
Building better genome annotations across the tree of life
Abstract
Recent technological advances in long-read DNA sequencing accompanied by reduction in costs have made the production of genome assemblies financially achievable and computationally feasible, such that genome assembly no longer represents the major hurdle to evolutionary analysis for most nonmodel organisms. Now, the more difficult challenge is to properly annotate a draft genome assembly once it has been constructed. The primary challenge to annotations is how to select from the myriad gene prediction tools that are currently available, determine what kinds of data are necessary to generate high-quality annotations, and evaluate the quality of the annotation. To determine which methods perform the best and to determine whether the inclusion of RNA-seq data is necessary to obtain a high-quality annotation, we generated annotations with 12 different methods for 21 different species spanning vertebrates, plants, and insects. We found that the annotation transfer method TOGA, BRAKER3, and the RNA-seq assembler StringTie were consistently top performers across a variety of metrics including BUSCO recovery, CDS length, and false-positive rate, with the exception that TOGA performed less well in some monocots with respect to BUSCO recovery. The choice of which of the top-performing methods will depend upon the feasibility of whole-genome alignment, availability of RNA-seq data, importance of capturing noncoding parts of the transcriptome, and, when whole-genome alignment is not feasible, the relative performance in BUSCO recovery between BRAKER3 and StringTie. When whole-genome alignment is not feasible, inclusion of RNA-seq data will lead to substantial improvements to genome annotations.
The reporting in 2001 of the first draft of the human genome sequence (Lander et al. 2001; Venter et al. 2001) ushered in a new era of genome-scale analysis, with a concomitant, rapid increase in the development of bioinformatics tools and resources to interrogate genomes for evolutionary patterns and features of biomedical interest. But even as genomes became available for other model organisms such as mouse (Mus musculus) (Mouse Genome Sequencing Consortium et al. 2002) and rhesus macaque (Macaca mulatta) (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007)—and had been previously published for smaller genomes such as Drosophila melanogaster (Adams et al. 2000)—the high cost of generating genome assemblies at the time made it prohibitive to research groups working on nonmodel organisms. Absent genome assemblies and annotations, such groups were forced to embark on time-consuming efforts to sequence small sets of conserved genes with Sanger sequencing using primers designed with other genomes, target anonymous loci such as AFLPs or de novo assembled RAD-seq reads. These methods imposed an analytical glass ceiling on the types of inferences that could be made and prevented the framing of research findings in a genomic context. Although the advent of RNA-seq inched nonmodel organism research closer to understanding patterns at functional loci, de novo assembled transcriptomes presented novel analytical challenges and potential distortions of evolutionary patterns relative to what could be obtained with access to a genome assembly (Freedman et al. 2021).
Beginning with the advent of widespread access to high-throughput, low-cost short-read sequencing (Alkan et al. 2011) and continuing with recent technological advances in long-read DNA sequencing such as Pacific Biosciences HiFi (Wenger et al. 2019) and Oxford Nanopore Technologies (Jain et al. 2018), substantial reduction in costs have made the production of genome assemblies financially achievable and computationally feasible, such that genome assembly no longer represents the major hurdle to evolutionary analysis for most nonmodel organisms. Now, the more difficult challenge is to properly annotate a draft genome assembly once it has been constructed. The challenge is not so much the difficulty or computational resources required to run any one genome annotation tool, but how to (1) select from the myriad gene prediction tools that are currently available, (2) determine what kinds of data are necessary to generate high-quality annotations, and (3) evaluate the quality of the predicted transcript and gene models.
Currently available tools approach the annotation problem in very different ways. Early tools used hidden Markov models (HMMs) to scan genomes for sequences representing protein-coding intervals, with AUGUSTUS (Stanke and Waack 2003) being the most widely used example. Recent implementations of this approach, such as BRAKER1 (Hoff et al. 2016) and BRAKER2 (Brůna et al. 2021) wrap optimized implementations of AUGUSTUS, using protein and RNA-seq evidence, respectively—and with the latest release of BRAKER3 (Gabriel et al. 2024), both—to train HMMs. Transcript assemblers such as StringTie (Pertea et al. 2015) implement a graph-based framework to directly assemble transcripts from splice-aware alignments of RNA-seq reads to the genome. Tools such as Comparative AUGUSTUS (CGP) (Nachtweide and Stanke 2019), TOGA (Kirilenko et al. 2023), and Liftoff (Shumate and Salzberg 2021) use whole-genome alignments (WGAs) to transfer annotation evidence between genomes, with CGP involving multiway transfer of HMM-based gene predictions, and the TOGA/Liftoff lifting over annotations from a high-quality reference annotation in an exon-aware fashion.
A large part of the difficulty in determining what strategy will work for “my genome” is that annotation methods have, for the most part, been benchmarked and optimized with genomes from a very small slice of the tree of life—small genomes such as D. melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae—or with emphasis on vertebrates (Cantarel et al. 2008; Pertea et al. 2015; Hoff et al. 2016; Shao and Kingsford 2017). A related problem is that, although centralized and automated annotation pipelines maintained by organizations such as Ensembl and NCBI provide a simple and high-quality option, these groups cannot annotate more than a fraction of assembled genomes. As of December 12, 2024, only 1101 of 9651 reference genomes from Metazoans have been annotated by NCBI and only 169 of 1913 reference genomes from flowering plants (https://www.ncbi.nlm.nih.gov/datasets/genome/). NCBI has annotated <50% of all of its hosted eukaryotic reference genomes, amounting to <20% of all eukaryotic genomes (https://www.ncbi.nlm.nih.gov/datasets/genome/). Given the current rate of genome annotation by NCBI, it is hard to imagine that it will keep pace with the rate at which new genomes are assembled. Additional obstacles to researchers’ use of public annotation resources take the form of specific requirements for their use. For example, NCBI requires that a submitted genome be a chromosome-level assembly and that RNA-seq data must be available for the species in question. This will impose further limits on which research groups will be able to annotate their new genome assembly, unless they proceed to generate an annotation themselves.
Previous studies assessing genome annotation methods have investigated various aspects of the annotation problem, including assessing the quality of reference annotations (Park et al. 2023), focusing on plants (Vuruputoor et al. 2023), or comparing stand-alone methods to workflows that integrate evidence from multiple annotation tools (Banerjee et al. 2021). Lacking are studies that broadly survey current annotation tools and their application across the taxonomic diversity likely to affect tool performance. Motivated by the practical challenges facing research groups seeking to annotate new genome assemblies, here we evaluate the contents of genome annotations produced by 12 different methods across a broad swath of the tree of life. These methods include the tools cited above, including the pairing of StringTie and Scallop (Shao and Kingsford 2017) with two different RNA-seq aligners, as well as MAKER (Cantarel et al. 2008). Our taxonomic sampling includes two vertebrate clades (birds and mammals), two insect clades (Drosophila spp., and lepidopterans), and two plant clades (rosids and monocots), totaling 21 species (Table 2, below). Although “genome annotation” is often treated as an omnibus term that includes both the prediction of the genomic positions of genes and constituent isoforms and the assigning of gene symbols and functions, we focus on the first of these two components. Our goals are to determine (1) which methods are consistently top performers with respect to various sensitivity and specificity metrics, (2) the contents of individual annotations, (3) whether the inclusion of RNA-seq data is essential for producing a high-quality annotation, and (4) whether species and taxonomic group affect annotation method performance. We focus more attention on results for protein-coding genes than on the entire transcriptome that may include noncoding sequences, as organismal biologists tend to have deeper interest in evolution at and the functions of protein-coding loci, especially where genome assemblies are relatively new and genome-enabled research in a particular system has only just begun.
Results
We evaluated 12 different methods for genome annotation (Table 1), testing each method in 21 species across vertebrates, insects, and plants (Table 2). We selected species that broadly span much of the eukaryotic tree of life, covering six taxonomic groups: leipidopterans, dipterans, birds, mammals, rosid plants, and monocots. For each group, we selected a “reference” species known to have an extremely high-quality genome and annotation and then added two to three additional species representing varying levels of divergence from the reference species.
