino acid sequences encoded by the final coding exons were similar to ORF1p. (D) Protein structures of the ORF1p-like amino acid sequence encoded by the noncanonical exons predicted by AlphaFold2. Coiled-coil (CC), RNA recognition motif (RRM), and C-terminal domain (CTD). (E,F) UCSC Genome Browser views of ORF1p-like isoform with RepeatMasker tracks. Red arrows indicate the ORF1p-like exons. The UCSC Genome Browser was accessed on June 25, 2024. NUP42 in the Elephas maximus indicus (Indian elephant) genome assembly (GCF_024166365.1; E), and USP4 in the Mirounga angustirostris (Northern elephant seal) genome assembly (GCF_021288785.2; F).
Evolutionary history of other L1 ORF1-derived exons
To further explore the evolution of L1 ORF1p fusion isoforms other than the Lyosin protein, we extracted intact L1 ORF1-derived exons from the genome assemblies as with the Lyosin protein. First, we performed a BLAT search for the identified fusion proteins. Next, overlaps with the BLAT best hits against the canonical isoforms were examined. Finally, L1-derived ORFs of more than 200 amino acids were extracted. As a result, the L1-RFX5 protein sequence with intact exons was obtained from eight Siluriformes species. In the NUP42-L1 protein, ORF1-derived exons were obtained from five species of Afrotheria, and the USP4-L1 protein sequence was identified in 13 species of Carnivora (Fig. 7A,B). All these isoforms with intact exons were located within 100 kb of their neighboring genes (MINDY1 for L1-RFX5, GPNMB for NUP42-L1, and IHO1 for USP4-L1) (Supplemental Table S5–S7). Purifying selection for amino acid sequences of ORF1-derived exons was detected for the L1-RFX5 protein (dN/dS = 0.3151, P = 1.253 × 10−28) and the NUP42-L1 protein (dN/dS = 0.4116, P = 1.198 × 10−9), but not for the USP4-L1 protein (dN/dS = 0.8646, P = 0.4931). Although the evolutionary conservation trends were divergent, this suggests that at least the L1-RFX5 and NUP42-L1 proteins have been conserved.

Evolutionary history of other ORF1p fusion isoforms. (A) Results of the detection of intact L1 ORF1-derived exons on the host phylogenetic trees. The phylogenetic trees with divergence times were based on the TimeTree (Kumar et al. 2022). Otophysi (a group of bony fishes), Siluriformes (catfishes); Musteloidea (weasels and relatives); Pinnipedia (seals and relatives); Ursoidea (bears and relatives). (MYA) Million years ago. (B) Maximum likelihood-based phylogenetic tree of ORF1p-derived amino acid sequences of fusion proteins and L1 ORF1ps obtained from Dfam database (77 proteins). Each ORF1-derived exon was clustered into a single clade. Ultrafast bootstrap support values (1000 replicates) of internal nodes are shown for the clades of the ORF1-derived exons.
To investigate the evolutionary origins of the L1 ORF1-derived exons, we constructed a maximum likelihood-based phylogenetic tree of L1 ORF1ps (77 sequences) from the Dfam curated database. The ORF1-derived exons of the same loci were clustered into a single clade. The branch length within the Lyosin clade was longer than that of other L1 exons. This may be because of the relatively older origin of the Lyosin protein, which led to sequence diversification. The NUP42-L1 and USP4-L1 proteins are phylogenetically close. This is because both are derived from the mammalian L1. Taken together, these data suggested that the emergence of novel splicing isoforms by the insertion of the L1 ORF1 and their subsequent domestication has occurred multiple times in vertebrate evolution.
