
Quality assessment of long read data in multisample lrRNA-seq experiments using SQANTI-reads
- 1Department of Molecular Genetics and Microbiology, University of Florida, Gainesville, Florida 32610, USA;
- 2University of Florida Genetics Institute, University of Florida, Gainesville, Florida 32610, USA;
- 3Institute for Integrative Systems Biology (I2SysBio), Spanish National Research Council (CSIC), Paterna 46980, Spain;
- 4UF Health Cancer Center, University of Florida, Gainesville, Florida 32610, USA
Abstract
SQANTI-reads leverages SQANTI3, a tool for the analysis of the quality of transcript models, to develop a read-level quality control framework for replicated long-read RNA-seq experiments. The number and distribution of reads, as well as the number and distribution of unique junction chains (transcript splicing patterns), in SQANTI3 structural categories are informative of raw data quality. Multisample visualizations of QC metrics are presented by experimental design factors to identify outliers. We introduce new metrics for (1) the identification of potentially under-annotated genes and putative novel transcripts and for (2) quantifying variation in junction donors and acceptors. We applied SQANTI-reads to two different data sets, a Drosophila developmental experiment and a multiplatform data set from the LRGASP project and demonstrate that the tool effectively reveals the impact of read coverage on data quality, and readily identifies strong and weak splicing sites.
Short-read RNA sequencing (srRNA-seq) is the most common and cost-effective approach for studying the transcriptome. In srRNA-seq, transcripts must be inferred computationally, which can lead to inaccuracies in transcript identification (Liu et al. 2016; Newman et al. 2018). Recent advances in single-molecule long-read sequencing (LRS) technologies have opened new avenues for transcriptome analysis (for reviews, see Marx 2023; van Dijk et al. 2023). In long-read RNA sequencing (lrRNA-seq), full-length transcripts can be observed as single sequencing reads, allowing for direct transcript detection without the need for an assembly step. As with any technology, lrRNA-seq is not without errors, and factors such as mRNA degradation, library preparation failures, and sequencing inaccuracies can result in bias in the data.
A database tracking bioinformatic tools for LRS (Amarasinghe et al. 2021) identifies numerous tools for the initial processing of lrRNA-seq data primarily assessing the accuracy of basecalling and the length of the reads. These include pycoQC (Leger and Leonardi 2019), LongQC (Fukasawa et al. 2020), and nanoQC (De Coster et al. 2018), which offer a first-pass analysis of lrRNA-seq data. Other tools, such as SQANTI3 (Pardo-Palacios et al. 2024a), TALON (Wyman et al. 2020), FLAMES (Holmqvist et al. 2021), Iso-Seq (https://isoseq.how/), and IsoTools (Lienhard et al. 2023), focus on evaluating transcript models inferred from the data. However, most current tools for lrRNA-seq read quality control were developed during the early stages of these technologies and are generally limited in the number of evaluated features and/or samples. As LRS technologies rapidly improve in quality and decrease in cost, the experimental scope possible with these technologies has expanded. The need for a comprehensive and comparative read quality assessment tool capable of analyzing millions of reads and dozens (or more) samples is critical.
The rapid decline in costs implies that the use of lrRNA-seq will continue to expand, with experimental designs involving multiple samples becoming more common (Glinos et al. 2022; Joglekar et al. 2024; Mahmoud et al. 2024; Patowary et al. 2024). From a quality control perspective, this necessitates that data sets are homogeneous, without systematic bias associated with experimental groups, and free of outliers. Moreover, the generated data must be sufficient to address the research questions that motivated the experiment. The increase in throughput now makes it possible to design experiments that include barcoding and multiplexing to balance library preparation and sequencing across experimental groups (Auer and Doerge 2010). This approach helps avoid confounding technical variation with the treatments of interest and facilitates discriminating between failed technical replicates (TRs) and failed samples. Finally, technological advancements such as more accurate basecallers (https://github.com/nanoporetech/dorado) and the availability of novel library preparation methods such as MAS-ISO-seq (Al'Khafaji et al. 2024), CapTrap (Carbonell-Sala et al. 2024), R2C2 (Volden et al. 2018), Nano3P-seq (Begik et al. 2023), or FLAM-seq (Legnini et al. 2019) motivates the need for tools that can easily evaluate how these improvements impact data quality.