Summary of genome annotation methods evaluated in this study
Information on genomes for which annotations were generated in this study
We chose well-established genome annotation methods that sample from the three major approaches to the genome annotation problem. At the core of six of these—MAKER (Cantarel et al. 2008) with both protein and RNA-seq evidence; BRAKER1 (RNA-seq evidence only); BRAKER2 (protein evidence only); BRAKER3 (protein and RNA-seq evidence); CGP using protein evidence; and CGP with RNA-seq evidence—are HMM-based ab initio predictions by AUGUSTUS, with MAKER and BRAKER including predictions from additional ab initio tools. CGP enables evidence-based prediction within species while leveraging cross-species information via a WGA. Liftoff transfers annotations from a related genome that, ideally, has a complete, high-quality genome annotation. TOGA performs annotation transfer in an exon-aware fashion, conducting orthology inference relative to the source annotation as part of the process. For annotation transfer methods, we use the “reference” species (in bold in Table 2) as the annotation source to project to the other species in the clade; to generate predictions for the reference species itself, we use the closest relative (as indicated in Table 2). For lepidopterans, we initially included Heliconius melpomene as a reference species for generating annotations with CGP, but as it failed to produce more than a handful of predictions, which we did not include, and as the lepbase annotation for H. melpomene was generated with BRAKER1, no additional reference-based analyses were conducted for lepidopterans. Four methods assemble transcripts directly from RNA-seq alignments to a genome with one of two aligners: StringTie with either HISAT2 (Kim et al. 2019) or STAR (Dobin et al. 2013) as the aligner, and Scallop (Shao and Kingsford 2017) with either of these aligners. We chose these methods because they sample the current state of the art for three distinct approaches to the annotation problem (ab initio prediction, assembly from RNA-seq reads, and annotation transfer); they are highly cited, commonly used; and they have publicly accessible code repositories, most of which show recent contributions by developers. We initially sought to include the Comparative Annotation Toolkit (Fiddes et al. 2018) but found it to be infeasible owing to the computational and technical requirements to run on the large number of species we tested.
We calculate a small number of genome annotation quality metrics (Supplemental Tables S3 and S4). Some are aimed at estimating sensitivity such at BUSCO recovery and RNA-seq alignment rates to annotated transcripts. Others focus on precision and accuracy, such as the gene-level false-positive rate (FPR), and the coverage of reference proteins by predicted proteins. For a small subset of performance metrics, we use NCBI annotations as a putative benchmark, for example, identifying the proportion of predicted protein-coding genes that fall entirely outside of NCBI protein-coding gene intervals. Although not all of the NCBI annotations for the species we generate predictions for are necessarily complete, our target species are either well-established model organisms or those with a long history of study and substantial genomic resources, such that their NCBI annotations are likely to be of reasonably good quality. To the extent that this is true, we view them as useful to highlight tools that generate predictions that deviate substantially from those produced by NCBI in a manner consistent with what would be expected for a lower-quality annotation. The annotations for our designated “reference” species for each taxonomic group—Homo sapiens, D. melanogaster, Arabidopsis thaliana, Zea mays, and Gallus gallus—have undergone substantial manual curation and are likely complete or nearly so. Because of this, we place more emphasis on results from these species—and on M. musculus, given that its annotation has comparable curation and completeness—when we contrast annotation tool outputs with NCBI annotations.
Of the 12 different methods we assess here, seven (three BRAKER variants, two CGP variants, MAKER, TOGA) produce annotations of protein-coding genes only; four (two StringTie variants, two Scallop variants) produce annotations of transcripts but do not annotate coding sequence (CDS) directly; and one (Liftoff) produces annotations of both transcripts and CDSs. We report assessments of protein-coding and transcriptome annotations separately. To add CDS annotations to StringTie and Scallop annotations, we used TransDecoder (https://github.com/TransDecoder/TransDecoder) to predict open reading frames (ORFs) and explicitly label features as CDS or UTR. Although there are other published ORF-detection tools for use with transcript assemblies derived from RNA-seq data, for example, CodAn (Nachtigall et al. 2021), we used TransDecoder because it is by far the most widely used tool for this purpose, with 80,011 downloads from bioconda (https://bioconda.github.io/) as of December 11, 2024, and it is ranked in the top 10% of all packages downloaded from bioconda. The necessity of using TransDecoder means that comparison of CDS predictions from StringTie and Scallop with those of other tools that directly predict CDS partly confound the performance of the assembler and the ORF-detection method.
Our second level of analysis is for the “transcriptome,” consisting of the entirety of features that tools predict, agnostic to whether sequences are coding and their reading frames. For tools that only produce protein-coding predictions (BRAKER, CGP, TOGA, and MAKER), the proteome and transcriptome are equivalent in our assessment; although as these tools make no attempt to predict features such as noncoding RNAs or UTRs, we presume these will be missing. At this level of analysis, the extracted transcript sequences are extracted without regard to their coding status, such that the performance confounding between TransDecoder and the RNA-seq assemblers is removed. In reporting our results, we are careful to distinguish differences among methods that reflect intrinsic design differences versus those that reflect differences among methods that were designed to achieve the same objective, for example, among tools that solely produce protein-coding predictions.
Proteome: BUSCO recovery
The ability of a gene prediction method to recover protein-coding genes known to be conserved across a wide array of taxa is a good approximation for its ability to recover at least some subsequence of any gene, including those showing less conservation. We used compleasm (Huang and Li 2023) to assess the recovery of conserved, single-copy orthologs, or BUSCOs (Simão et al. 2015), and define a “compleasm score” measuring the proportion of BUSCO targets in a search that have matches to query transcripts (Supplemental Table S4). Compleasm is a reimplementation of the BUSCO software that substitutes miniprot (Li 2023) for MetaEuk (Levy Karin et al. 2020) as the protein-to-genome aligner, leading to a substantial increase in speed as well as an increase in accuracy. Across the vertebrate genomes, TOGA produced the highest proteome compleasm score in all species except Canis familiaris, followed by one of the BRAKER or CGP methods; for C. familiaris, BRAKER3 performed best (Fig. 1A). For dipterans and rosid plants, BRAKER and CGP produced either higher proteome compleasm scores than other methods or scores effectively tied with TOGA (Fig. 1B,C; Supplemental Fig. S1). In all vertebrates, BRAKER2 (with protein evidence) had higher compleasm scores than BRAKER1 (RNA-seq evidence), and in all species except M. musculus, combining RNA-seq and protein evidence with BRAKER3 led to clear improvements in BUSCO recovery relative to BRAKER2 (Fig. 1). Liftover methods performed inconsistently in monocots, for which BRAKER3 typically had the highest compleasm scores followed very closely behind by the RNA-seq assemblers (Fig. 1B). In two of the four monocots, Liftoff outperformed TOGA (Fig. 1B). There was a tendency for liftover methods to recover fewer BUSCOs with increasing divergence from the reference species. The greater evolutionary distances of Oryza sativa and Brachypodium distachyon from Z. mays (i.e., the annotation used as the source to be lifted over) than between Z. mays and Setaria italica (each the other's source), combined with the difficulty performing WGAs in plants, are the likely joint cause of poor BUSCO recovery in O. sativa and B. distachyon. In many species, CGP tended to perform less well than implementations of BRAKER and, in a few cases, performed poorly. CGP failed to produce more than a handful of transcript predictions for lepidopterans, and BRAKER2 consistently failed with Brassica oleracea, which is why these results are not included. For Drosophila pseudoobscura and B. oleracea, BUSCO recovery with CGP was poor, with compleasm scores of ∼50% or less. MAKER, a putative full annotation workflow that leverages both protein and RNA-seq evidence, consistently lagged in performance behind other standalone methods. These patterns are not influenced by variation in the presence of BUSCOs in the underlying genome assemblies, which might produce taxonomic effects (proteome compleasm score vs. proteome compleasm score/genome compleasm score, Pearson's ρ = 1.0, P = 2.2 × 10−16).
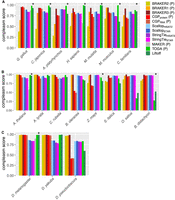
Proteome compleasm scores for predicted transcripts by annotation method for vertebrates (A), plants (B), and dipteran insects (C). The score is calculated as 1 − (number missing BUSCOs/total number of BUSCOs searched). For results on lepidopterans, see Supplemental Figure S1. Asterisks indicate Liftoff annotations at which the “polishing” method was successful. Within groups, species are ordered from left to right in order of increasing evolutionary distance from the reference species. P's in parentheses indicates that the method only makes protein-coding predictions.