Discussion
LINEs are some of the most active transposons in vertebrates. Among them, L1 is the only LINE that remains active in the human genome (Hoyt et al. 2022). L1 ORF1p is an RNA-binding protein that assembles to package L1 RNA (Hohjoh and Singer 1996). ORF1p then facilitates the rearrangement of nucleic acid structures and is thought to function as a nucleic acid chaperone (Martin and Bushman 2001). Structural analysis has suggested that both RRM and CTD are required for L1 ORF1p to bind to RNA (Khazina and Weichenrieder 2009). The Lyosin protein, however, lacked CTD (Fig. 1D). Thus, the Lyosin protein probably lost its native RNA-binding ability. In contrast, amino acids for the RRM domain were selectively conserved in Lyosin (Fig. 3B). Homo-trimer formation is also important for L1 ORF1p function (Martin et al. 2003). For trimer formation in L1 ORF1p, the C-terminal half of the CC domain is necessary and sufficient (Khazina and Weichenrieder 2009). In the Lyosin protein, the C-terminal half of the CC domain is less conserved than the N-terminal half. More experimental studies are required to identify the molecular function and structural features of the Lyosin protein.
We confirmed the independent truncation of the coding sequences of exon L in several genome assemblies (Supplemental Fig. S2). There is a concern that some truncation mutations may be errors in genome assembly, but recent attempts to create more accurate genome assemblies will gradually resolve this issue (Rhie et al. 2021). The recurrent gene losses have been observed in L1TD1, a mammalian L1 ORF1-derived gene. The L1TD1 gene was acquired in the last common ancestor of eutherians but was lost independently in the lineages of Afrotheria, ruminants, and bats (McLaughlin et al. 2014). The loss of the L1TD1 gene has been observed in megabat lineages, in which active L1 elements are extinct. This correlation is consistent with the hypothetical scenario, in which the L1TD1 protein is a LINE suppressor; when the target LINE is extinct, the L1TD1 gene becomes unnecessary and is subsequently lost by random mutations (McLaughlin et al. 2014). This hypothetical model was also proposed in the viral restriction factors derived from ERVs (Johnson 2019). Nonetheless, a recent reporter-based assay showed that the human L1TD1 protein is not capable of suppressing human L1 activity (Jin et al. 2024). It is also possible that the L1TD1 protein has the potential to function as both an L1 repressor and a translational regulator and that the balance of these dual functions differs between lineages (McLaughlin et al. 2014). The Lyosin transcript was expressed in the testis (Fig. 4; Supplemental Fig. S4). The hypothesis that the Lyosin protein is a suppressor of L1 activity is plausible because the repression of transposon in germ cells is advantageous for avoiding transposon insertion into germline DNA. However, there is currently no evidence supporting this hypothesis, as expression of the Lyosin protein did not affect the L1 activity in human cells (Fig. 5). Further studies in reptilian or avian experimental systems will be needed to validate whether the Lyosin protein represses the L1 retrotransposition. Currently, we have not detected the endogenous Lyosin protein in the testis. The protein experiments using the antibody recognizing the reptilian MYL4 or Lyosin protein are also needed.
The transcriptional regulation of Lyosin remains to be elucidated. Considering the origin of Lyosin and its expression in the testis, it is plausible that Lyosin expression might reflect the transcriptional pattern of L1 elements. Among transcription factors previously reported to promote human L1 transcription such as RUNX3, SP1, SOX2, and YY1 (Luqman-Fatah and Miyoshi 2023), RUNX3 binding sites were identified near the transcription start site (TSS) of the Lyosin isoform but not in the canonical MYL4 isoform in Anolis carorinensis (Supplemental Fig. S7A). However, conserved upstream sequences near exon L were not detected among the three species (A. carorinensis, P. sinensis, and A. mississippiensis) in which Lyosin expression was confirmed (Supplemental Fig. S7B). Future studies employing in vitro reporter assays and chromatin immunoprecipitation analyses will identify responsible transcription factors responsible for Lyosin expression.