Proteome composition: number and length of CDSs
The constituent CDS predictions that underlie compleasm scores varied widely among methods. Considering our reference species (and M. musculus) for which their high-quality annotations justify direct comparison, TOGA, Liftoff, and StringTie + TransDecoder generate CDS predictions that most closely approximate CDS length and count distributions of NCBI annotations (Fig. 2). Scallop + TransDecoder and ab initio methods tend to be composed of shorter CDSs relative to those three methods and NCBI annotations, and in some cases, large numbers of shorter CDSs are predicted, for example, by both versions of CGP for H. sapiens. Although ab initio methods do not consistently generate larger numbers of CDSs than other methods, they frequently produce shorter CDS predictions (Supplemental Figs. S2, S3). Taken together with the direct comparisons to reference annotations hosted by NCBI, this global pattern suggests that CGP, BRAKER, MAKER, and, to a lesser extent, Scallop+TransDecoder will often produce CDSs composed of fragmented gene models.
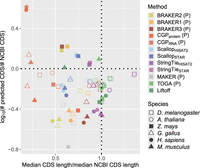
Joint distributions of number of predicted CDSs (normalized by number of NCBI predictions) over median predicted CDS length (normalized by median NCBI CDS length) for the reference species for each taxonomic group we examined, as well as M. musculus. Dotted lines indicate equivalence to NCBI annotation, such that methods that are closest to the intersection of those lines best approximate CDS length and number of NCBI annotations. A “P” indicates that the method only makes protein-coding predictions.
Proteome false positives: intergenic predictions
To quantify the frequency of false positives, we employ a conservative, gene-level metric, specifically the frequency of predicted protein-coding genes that fall entirely outside the CDS boundaries of NCBI protein-coding gene coordinates. Because treating NCBI annotations as a truth set for species other than our reference species might confound NCBI false negatives with prediction method false positives, we first evaluated whether this confounding was likely to introduce bias or, at the least, to obscure underlying patterns. For our reference species, we inferred a lack of bias, and thus the appropriateness of using NCBI annotations as a truth set, in the strong correlation between predictions unique to classes of methods and the intergenic FPR (relative to NCBI) for those classes (Supplemental Results S1; Supplemental Fig. S4).
For all but A. thaliana, the FPR at which predicted genes fell entirely within intergenic regions relative to the respective NCBI annotations was lowest for liftover methods, between which there were minimal differences; for A. thaliana, StringTie (with STAR alignments) had the lowest FPR but was minimally lower than for TOGA and Liftoff (Fig. 3). With few exceptions, RNA-seq assembler FPRs were lower than that for ab initio methods and were often minimally greater than for liftover methods (Fig. 3). For all species, FPRs for the best-performing method were ≤10%, and for many species, those predictions occurred <5% of the time (Fig. 3). Regardless of the species and evidence type, FPRs for CGP were much larger, often >50% (Fig. 3). Although BRAKER FPR was typically less than that of CGP, regardless of the evidence type used, BRAKER FPR was higher than the best-performing RNA-seq assembler in 16 of 18 species and frequently exceeded 40%. The observed FPRs suggest that, even for the best-performing methods, hundreds or even thousands of gene predictions will fall outside of the genomic intervals for known real CDSs.
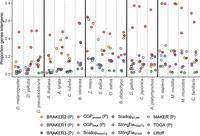
Proportion of predicted protein-coding genes that fall entirely outside the intervals for NCBI protein-coding gene, organized by species and method. Species within taxonomic group are arranged from left to right in order of increasing divergence from the reference species. A “P” indicates that the method only makes protein-coding predictions.
Proteome accuracy: reference protein coverage
To the extent that protein-coding transcripts in our reference species represent validated, real proteins, the first way we assessed the accuracy of protein coding predictions was to quantify the frequency of high-coverage BLASTP (Camacho et al. 2009) hits to the proteins of the reference species for the taxonomic group in question. For our reference species for which the NCBI target database corresponds to the same species, relative performance differed between species with smaller versus larger, more complex genomes. In the species with smaller genomes, liftover methods performed best in D. melanogaster but lagged slightly behind BRAKER and CGP in A. thaliana, whereas BRAKER and CGP proteins consistently obtain greater coverage or NCBI reference proteins than RNA-seq assemblers (Fig. 4). Consistent with the difficulty of performing WGA in large plant genomes, in Z. mays, liftover methods obtained reference protein coverage well below that achieved with BRAKER3 and RNA-seq assemblers (Fig. 4). Globally, TOGA achieved greater reference protein coverage than Liftoff, and in vertebrates, TOGA achieved the highest reference protein coverage (H. sapiens) or was virtually tied with BRAKER3 (G. gallus) (Fig. 4). In vertebrates, the RNA-seq assemblers obtained slightly lower reference protein coverage than the top-performing ab initio method (Fig. 4). In the nonreference species, we observed similar patterns (Supplemental Fig. S5), but with TOGA consistently obtaining the greatest reference protein coverage in the other vertebrate species (Supplemental Fig. S5A) and with liftover methods performing poorly in additional monocot species (Supplemental Fig. S5B).
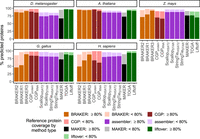
BLASTP hit frequency by coverage of predicted proteins to NCBI proteins for reference species proteomes.
Proteome accuracy: PSAURON scores
The second way in which we assessed the accuracy of proteome predictions was to leverage PSAURON (Sommer et al. 2025), a recently developed machine-learning-based approach. PSAURON uses a model trained on more than 1000 species to predict the likelihood that a sequence is generated by a true protein-coding interval and also calculates a global score for the entire set of proteins. Across all species, TOGA obtained the top PSAURON score or was virtually tied with an alternate top-performing method (Fig. 5). Scores were mostly low for CGP, with Liftoff also underperforming relative to other methods (Fig. 5). In dipterans and rosid plants, scores were similar among other methods, and in monocots, an ab initio method achieved the second highest score (Fig. 5). In vertebrates, StringTie + TransDecoder achieved the second highest score close behind TOGA, although BRAKER3 scores were often nearly as high (Fig. 5). Although the veracity of NCBI protein predictions across all of our study species undoubtedly vary in quality, it is notable that PSAURON scores are highly correlated with the fraction of predicted proteins that have BLASTP hits to a database composed of the NCBI proteins for our study species from the relevant taxonomic group (within group correlations, Pearson's ρ = 0.918–0.996, P ≤ 7.25 × 10−19) (Supplemental Figure S6).
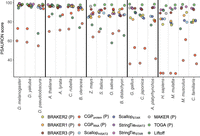
PSAURON scores for annotation method proteomes, with species ordered within taxonomic group from left to right in increasing divergence from the reference species. A “P” indicates that the method only makes protein-coding predictions.
Transcriptome: BUSCO recovery
Compleasm scores obtained at the transcriptome level indicate, regardless of whether ORFs have been called by an annotation pipeline, the extent to which the predicted transcripts recover conserved protein-coding sequences. In other words, these scores estimate the predicted transcriptome's recovery of the proteome. Comparing transcriptome to proteome scores can thus be informative about issues with workflows that separate functional sequence discovery and ORF detection, for example, pairing an RNA-seq assembler with an ORF detection tool. Transcriptome and proteome compleasm scores were highly correlated, with one universal exception: For StringTie, Scallop, and Liftoff, proteome scores were lower than transcriptome scores (Fig. 6; Supplemental Fig. S7). For ab initio methods, minimal differences between proteome and transcriptome scores undoubtedly reflect differences in how protein and DNA (i.e., CDS) query-based searches are conducted, given that the former is directly derived from the latter. For Liftoff and the RNA-seq assemblers, causes of information loss likely differ, the former presumably owing to errors in genome alignment that disrupt ORFs. For RNA-seq assemblers, TransDecoder does not retain transcripts for which it cannot find a valid ORF, raising the possibility that the lower proteome scores result from false negatives owing to BUSCOs that are recovered with transcripts for which no ORF was detected, such that they are excluded from the proteome. In our reference species, although such false negatives do occur, larger fractions of missing BUSCOs are often caused by putatively incorrect (including truncated) ORF predictions, instances in which a transcript in the transcriptome matches a BUSCO that is not detected in the proteome but that transcript is present in the proteome (Supplemental Fig. S8). The relative contribution of such errors to missing BUSCOs in the proteome is greater for StringTie than Scallop (Supplemental Fig. S8).