The multiple gene losses can be alternatively explained by the gene replacement hypothesis, in which the functional replacement of one gene with another makes the existing gene dispensable and leads to its loss. Importantly, such evolutionary replacement has been proposed for the evolution of syncytin genes, which are derived from ERVs, another group of retrotransposons (Imakawa et al. 2015). Functional syncytin genes have different viral origins in mammalian lineages (Lavialle et al. 2013), and functional reduction of several syncytin genes has been reported in different mammals (Nakaya et al. 2013; Shoji et al. 2023). This can be explained by the scenario in which a new syncytin gene inherits placental functions from the older syncytin gene like “baton-pass” (Imakawa et al. 2015). In the case of the Lyosin isoform, L1 ORF1 is interspersed in the vertebrate genomes, and functional replacement of the Lyosin protein with a new Lyosin-like protein may occur. Our analysis identified at least three ORF1p fusion proteins other than the Lyosin protein (Fig. 6). Further identification of ORF1p fusion proteins, as well as characterization of shared features such as tissue-specific expression patterns, will provide a deeper insight into the evolutionary mechanisms underlying the emergence of ORF1p-derived protein isoforms.
Co-option of transposable elements as protein-coding exons has been well documented in DNA transposons and LTR retrotransposons (Cordaux et al. 2006; Volff 2006; Abascal et al. 2015; Cosby et al. 2021). Even taking our study into account, the number of LINE fusion isoforms remains few, given their abundance. However, we believe that the number of conserved LINE fusion proteins will increase with future studies. First, the evolutionarily conservation of fusion proteins with other LINE ORFs, including ORF2 and primate L1 ORF0 (Denli et al. 2015), remains to be elucidated. Second, improved gene annotation in genome assemblies of nonmodel organisms will also facilitate the identification of novel transposon fusion proteins. This is because the current RefSeq annotation may overlook noncanonical protein isoforms. Therefore, incorporating more RNA-seq data from diverse species into annotation pipelines will improve the detection of noncanonical protein isoforms. Also, investigating the splicing patterns with single-cell resolution by single-cell RNA-seq will help to guess their physiological functions.
Transposon exonization does not always result in the transposon fusion proteins. More generally, transposon insertion can alter splicing patterns by providing intrinsic splicing sites and can generate new protein isoforms by partially altering the reading frames. Recently, proteomics and ribosome profiling have revealed the noncanonical protein isoforms caused by transposon exonization in the human genome (Arribas et al. 2024). Long-read sequencing technologies revealed a shortened protein isoform by transposon exonization, which is involved in the primate-specific immune response (Pasquesi et al. 2024). Thus, the emergence of transposon-derived exons has been revealed with higher resolution. Future comparative genomic analyses will elucidate the extent to which these exons are fixed in populations and determine their degree of evolutionary conservation over time.
In this study, we identified the exonized L1 elements that give rise to ORF1p fusion proteins and are evolutionarily conserved in vertebrate genomes for periods ranging from tens of millions to more than 280 million years. These findings provide valuable insights into how transposons contribute to the generation of lineage-specific splicing isoforms—a critical yet unresolved question in the field of genomic biodiversity.
Methods
Similarity search of the RefSeq proteins of American alligator against LINE ORF1ps
The RefSeq proteins of the American alligator (GCF_000281125.3, ASM28112v4) were subjected to a sequence homology search using MMseqs2 (Steinegger and Soding 2017) against 147 LINE ORF1ps of Dfam3.6 (Storer et al. 2021).
Protein structure analysis
The protein structure of American alligator Lyosin (XP_019356465.1) was predicted using AlphaFold2 implemented in ColabFold v1.5.2 (Jumper et al. 2021; Mirdita et al. 2022). The obtained Protein Data Bank (PDB) files of the whole Lyosin and the RRM were analyzed and visualized using PyMOL v2.5.0 (https://www.pymol.org/). The “super” command implemented in PyMOL was performed for the superposition and the RMSD calculation of α-carbon atoms.
BLASTP search in the NCBI database
The amino acid sequence of American alligator Lyosin (XP_019356465.1) was used as a query. The BLASTP search was performed against the database of “nr All nonredundant GenBank CDS translations+PDB+SwissProt+PIR+PRF excluding environmental samples from WGS projects” with default parameters on the NCBI web server as of November 29, 2023 (Supplemental Table S1). To construct the phylogenetic trees of the putative Lyosin proteins, these protein sequences were aligned using MAFFT v7.487 with the “L-INS-i” option (Katoh and Standley 2013). IQ-TREE2 v2.0.8 (Minh et al. 2020) was used to construct a phylogenetic tree with 1000 replicates generated by an ultrafast bootstrap approximation (Hoang et al. 2018).