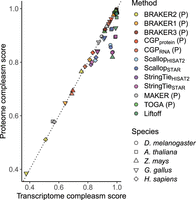
Correlation between proteome and transcriptome compleasm scores for the five reference species. P's in parentheses indicate a method only produces protein-coding predictions.
Transcriptome: RNA-seq alignment rate
When researchers plan to use their annotation to analyze patterns of gene expression, an important question to ask is, to what extent does the annotation capture the variation observed through sequencing? The alignment rate of RNA-seq reads to the set of annotated transcripts gets at this question. Because RNA-seq assemblers and Liftoff will include noncoding transcripts, as well as UTRs associated with CDS, our a priori expectation is that alignment rates for these tools will be higher than for ab initio methods and TOGA. Thus, we focus on answering two questions. First, are the alignment rates for tools that include noncoding sequences truly greater than those for annotations solely composed of CDS, and if so, what is the magnitude of the alignment rate difference? Second, do some tools that only predict CDS do a better job than others of capturing proteome variation captured in RNA-seq reads? There is a large disparity between alignment rates for RNA-seq assemblers versus tools that only predict CDS, in some cases exceeding a difference of 50% (Fig. 7). Alignment rates for Liftoff lag slightly behind those for the assemblers for most taxa, except in monocots, in which Liftoff alignment rates are comparable to those for ab initio tools (Fig. 7). Alignment rates for StringTie and Scallop are consistently better than those for NCBI annotations (Fig. 7). There are no consistent top performers for CDS-only tools, but for the ab initio tools (CGP, BRAKER, and MAKER), in 13 of 18 species tools that use RNA-seq evidence have higher alignment rates than those that do not (Fig. 7). It is worth noting that compared with CDS-only tools, RNA-seq assemblers (paired with TransDecoder) appear to capture comparable amounts of the protein coding variation found in RNA-seq reads. That is, alignment rates to CDS sequences are much more similar among methods, even if alignment rates for assemblers often rank near the top (Supplemental Fig. S9).
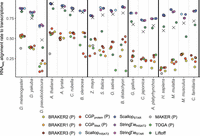
RNA-seq alignment rate to transcriptomes. P's in parentheses indicate that a tool only generates CDS predictions, and X's indicate alignment rate to NCBI annotations. Species are ordered from left to right in increasing divergence from the reference species for the taxonomic group.
Because these comparisons were based upon alignment of the same reads that were assembled by StringTie and Scallop, we considered the possibility that this would provide an unfair advantage to the assemblers relative to tools that only used RNA-seq data to generate splice hints (BRAKERRNA and CGPRNA) or to filter HMM-based predictions post hoc (MAKER) and, even more so, for methods that did not use RNA-seq data (BRAKERprotein and CGPprotein). We found that recycling of RNA-seq data used to generate annotations to calculate alignment rates introduced negligible, if any, bias (Supplemental Results S2; Supplemental Fig. S10).
UTRs in RNA-seq assemblers
Our finding that failure to detect or incorrect prediction of ORFs in StringTie and Scallop assemblies led to BUSCO false negatives made us speculate that the reduction in RNA-seq assembler alignment rate for the proteome relative to the transcriptome might have three nonexclusive causes: exclusion of real noncoding transcripts, ORF assembly errors, and the exclusion of UTRs. With respect to UTRs, we assessed whether CDS intervals may have incorrectly been classified as UTRs. First, for StringTie and Scallop, we looked at the proportion of predicted transcripts composed of UTR and how those compared to the NCBI annotations. Ratios of UTR to CDS length were greater for Scallop and StringTie assemblies than for NCBI annotations (Supplemental Fig. S11). These disproportionately long (relative to NCBI) UTRs constitute an excess of target sequence for alignment, such that their exclusion will contribute to a reduction in alignment rates. We next considered the possibility that the greater proportional length of UTRs for RNA-seq assemblers relative to NCBI annotations could be because of the NCBI pipeline computationally truncating UTRs when mitigating for cases of putative transcriptional readthrough past stop codons. In this case of our reference genomes (H. sapiens, M. musculus, D. melanogaster, Z. mays, and A. thaliana) for which there has been extensive curation would be expected to have a lower ratio of UTR to CDS length. However, plotting the ratio of predicted UTR-to-CDS ratios for the assemblers over that for NBCI predictions produces the opposite pattern, in which these ratios of ratios are lower for our reference genomes (Supplemental Fig. S11). Next, we looked for evidence of CDS in predicted UTR features. If this were a pervasive problem, then we would expect a large fraction of UTRs to have BLASTX hits to an NCBI protein database for the same species. As would be expected if undetected CDS occur in the UTR intervals for RNA-seq assemblers, the percentage of transcripts with a UTR BLASTX hit increases as the length of UTRs relative to CDS for assemblers increases relative to that observed for NCBI annotations (Fig. 8). Depending upon the species and particular assembler-aligner combination, up to 60% of transcripts may potentially contain undetected CDS in regions annotated as UTRs. Although it is beyond the scope of this study to decompose alignment rate differences between proteome and transcriptome further, these results and those for BUSCO missingness from assembler proteomes suggest that ORF false negatives and the incorrect classification of CDS as UTR may mask a persistent advantage for RNA-seq assemblers over other methods with respect to alignment of RNA-seq data to protein-coding loci that are important for downstream expression analyses.
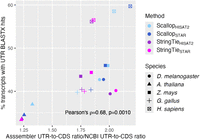
Evidence for undetected CDS in predicted UTR intervals for StringTie + TransDecoder and Scallop + TransDecoder. Increasing percentage of RNA-seq assembler transcripts with UTRs that have a BLASTX hit to the NCBI protein database of the same species, as a function of an increase in the assembler UTR-to-CDS ratio relative to that for NCBI annotations.
Discussion
With genome assembly and annotation increasingly becoming part of the workflow for researchers studying nonmodel organisms, the choice of an annotation method depends upon knowing whether a particular method will perform well in the species in question, as well as what data will need to be generated to generate the best-quality annotation possible. Previous efforts to evaluate and compare methods have sampled a small slice of the tree of life and have often focused on small, tractable genomes with extensive genomic resources or on other model organisms such as H. sapiens or M. musculus. This can make choosing a method more difficult, because the species used to benchmark annotation tools may be evolutionary distant from a newly assembled genome of interest. Our investigation overcomes this problem by evaluating a large set of methods across a broad taxonomic swath, revealing both cross-species and taxon-restricted patterns. By identifying methods that performed well across diverse species, we believe that researchers using those methods will likely be able to generate reasonably high-quality annotations for their newly assembled genome of interest. Our findings have implications for study design, data collection, and annotation method choice, and they highlight ongoing challenges that require further methods development.
First and foremost, when the primary focus of a research program is the proteome and a high-quality well-annotated genome is available for an organism that is not too distantly related to the genome being annotated, CDS-aware annotation with TOGA can, in most cases, produce an extremely high-quality annotation. Although our findings highlight the power of annotation transfer from another species with a high-quality annotation, they also highlight its limitations. Although TOGA often had high sensitivity, low rates of intergenic predictions, and high accuracy of protein-coding transcripts, we found that sensitivity could be much lower in plants, particularly in those with larger genomes. This undoubtedly stems from known difficulties performing WGA with plant genomes (Song et al. 2024), which, as we observed with monocots, is exacerbated at greater evolutionary distances between the source and target genomes. For some taxa, there may be few if any closely related species with high-quality annotations, or genome alignments may be fragmented or contain many missing intervals in the target species.
If a source genome and annotation are unavailable or if annotation transfer leads to a low proteome compleasm score, our findings suggest an RNA-seq assembler is the next best alternative. In particular, the StringTie proteome will achieve an accuracy comparable to that of TOGA, and the length distribution of StringTie CDS suggests a larger proportion of them will be closer to full-length then those predicted by Scallop. Although StringTie does not do quite as good a job at BUSCO recovery compared with the top-performing BRAKER implementation (usually BRAKER3), both RNA-seq assemblers and liftover methods have a major advantage over ab initio annotation tools, in that the latter typically produce thousands to tens or even hundreds of thousands of false-positive predictions that need to be filtered but may be difficult to identify. The reasons for such predictions undoubtedly differ among types of annotation tools. Model-based ab initio tools look for sequence patterns that are consistent with ORFs, even if there is no evidence of transcription. Conversely, RNA-seq assemblers are heavily evidence dependent, such that if enough RNA-seq reads align to a region that a transcript can be assembled, a gene model will be reported. Determining whether any of the intergenic protein-coding genes assembled with StringTie or Scallop are the product of assembling transcriptional noise or of a novel protein-coding locus is beyond the scope of this study, but at least for our high-quality reference genomes, the latter scenario seems far less likely. Filtering out spurious predictions for ab initio tools remains a larger challenge, given that they tend to predict shorter CDS predictions, which exacerbates the problem of distinguishing spurious predictions from real, albeit fragmented constituents of true underlying CDS. Overall, and despite the issues we discovered with ORF detection in RNA-seq assembler annotations, assemblers are particularly valuable for larger, more complex genomes and consistently rank highly for both proteome- and transcriptome-level metrics. One final consideration in choosing between TOGA and StringTie relates to the recovery of the true underlying splice-site variation. If researchers have a keen interest in the evolution of alternative splicing or the evolution of novel, lineage-specific protein-coding sequences, TOGA will be less useful in that it will only recover transcripts that reflect the observed splicing patterns in the source genome.
If researchers’ interests extend to the full transcriptome (including noncoding RNAs and UTRs), an assembler such as StringTie is a reasonable choice, as HMM-based methods and TOGA do not predict such sequences, and StringTie typically outperforms Liftoff with both respect to proteome accuracy and to representation of the expressed transcriptome as reflected in the RNA-seq alignment rate. However, after comparing StringTie and Liftoff annotations with respect to performance metrics such as those we evaluate in this study, there may be cases in which Liftoff offers advantages over StringTie. For example, although the RNA-seq alignment rate may be lower for Liftoff than for StringTie in most cases, when suboptimal WGA does not impact annotation, Liftoff will have higher sensitivity with respect to recovering BUSCOs. In other words, as a tool to recover the entire transcriptome, it may not do as well as StringTie, but it will likely do a better job of recovering the proteome. Nevertheless, prioritizing the inclusion of ncRNA comes with an inherent tradeoff, as liftover methods and the best-performing ab initio method will typically do a better job of BUSCO recovery than StringTie; the exception with respect to liftover methods is for taxa in which WGA negatively impacts annotation transfer methods.
Although liftover methods and BRAKER may appear to have advantages over assemblers for proteome recovery, we found reason to believe that advantage may in fact be overestimated. Our discovery that predicted UTR features in StringTie+TransDecoder and Scallop+TransDecoder annotations have high fractions of sequence that with BLASTX hits to NCBI proteins suggests that both failed to recover many CDS exons. Our results suggest that TransDecoder may have a harder time correctly classifying CDS exons at the termini of a transcript than those within the transcript body. Although long-read technology might help overcome this deficiency, we suggest that there is room for method development to improve ORF detection from predicted CDS transcripts. Improved ORF detection and CDS exon boundary delineation would lead to improved performance with respect to the metrics we used to assess the proteome in this study.
Although we did not assess the utility of cDNA long reads for annotation, we expect our findings to be robust to their adoption, and the performance edge advantages, however modest, of BRAKER over RNA-seq assemblers with respect to proteome completeness will almost certainly diminish if the assemblers do not ultimately surpass BRAKER (and perhaps even TOGA). The direct evidence of splicing patterns across full-length reads will enable reconstruction of full-length transcripts, whereas in the HMM context, longer reads will simply lead to more accurate detection of splice sites and more accurate model parameterization, to the extent that any model can capture the diversity of sequence composition and splicing patterns observed in higher organisms. We suspect that increasingly accurate model parameterization will lead to diminishing returns relative to direct assembly of transcripts from reads, predicated on the assumption that ORF prediction on full-length cDNA reads will become trivial to undertake with high accuracy.
There is one important step in genome annotation that we did not address in our study, namely, post-hoc filtering of annotation. There are many potential metrics to guide such filtering, such that one could remove transcripts that are very short, that are lowly expressed, or that do not have a hit to a BLAST database or some other database that enables gene symbol assignment, providing an indication that the transcript encodes a real protein. Such filtering will be particularly important when an annotation method is known to produce many false-positive predictions, and yet, the choice of metrics and thresholds may be ad hoc. The best approach will likely involve selecting metrics and tuning thresholds to maximize performance with respect to compleasm scores, PSAURON scores, and recovery of functional information with tools such as eggNOG-mapper (Cantalapiedra et al. 2021).
Although a subset of the methods we evaluated performed extremely well with respect to a subset of performance metrics, we suspect that, in many cases, combining annotations from multiple sources will be required to obtain a reasonably complete and accurate annotation, especially if one seeks to include accurate protein-coding predictions and noncoding features. There are a small number of currently supported tools that merge annotations generated from different pipelines; for example, Mikado (Venturini et al. 2018) and EVM (Haas et al. 2008) offer flexible integration from diverse annotation sources, whereas tools such as gFACs (Caballero and Wegrzyn 2019) and FINDER (Banerjee et al. 2021) integrate annotations but accept as input a more restricted set of gene predictions, some of which we found to perform poorly as stand-alone methods. Although beyond the scope of this paper, a fruitful area of future research would be a systematic evaluation of these tools to determine their relative strengths and weaknesses.
Even as much work remains to be done, our findings suggest some general guidelines for a researcher deciding how to annotate their newly assembled genome.
-
If a high-quality, well-annotated genome is available for a close to modestly divergent species from the newly assembled genome and if annotating protein-coding genes is the primary interest, generate a TOGA genome annotation. Generate a proteome compleasm score to make sure that issues with WGA or evolutionary divergence do not lead to noticeably reduced sensitivity to what is expected relative to other candidate methods, with alternative methods being determined by the availability of RNA-seq data.
-
If annotation transfer with TOGA is not feasible, generate RNA-seq data for at least the tissues related to the most pressing project needs but, ideally, across as many tissues as necessary to capture the species’ transcriptional complexity. Generate annotations with BRAKER3 and StringTie. To make an objective comparison of BUSCO recovery that avoids false negatives owing to TransDecoder making incorrect ORF predictions, generate transcriptome compleasm scores for both annotations. If BRAKER3 clearly outperforms StringTie, use that annotation with the expectation that much filtering will be needed in order to reduce intergenic false positives. If compleasm scores are comparable or StringTie recovers more BUSCOs, use the StringTie-TransDecoder annotation. Then, merge the transcripts for which no ORF could be detected back into the final annotation; this particular task or the entire StringTie annotation can be performed using code found at GitHub (https://github.com/harvardinformatics/AnnotationRNAseqAssembly).
-
If the goal is to annotate the entire transcriptome (CDS, UTRs, and noncoding RNA transcripts) and if a well-annotated genome is available for a close to modestly divergent species, consider using Liftoff. If RNA-seq data are available, generate annotations with both StringTie and Liftoff, and use the annotation quality metrics we have employed in this paper in making a choice between the two options.
-
If it is not possible to use TOGA and RNA-seq data are not available, use BRAKER2.
-
If there appears to be complementarity in the recovery of real protein-coding sequences between the methods (based upon BUSCO recovery), consider using an approach to integrate predictions.
In conclusion, the longer-term challenge for building genome annotations across the tree of life is to make methodological advances suggested above and to integrate them into reproducible, automated workflows that can be deployed with minimal headaches for biologists. When this happens, population and comparative genomics studies will be easy to scale to hundreds, and even thousands, of species, unleashing unprecedented power to tackle long-standing questions regarding the genetic architecture of phenotypic variation and the evolutionary mechanisms that generate and maintain biodiversity.
Methods
Additional details on bioinformatics package command lines and custom Python scripts used to implement analyses described below are available in the relevant GitHub repositories detailed in the Software availability section.