Tetrapoda genomic search
The genome assemblies of Tetrapoda were downloaded from the NCBI assembly via GenomeSync (Kryukov et al. 2023; https://genomesync.org/) accessed on January 12, 2022 (Supplemental Table S3). The representative Lyosin amino acid sequences from the RefSeq database were used as queries as follows: XP_019356465.1 (A. mississippiensis), XP_025041760.1 (P. sinensis), XP_025915001.1 (A. rowi), and XP_033026289.1 (Lacerta agilis) for the search using BLAT v35 (Kent 2002). The best hit with query cover rates ∼10% above the proportion of the canonical isoform was used as a cut-off (Lyosin: >50%; RFX5: >75%; NUP42-L1: >45%; USP4-L1: >60%). To examine overlap with canonical isoforms and proximity to neighboring genes, the BLAT search was performed using the following sequences as queries: XP_027013259.1 (RFX5), XP_027013275.1 (MINDY1), XP_049750115.1 (NUP42), XP_049750112.1 (GPNMB), XP_045718342.1 (USP4), and XP_045718351.1 (IHO1). To investigate the intactness of exon L of the Lyosin transcript, the sequences of BLAT hits were retrieved with the upstream and downstream 600 nucleotides. The coding sequences more than 200 codons (between start and stop codons for Lyosin and L1-RFX5, and between stop codons for NUP42-L1 and USP4-L1) were then extracted using the getorf program in EMBOSS (Rice et al. 2000). The resulting sequences were aligned and manually checked to retrieve the intact exon L meeting the following criteria: (1) aligning with the previously identified L1 ORF1-derived exons, (2) including the splice site in frame, and (3) retaining more than 200 codons after trimming of putative introns. Taxonomic trees used in the analysis were retrieved from TimeTree 5 on December 7, 2023 (Kumar et al. 2022). The obtained trees were visualized with the annotations of the BLAT result and the intactness of exon L using ggtree v3.10.0 (Yu et al. 2017).
Molecular evolutionary analysis
The dN/dS ratio (ω) was estimated using the codeml program in PAML v4.8 (Yang 2007) based on the codon alignments of the ORF1-like exon L sequences under the one-ratio ω model: M0. The likelihood ratio tests were conducted by comparing models of ω = 1 and estimated ω to test the purifying selection. To detect positive selection, we performed the likelihood ratio tests to compare two pairs of site-specific models (neutral model vs. positive selection model): M1a versus M2a and M7 versus M8. The sites under positive selection were identified by the Bayes empirical procedure on M8.
Transcriptome analysis
The tissue transcriptomic data were downloaded from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (Supplemental Table S8; St John et al. 2012; Marin et al. 2017; Xu et al. 2019; Zhu et al. 2022). The FASTQ files were trimmed using fastp v0.23.2 (Chen et al. 2018) and mapped to the green anole reference genome (rAnoCar3.1.pri, GCA_035594765.1) using STAR v2.5.2b (Dobin et al. 2013) with the “‐‐outSAMattributes NH HI NM MD XS AS ‐‐outFilterMultimapNmax 500” options. Genome-guided transcript assemblies using StringTie2 v2.1.6 (Kovaka et al. 2019) were conducted for each sample. The generated GTF files were merged using StringTie2 with the “‐‐merge” option. Reads mapped to features in the merged GTF file were counted using featureCounts v2.0.1 (Liao et al. 2014) with the “-T 8 -s 2 -t exon -g gene_id -J” options. The transcripts of MYL4 and Lyosin were identified by the sequence similarity search. To quantify the splicing, the number of reads spanning exons 2 to 3 (CM069372.1:35022971–35028148) or exons L to 3 (CM069372.1:35026283–35028148) was obtained from the splicing junction tables included in STAR outputs.