Target taxa
Genome annotation tools are typically developed and optimized using high-quality genome assemblies from a small suite of model organisms, for example, H. sapiens, C. elegans, and D. melanogaster. As a result, it is difficult to generalize their performance in this narrow context to taxonomic groups that are highly divergent from those focal taxa and for which the genome assemblies may not be of comparable quality. To facilitate more accurate generalizations regarding the performance of annotation methods and, conversely, to explore whether there are effects of taxonomy and genome structure on annotation quality, we generated genome annotations for 21 species spanning six taxonomic groups: three species of lepidopteran butterflies, three Drosophila species (dipterans), three birds, four mammals, four rosids, and four monocots (Supplemental Table S1). With the exception of the butterflies, we included as a “reference” a species for which both a high-quality genome assembly and annotation were available, and downloaded the soft-masked assemblies and annotations from NCBI. The NCBI Eukaryotic Annotation Pipeline combines alignment of RefSeq sequences (when available), alignment of RNA-seq and protein data, and ab initio predictions with NCBI's Gnomon gene prediction tool (for more details, see https://www.ncbi.nlm.nih.gov/refseq/annotation_euk/process/#gnomon). Each group contains at least one species that is relatively closely related to this reference. Because the NCBI genome versions and annotations for the butterflies we selected are older than those widely used by the research community, we used lepbase (Challi et al. 2016) assemblies that were filtered to remove all scaffolds <1 kb in length (Edelman et al. 2019). High-quality annotations for these species were either unavailable or were generated by tools we evaluated and thus inappropriate to serve as a truth set. For example, the annotation for H. melpomene was generated with BRAKER. Because of the unavailability of high-quality annotations for lepidopterans, for these species, we only generated a subset of performance metrics. Furthermore, we used H. melpomene as a reference assembly in WGAs (see below), but we did not generate annotations for this species.
Genome assembly and reference annotation quality
To determine if BUSCO recovery in transcriptomes and proteomes (see below) was impacted by the completeness of the genome assembly, we quantified the number of single-copy orthologs (BUSCOs) contained in a genome (Simão et al. 2015) using compleasm v. 0.2.6 (Huang and Li 2023), and calculated a compleasm score as 1 – (number missing BUSCOs/total number of BUSCOs searched). These scores and information on genome assembly version, genome size, contig N50, and scaffold N50 that were pulled down from NCBI are recorded in Supplemental Table S1. We also calculated length statistics for CDS using a custom Python script, GenerateFastaSeqLengthTable.py (see Supplemental Table S4; Supplemental Code S1), and the median RNA-seq alignment rate across the libraries for each species’ annotation, using GetMedianAnnotationAlignRate.py (Supplemental Table S4; Supplemental Code S1). For compleasm analyses, we use the following lineage databases: arthropoda_odb10 for dipterans, lepidoptera_odb10 for lepidopterans, viridiplantae_odb10 for roids and monocots, aves_odb10 for birds, and mammalia_odb10 for mammals.
RNA-seq data acquisition and processing
For use with annotation methods that either build transcripts from RNA-seq reads or use read alignments to generate splice hints, for each species we downloaded 15–20 FASTQ file accessions from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra). We used the following criteria to choose accessions. We only considered (1) paired-end reads with a release data of 2011 or later; (2) those with at most one run per biosample; (3) Illumina reads sequence on HiSeq 2000, NextSeq, or newer instruments (i.e., no Genome Analyzer II). We excluded experimental treatments such as knockdowns, infections, and CRISPR modifications. If these criteria resulted in more than 20 possible SRA biosamples, we further required a minimum read length of 100 bp and, for Metazoans, preferentially selected brain or head samples. If fewer than 20 samples were available, we relaxed read length, release date, and instrument criteria with the goal of retaining 15 SRA biosamples. With the exception of the lepidopteran Danaus plexippus (for which eight of 19 libraries had 36 bp reads), we strictly excluded paired-end libraries in which the read length was <50 bp. In a small number of cases in which libraries contained hundreds of millions of reads, we down-sampled libraries to about 20 million read pairs with seqtk (https://github.com/lh3/seqtk).
To process the reads prior to sequence alignment, we stripped adapters with Trim Galore! (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), wrapping cutadapt v. 2.10 (Martin 2011). We did not trim low-quality bases at the ends of reads because the short-read aligners used in this study soft-clip such bases so they do not impact sequence alignment and because such trimming can bias expression estimates (Williams et al. 2016). To avoid having to make inferences from SRA metadata regarding the strandedness of RNA-seq libraries and to avoid the likely variable effectiveness of stranding protocols, we treated all libraries as unstranded, under the assumption that such information, if used, would result in modest improvements in performance for annotation methods that leverage RNA-seq alignments. Accessions used in this study are provided in Supplemental Table S2.
Annotation tools
A general overview of the annotation tools evaluated, including required input data, output format, and the constituent features of predictions, can be found in Supplemental Table S5.
RNA-seq assembly
We used RNA-seq reads to directly assembly transcripts with StringTie v. 2.1.2 and Scallop v. 0.10.5. These assemblers take as input spliced alignments of reads to the genome. We evaluated the impact of aligner by generating assemblies with two different aligners: HISAT2 v. 2.2.1 and STAR v. 2.7.9a. Following best practices and to leverage evidence for splice sites across multiple samples, we used a two-pass approach to generate STAR alignments. In the first pass, we generated an initial set of alignments for each sample. In the second pass, we concatenated the splice site tables generated for each sample's first-pass alignment, and supplied the concatenated table as splice-site evidence for the second pass alignment of each sample. To generate a merged transcript assembly combining information across all individual-level assemblies, for StringTie and Scallop, we used the stringtie-merge function and TACO v. 0.7.3 (Niknafs et al. 2017), respectively. With our lepidopteran species, we initially evaluated two additional assemblers: PsiCLASS (Song et al. 2019) and Scallop2 (Zhang et al. 2022). Poor performance relative to StringTie and Scallop, as well as excessive run times for PsiCLASS, led us to not consider these two tools further.
StringTie and Scallop annotations contain transcript and exon features; they do not predict CDS. Therefore, to incorporate CDS predictions into the merged annotations, we used TransDecoder v. 5.5.0 (https://github.com/TransDecoder/TransDecoder) and an associated workflow (https://github.com/TransDecoder/TransDecoder/wiki) that predicts ORFs and then leverages ORF predictions to predict CDS and UTR intervals associated with the GTF format input annotation file. After initial prediction of likely candidate ORFs, we ran BLASTP v. 2.12.0 (Camacho et al. 2009) searches against a protein database consisting of UniProt and Trembl entries from all the species that we are attempting to annotate in the species group; for example, for dipterans, the database consists of entries for D. melanogaster, D. pseudoobscura, and Drosophila yakuba. We provided the search results as an input to transDecoder-predict, such that given two similarly scoring orfs, we preferentially kept the one with a BLASTP hit. In the interest of minimizing the filtering out of real ORFs, we set the maximum e-value threshold for these BLASTP searches to 1 × 10−4. It should be noted that the workflow as described filters out of the final annotation any transcript without a retained ORF prediction. The filtered ORFs contain an unknown fraction of real ORFs that TransDecoder failed to discover, as well as ncRNAs. For proteome-level analyses, we used the CDS transcripts extracted from the TransDecoder annotation using the version of gffread provided with cufflinks (Trapnell et al. 2010). For transcriptome-level analyses, we used the original StringTie and Scallop annotations prior to their processing with TransDecoder. Although the focus of our research is on prediction of protein-coding genes, at GitHub (https://github.com/harvardinformatics/AnnotationRNAseqAssembly) we employ as part of our workflow a Python script for adding back into the final annotation these putative false negatives and ncRNA annotations.