Animals
An adult male Madagascar ground gecko (P. picta) was provided by the animal resource development unit, RIKEN CLST. The experiments were performed in accordance with the regulations for animal experiments approved by the Nagoya University animal experiment committee and the guidelines for the proper conduct of animal experiments (Science Council of Japan).
RT-PCR
Total RNA from the heart and testis of the Madagascar ground gecko was purified using ISOGEN (Nippon Gene 311-02501) and a direct-zol RNA miniprep kit (Zymo Research R2050). The cDNA was synthesized from total RNA using Verso cDNA synthesis kit (Thermo Fisher Scientific AB1453A). PCR was performed with 30 cycles of amplification (10 sec at 98°C, 5 sec at 60°C, and 5 sec at 68°C) by KOD One master mix (Toyobo KMM-101). A set of primer 1 (5′-AAGCAGGCTGCCACCATGGCCCCCAAAAAGCCGGA-3′) and primer 2 (5′-ACAAGAAAGCTGGGTTTAAGCGTAATCCGGAACATCGTATGGGTAGCCAGACATGATGTGTTTGAC-3′) and a set of primer 3 (5′-AAGCAGGCTGCCACCATGAAAATGCCAAACAAGTCCAC-3′) and primer 2 were used for amplification of MYL4 and Lyosin, respectively.
Plasmids
To construct the MYL4 and Lyosin expression plasmids (pPB-MYL4-HA and pPB-Lyosin-HA), the C-terminally HA-tagged MYL4 and Lyosin were amplified from cDNA. The mammalian expression plasmid (VectorBuilder VB900088-2265rnj) was linearized by inverse PCR, and the PCR product of MYL4 or Lyosin was inserted into the linearized plasmid. The nucleotide sequences of inserts were determined by Sanger sequencing (Eurofins Genomics). For construction of the reporter plasmid for L1 retrotransposition assay (pL1-spPuro), fragments of human L1 ORF1 and ORF2 from the EF06R plasmid were amplified by PCR and cloned into pcDNA3.1(+) (Invitrogen V79020) with the puromycin-resistant gene split by an intron. EF06R was a gift from Eline Luning Prak (Addgene 42940) (Farkash et al. 2006). All PCRs described above were carried out using KOD One master mix (Toyobo KMM-101), and all fragment ligation reactions were performed using NEBuilder HiFi DNA assembly master mix (New England Biolabs M5520AA).
Western blotting
293T human embryonic kidney cells (Riken BioResource Research Center RCB2202) were seeded on 24-well plates. The next day, cells were transfected with pPB-MYL4-HA or pPB-Lyosin-HA using Avalanche everyday transfection reagent (EZ Biosystems EZT-EVDY-1). The cells were lysed in sample buffer for SDS-PAGE (Nacalai Tesque 09499-14) 24 h after transfection. SDS/PAGE was performed, and peptides were transferred from the gel to polyvinylidene difluoride membranes. The membranes were reacted with rabbit anti-HA antibody (Medical & Biological Laboratories 361), and mouse anti-alpha-tubulin antibody (Proteintech 66031-1-Ig) as primary antibodies. Goat antirabbit antibody (Invitrogen 32460) and goat antimouse IgG antibody (Invitrogen 32430) were used as second antibodies. Signals were detected using SuperSignal west femto system (Thermo Fisher Scientific 34095).
Immunofluorescence assay
One of the HA-tagged protein expression plasmids (pPB-MYL4-HA, pPB-Lyosin-HA, or pPB-L1ORF1p-HA) and pPB-L1ORF1p-FLAG were transfected into 293T cells seeded into a slide chamber (Watson 192-008) using Avalanche everyday transfection reagent. At 24 h after transfection, the cells were fixed with 4% paraformaldehyde phosphate buffer for 15 min at room temperature. The cells were incubated with blocking buffer (PBS; 5% [w/v] BSA, 0.5% [v/v] Triton X-100, 0.1% [v/v] Tween 20) for 15 min at room temperature. The cells were reacted with rabbit anti-HA antibody and mouse anti-FLAG antibody (Sigma-Aldrich F3165) diluted with antibody reaction buffer (PBS, 1% [w/v] BSA, 0.1% [v/v] Tween 20) for 1 h at room temperature. After washing with PBS, the cells were incubated with goat antirabbit IgG with Alexa Fluor 594 (Invitrogen A11012) and goat antimouse IgG with Alexa Fluor 488 (Invitrogen A32723) diluted with the antibody reaction buffer for 1 h at room temperature. After washing with PBS, the cells were observed with fluorescence microscopy (Keyence BZ-X810).
L1 retrotranspositon assay
293T cells were seeded on a 24-well plate, and the next day, the reporter plasmid, pL1-spPuro, was transfected into cells with one of the protein expression plasmids (pPB-EGFP, pPB-MYL4-HA, pPB-Lyosin-HA, or pPB-MOV10) using Avalanche everyday transfection reagent. After 72 h of transfection, cells were passaged to a six-well plate with 1 µg/mL of puromycin. After 14 days, the cells were fixed with 4% paraformaldehyde phosphate buffer and stained with 1% crystal violet.
Screening vertebrate RefSeq proteins for ORF1p fusion isoforms
To identify vertebrate genes with alternative splicing isoforms containing ORF1p-like domains, vertebrate RefSeq proteins (release 220) were downloaded from NCBI on October 12, 2023. Proteins labeled “isoform” were retrieved and were then subjected to a sequence homology search using MMseqs2 (Steinegger and Soding 2017) against 147 LINE ORF1ps of Dfam3.6 (Storer et al. 2021). The E-value cutoff was set at 1 × 10−10, and the alignment coverage to ORF1p was required to be >50%. Then, protein-coding genes with both ORF1p-like isoforms and non-ORF1p-like isoforms (i.e., isoforms that are not similar to ORF1p) were collected from each species. To identify evolutionarily conserved transposon fusion protein isoforms, we retrieved these protein-coding genes that were commonly identified in two or more genera. At this point, we identified three protein genes (MYL4, RFX5, UNC13C) from nonmammalian RefSeq proteins and three protein genes (NUP42, USP4, ERVFC1) from mammalian RefSeq proteins (Supplemental Table S4). Of these, ERVFC1 was identified in the marmoset (Callithrix jacchus) and vampire bat (Desmodus rotundus); however, those evolutionary relationships were not considered to be orthologous based on their flanking genes. Presumably the different L1 ORF1 loci were annotated as genes with the same name. Next, we used the NCBI Gene browser (https://www.ncbi.nlm.nih.gov/gene/) to investigate whether the ORF1p-like isoforms were derived from an alternative exon (Supplemental Fig. S5). Unexpectedly, the ORF1p-like exon in UNC13C was a canonical coding exon that is also found in human and mouse. In three species, UNC13C contained the isoforms lacking this ORF1p-like exon and was identified as a gene with an alternative ORF1p-like isoform in our workflow (Supplemental Fig. S8A). Thus, UNC13C is likely to have a canonical coding exon homologous to L1 ORF1 (Supplemental Fig. S8B). This is interesting; however, UNC13C was outside of the scope of this study, and no further analysis was performed. As a result, four genes—MYL4, RFX5, NUP42, and USP4—were identified as genes having an alternative ORF1p-like isoform.
Data access
The data sets and scripts generated in this study are available as Supplemental Data.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Hiroshi Kiyonari (RIKEN CLST, Japan) for kindly providing of an adult male Madagascar ground gecko (Paroedura picta). This work was supported by Grant-in-Aid for Japan Society for the Promotion of Science (JSPS) fellows JP23KJ1055 to K.K. and JSPS KAKENHI JP20K06775 to S.N. and 25K02265 to K.K. and S.N. The supercomputing resource was partially supported by the NIG supercomputer at ROIS National Institute of Genetics.
Author contributions: K.K. and S.N. designed research; K.K. performed research; K.K., K.I., and S.N. analyzed data; and K.K., K.I., and S.N. wrote paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280007.124.
- Received September 7, 2024.
- Accepted April 11, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