Single-species ab initio methods
In contrast to approaches of transcript assembly from reads, a long-established approach for predicting genes (and CDSs in particular) is to parameterize HMMs that are designed to traverse scaffolds, identify exon boundaries, and connect exons into transcript and gene-level features. The most sophisticated single-species versions of this approach use external evidence to parameterize HMMs and identify specific genomic locations where exon splice junctions are located. We evaluated BRAKER1 and BRAKER2 (both v. 2.1.6), which conduct iterative training and gene prediction using RNA-seq read and protein alignment evidence, respectively. Both BRAKERs flavor wrap ab initio prediction with AUGUSTUS (Stanke et al. 2006) and GeneMark, with BRAKER1 using GeneMark-ET (Lomsadze et al. 2014) and BRAKER2 using GeneMark-EP+ (Brůna et al. 2020, 2021). Following developer recommendations, we provided protein evidence to BRAKER2 in the form of a protein FASTA from OrthoDB v.10 (Kriventseva et al. 2019) for the relevant taxonomic group, generated from prepartitioned raw files as provided by the BRAKER developers (downloaded on September 14, 2018) (https://bioinf.uni-greifswald.de/bioinf). The specific databases we used were as follows: for birds and mammals, orthoDBv100_vertebrate; for dipterans and lepidopterans, odb10_arthropoda; and for rosids and monocots, odb10_plants. For BRAKER1, we provided a BAM file of RNA-seq STAR alignments merged across all libraries from the species being annotated. We also generated annotations with BRAKER3 (Gabriel et al. 2024), which integrates separate BRAKER runs using protein and RNA-seq evidence into an integrated annotation. For BRAKER1 and BRAKER2, following the developers’ recommendations, we used their selectSupportedSubsets.py script to identify annotations with at least some support from evidence and removed unsupported annotations with a custom Python script, FilterOutUnsupportedBrakerAugustusAnnotations.py. This is unnecessary for BRAKER3, which performs filtering internally.
Single-species exon-aware liftover: TOGA and liftoff
Using WGAs to transfer annotations across species from well-annotated to poorly or unannotated species has a long history, for example, with the UCSC Genome Browser LiftOver tool first becoming available in 2006 (Hinrichs et al. 2006). To perform such “liftovers,” we used two different tools. TOGA (Kirilenko et al. 2023) transfers CDS annotations across genomes in an exon-aware fashion that minimizes disruptions of ORFs. It takes as input a WGA and involves several steps. We provide a detailed description of our workflow, including links to custom Python scripts used at various steps at GitHub (https://github.com/harvardinformatics/GenomeAnnotation/tree/master/TOGA). As part of our workflow, to remove potential spurious or bad annotation transfers, we filtered out any transcripts in the primary annotation output (query_annotation.bed) for which there was not a corresponding entry in orthology_classification.tsv, that is, transcripts for which TOGA could not determine an orthology class. Within each taxonomic group, we transferred annotations from the high-quality reference to all other species within the group, as well as from the species most closely related to the reference back to the reference species. For example, for dipterans we carried out three TOGA analyses, transferring D. melanogaster to both D. pseudoobscura and D. yakuba and from D. yakuba to D. melanogaster.
The second liftover method we evaluated was Liftoff v. 1.6.3, for which we specified reference genomes from which to transfer annotations in the same manner that we did for TOGA. Liftoff does not require a WGA as input, performing the alignment internally. It also transfers noncoding annotation features such as UTRs and noncoding RNAs. We used additional Liftoff options that are meant to account for evolutionary divergence between source and target genomes: -d, the “distance scaling factor,” and -flank, the number of flanking sequences to align as a fraction of gene length. Although the documentation does not provide guidance for suggested values for these options, we initially explored a range of values in generating annotations for C. familiaris and M. mulatta using humans as the source genome. For all subsequent analyses, we chose the pair of values that maximized BUSCO recovery: three and 0.2 for -d and -flank, respectively. Although Liftoff does not perform exon-aware annotation of CDS like TOGA does, it does have a “polish” option that will attempt to restore ORFs that are disrupted during liftover. Although we attempted to use this option for all Liftoff runs, it failed for several of them. We reported results for the polished annotation when possible, but at least for proteome BUSCO recovery (Fig. 1), polishing did not appear to have an impact comparable to the variation among annotation methods.
Multispecies ab initio annotation
In studies seeking to perform phylogenetic comparative analyses or annotate multiple genome assemblies from related organisms or in studies in which annotations or evidence (protein or RNA-seq) already exist for a subset of species of interest, methods that transfer evidence between lineages offer, in principle, a promising approach for performing genome annotation. We evaluated the most well-established approach for doing this, AUGUSTUS run in comparative mode (König et al. 2016), referred to hereafter as CGP. CGP relies on WGA. Thus, as a first step, for each taxonomic group of genomes, we used Progressive Cactus (Armstrong et al. 2020) to produce a WGA. We then used an AUGUSTUS accessory script, hal2maf_split.pl, to split the hal-format cactus output file into multiple subfiles in multiple alignment (MAF) format; in doing so, we set as the “reference” genome (with which to provide coordinate anchors), a species with both a highly contiguous assembly and a high-quality annotation, and split in such a way so as to avoid splits that bisect the genomic coordinates of annotated genes in the reference. For each taxonomic group of species, we ran CGP twice, once with splice site evidence from protein alignments and once from RNA-seq alignments. For analysis with protein evidence, similar to our analyses with BRAKER2, we used OrthoDB v.100 data representing the taxonomic group. For analysis with RNA-seq, we used the merged STAR alignments across samples. In both instances, following guidelines from the developers (Hoff and Stanke 2019), we generated splice hints files for each species using scripts and code provided as part of the AUGUSTUS package. In both modes, we did not predict UTRs or predict alternative isoforms; namely, one transcript prediction was made per putative gene. Detailed instructions regarding how we generated hints and ran CGP can be found at GitHub (https://github.com/harvardinformatics/GenomeAnnotation/tree/master/GenomeResearch/ComparativeAugustus).
MAKER
MAKER (Cantarel et al. 2008) is a genome annotation pipeline that has the ability to integrate multiple ab initio gene prediction packages and to use protein and RNA-seq-derived external evidence to perform post hoc curation of predictions. Because results with MAKER usually involve more than one run in order to retrain gene-prediction models, it is not a fully automated pipeline. Nevertheless, it has been used extensively owing to its purported ease of use. MAKER also has the option to perform quality-filtering and integration of annotations with EVidenceModeler (EVM) (Haas et al. 2008). For initial testing with three lepidopteran species, we ran MAKER v. 3.01.03 in four different ways that integrate predictions from AUGUSTUS (Stanke et al. 2006), SNAP (Korf 2004), and Genemark-ES (Lomsadze et al. 2005): (1) protein evidence only, without EVM; (2) protein and RNA-seq evidence, without EVM; (3) protein evidence only, with EVM; and (4) protein and RNA-seq evidence, with EVM. For protein evidence, we used the protein accessions associated with the lepbase (lepbase.org) Hmel2 genome assembly, which are proteins derived from BRAKER predictions. RNA-seq evidence was included as a gff3 file generated from the StringTie assembly using STAR alignments of the species’ RNA-seq samples. We used default settings for the EVM configuration scoring file. To produce annotations, we ran MAKER twice closely following Daren Card's detailed workflow (https://gist.github.com/darencard/bb1001ac1532dd4225b030cf0cd61ce2); see also the link to our workflow on GitHub in the Software availability section below. Because with these test runs the use of EVM frequently produced lower-quality annotations and because we wished to evaluate the potential of MAKER as a full-service annotation tool, runs for other taxa were only performed with both protein and RNA-seq evidence and without EVM. Furthermore, because MAKER is computationally intensive and can take a considerable amount of time to run, for the other taxonomic groups, we only generate MAKER annotations for the “reference” species of each group, with protein evidence being represented by the NCBI protein accession associated with the genome for the next closely related species in the set of species we annotated within each taxonomic group.
Annotation quality metrics
BUSCO recovery
For proteome and transcriptome-level analyses, database selection and compleasm score calculations were performed as explained above in the section Genome Assembly and Reference Annotation Quality. For the proteome analyses, we supplied protein FASTA files as input. For TOGA, we used the amino acid FASTA that is part of the software output, and for all other tools, we extracted the protein sequences with the version of gffread distributed with Cufflinks (Trapnell et al. 2010). For transcriptome-level analyses, we used the transcripts FASTA extracted by rsem-prepare-reference (see section below on expression calculations). Because we discovered that for RNA-seq assemblers, this utility can fail to include a small number of annotations, for these tools we first extracted transcripts with gffread (which were then used to build RSEM indices).
False positives: intergenic predictions
Motivated, in part, by some tools predicting far more CDS transcripts than are recorded in NCBI annotations, we assessed whether this could be owing to (presumably incorrect) intergenic predictions, in which intergenic is defined as falling entirely outside of the CDS intervals for all protein-coding genes annotated by NCBI. To do this, for each new annotation, we generated transcript-interval and gene-interval BED files, in which each entry represented the genomic boundaries of the transcript or gene (excluding UTRs), respectively. For StringTie and Scallop, we did this using WriteBrakerCdsTranscriptAndGeneIntervalBedFilesWithNoUTRs.py (Supplemental Table S4; Supplemental Code), and for BRAKER, MAKER, CGP, and Liftoff, we used WriteBrakerCdsTranscriptAndGeneIntervalBedFilesWithNoUTRs.py (Supplemental Table S4; Supplemental Code S1). We then used BEDTools v. 2.26.0 (Quinlan and Hall 2010) to intersect these files with a BED file consisting of UTR-stripped NCBI gene boundaries, recording the number of bases of overlap such that only same-strand overlaps were counted as overlaps, for example, intersectBed -s -wao -a newannotation_intervals.bed -b NCBI_gene_intervals.bed. Using awk, we then counted the number of predicted protein-coding genes lacking any overlap with NCBI gene coordinates.
To validate the use of NCBI as a benchmark for calculating intergenic FPRs, for our five reference species and M. musculus, we binned annotation methods into annotation method classes (CGP, BRAKER, StringTie, Scallop, liftover, and MAKER) and then examined the correlation between the frequency of predicted genes that are unique to a class and the frequency at which those genes fall outside of NCBI protein-coding gene intervals. We assume that predicted genes unique to a class of methods are likely to be false positives such that if this correlation is strong, then NCBI can be confidently used as a benchmark for individual methods. Specifically, for each species we created BED files of predicted genes for each annotation method, as well as NCBI annotations, which include a label for the method in the seventh column. Next, we concatenated these BED files into a single file and then sorted it with the BEDTools sortBed function. We then used BEDTools to merge intervals such that each row will be an interval for which a list of methods have overlapping genes, for example, mergeBed -i sorted.bed -c 7 -o distinct>merged.bed. Rows that do not have a label for NCBI are intergenic relative to NCBI; rows that only contain a single method class (without NCBI) are class-specific intergenic intervals; etc. Finally, we used a custom Python script, CalcGeneUniqueToMethodClassRate.py (Supplemental Table S4; Supplemental Code), to calculate the rate of unique predictions, intergenic (relative to NCBI) predictions, and the correlation between them.
Proteome accuracy
We first assessed the accuracy of our CDS predictions by calculating reference protein coverage of CDS predictions. To do this, we performed BLASTP v. 2.12.0 (Camacho et al. 2009) searches of NCBI protein sequences from the reference species for the taxonomic group against a protein database for each species–method combination, excluding lepidopterans, and set a maximum e-value of 1 × 10−5. We also customized the output format fields to enable easy calculation of coverage length of the reference proteins. We then use a custom Python script, CalculateBlastpFractionRefProteinsCovered.py, to calculate the fraction of predicted CDS that obtain high (≥80%) and low (<80%) coverage of a reference protein. To generate a second estimator of proteome accuracy for all species–method combinations, we used PSAURON v. 1.0.2 to generate a proteome-wide PSAURON score. We compared PSAURON scores to a separate estimator of accuracy, albeit one that is dependent upon NCBI annotations: the proportion of predicted proteins that have BLASTP hit to an NCBI protein in a database composed of the protein for all of the species that were used in our study for the taxonomic group of interest. For these, we set a maximum e-value of 1 × 10−5.
Expressed transcriptome recovery: RNA-seq alignment rate
To evaluate the extent to which predicted proteomes and transcriptomes capture expressed variation recovered in RNA-seq data, we used RSEM v. 1.3.3 (Li and Dewey 2011) to wrap Bowtie 2 (Langmead and Salzberg 2012) alignment of each RNA-seq library to the predicted set of transcripts. From these alignments, we calculated the median alignment rate (across the set of samples). Because using the same RNA-seq libraries to generate transcriptome assemblies with StringTie and Scallop may bias alignment rates upward relative to tools that do not leverage evidence from those RNA-seq libraries, we also ran RSEM on an additional test set of six RNA-seq paired-end SRA accessions for each of our five reference species.
Undetected CDSs in UTRs
Our RNA-seq assembly pipelines integrate ORF finding with TransDecoder such that exon features are decomposed further into CDS and UTR features. Although the substantial reduction in RNA-seq alignment rate we observed between StringTie and Scallop transcriptomes and their respective proteomes (CDS without UTRs) might be because of the absence of noncoding RNA transcripts in the proteomes, we also wondered if incorrectly calling CDS as UTRs might partly be responsible for the difference in alignment rates. Therefore, we examined our UTR annotations more closely, thinking that unusually large UTRs might hint at their containing CDS. First, because for relatively complete high-quality annotations the NCBI annotation pipeline will computationally truncate UTRs to prevent stop-codon readthrough, we contrasted the length-distribution RNA-seq assembler UTRs and those from NCBI, expecting that the disparity would be greater for the reference genomes for each of our taxonomic groups than for other, more recent genome assemblies for which truncation would not be as severe. Under this scenario, the reduction in alignment rate after UTR removal would be owing to an excess of reads originating from transcriptional readthrough (or because the NCBI UTR truncation was overly conservative).
Next, we considered the possibility that TransDecoder consistently fails to predict CDS ORFs at the terminal ends of transcripts, such that a large proportion of real CDS sequences is being incorrectly filtered out when we strip out UTRs. To do this, we focused on our reference species. With the exception of D. melanogaster, which is a high-quality, manually curated annotation, four of these species’ annotations are composed of RefSeq annotations and well-supported Gnomon predictions. All five are high-quality annotations with comparable completeness and thus useful benchmarks to evaluate the power of TransDecoder to recover CDS exons. To test our hypothesis, for our reference species, we extracted the UTR sequences from the StringTie and scallop annotations and used BLASTX v. 2.12.0 (Camacho et al. 2009) to search for matches against the NCBI protein sequences from the same species’ NCBI accession, with a maximum e-value of 1 × 10−5. We calculated the fraction of transcripts for which at least one of the UTRs had a hit to the protein database. We then compared this fraction to the median assembler UTR-to-CDS ratio/median NCBI UTR-to-CDS ratio. Values greater than one for this quantity indicate a larger fraction of UTR in an RNA-seq assembler relative to NCBI, and a positive correlation between it and the UTR BLASTX hit frequency would support false-negative CDS predictions. Steps for obtaining UTR/CDS ratios are elaborated in Supplemental Methods S1.
Software availability
Detailed explanation of steps in annotation pipelines, along with associated Python scripts for data processing are provided in the following repositories: RNA-seq transcript assembly (https://github.com/harvardinformatics/GenomeAnnotation/tree/master/GenomeResearch/RNA-seq and https://github.com/harvardinformatics/AnnotationRNAseqAssembly), BRAKER (https://github.com/harvardinformatics/GenomeAnnotation/tree/master/Braker and https://github.com/harvardinformatics/AnnotationBRAKER), TOGA (https://github.com/harvardinformatics/GenomeAnnotation/tree/master/TOGA and https://github.com/harvardinformatics/AnnotationTOGA), Liftoff (https://github.com/harvardinformatics/GenomeAnnotation/tree/master/GenomeResearch/Liftoff), CGP (https://github.com/harvardinformatics/GenomeAnnotation/tree/master/GenomeResearch/ComparativeAugustus), and MAKER (https://github.com/harvardinformatics/GenomeAnnotation/tree/master/GenomeResearch/Maker).
In addition, HPC slurm job scripts and specific command lines used to run annotation tools in this paper, as well as Python scripts to generate annotation quality metrics, are available at GitHub (https://github.com/harvardinformatics/GenomeAnnotation) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank members of the FAS Informatics Group for ongoing discussions regarding genome annotation. Within the Informatics Group, we specifically thank Nathan Weeks for troubleshooting software installation and execution and the many pull requests he submitted to fix bugs; we also thank Gregg Thomas for building a robust implementation of Cactus on the FAS Cannon compute cluster. We also thank Michael Hiller for feedback on an earlier version of this manuscript. This work was conducted on the traditional territory of the Wampanoag and Massachusetts peoples.
Author contributions: A.H.F. and T.B.S. conceived the research, A.H.F. analyzed the data with input from T.B.S., and A.H.F. wrote the paper with T.B.S.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280377.124.
- Received December 18, 2024.
- Accepted March 3, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