In this context, we present SQANTI-reads, an extension of SQANTI3 (Pardo-Palacios et al. 2024a), a tool originally designed for transcript model quality control, to jointly provide quality control metrics for long-read data and to analyze multiple samples for consistency and bias. We demonstrate that SQANTI3's structural categories and other quality control metrics, repurposed in SQANTI-reads, are highly effective for assessing the homogeneity in a lrRNA-seq multisample experiment, identifying read quality control failures, and detecting outliers. Additionally, we have added new metrics that provide insights into the potential utility and discovery power of the data, including variation at donor/acceptor sites and identification of potentially under-annotated genes and mis-annotated transcripts.
Results
SQANTI-reads can be used to evaluate long-read technology improvements
Long-read methods are rapidly improving, and both Nanopore and PacBio are updating their instruments, molecular protocols, and algorithms. Tools that can readily evaluate the broad impact on data quality of these technical improvements are highly needed to make informed decisions for downstream analyses. Our Drosophila data set contained the same set of samples processed on the MinION and PromethION instruments through several runs. In addition, the development of basecalling algorithms for ONT sequencing is a highly active and rapidly evolving field (Pagès-Gallego and de Ridder 2023; Diensthuber et al. 2024). We expect that as new basecallers emerge and algorithms are improved upon, users will want to evaluate the impact of different basecallers and algorithms on the quality of their experiments. This comparison can be done using SQANTI-reads. While Dorado is now the default basecaller for ONT, we demonstrate the utility of SQANTI-reads by comparing real-time Guppy basecalled reads and Dorado basecalled reads. We first evaluated whether Dorado effectively improved data quality without introducing biases and if data from several sequencing experiments could be merged. We compared the Guppy and Dorado basecallers using SQANTI-reads metrics. As anticipated, Dorado resulted in more reads with assignable barcodes, a higher number of mapped reads, more reads aligning to annotated genes, more reads aligning to annotated transcripts, and longer reads, without an increase in the proportion of reads with technical artifacts (Supplemental Fig. 1; Supplemental File 1). This confirms that Dorado improves basecalling accuracy without introducing unwanted biases and motivated the selection of Dorado basecalled reads for subsequent analyses.
In the Drosophila experiment, libraries were barcoded, pooled, and multiplexed across different MinION and PromethION runs, with a re-pooling step between the two machines. We used SQANTI-reads to compare the quality of the MinION and the PromethION runs, and to evaluate the consistency of the MinION and PromethION technology across TRs. The first MinION run (TR1) had higher percentages of reads classified as novel in catalog (NIC) and novel not in catalog (NNC) and with noncanonical junctions compared to the second and third MinION runs (TR2, TR3) and compared to the PromethION runs for the same libraries (Supplemental Fig. 2). NIC refers to reads with novel combinations of annotated junctions and NNC refers to reads with at least one unannotated junction. MinION runs TR2 and TR3 were similar in their quality metrics (described below) to the PromethION run of the same samples, and the TRs on the PromethION were similar. These results indicate that the technology performs consistently across instruments and runs. Based on these SQANTI-reads QC results, we aggregated data across TRs to further evaluate the quality of the lrRNA-seq experiment. Although TR1 had lower quality than the other TRs, the overall read numbers were low, and we decided to keep this TR in our evaluation of the samples.
SQANTI-reads metrics can be used to evaluate the global quality of the lrRNA-seq experiment
In a multisample lrRNA-seq experiment, all samples should be of similar quality. SQANTI3 uses the full-splice match (FSM) structural category to identify long-read sequences whose junctions are consistent with an annotated transcript model. However, for an lrRNA-seq experiment to accurately reflect the analyzed transcriptome, the reads should also capture the distribution of transcript lengths of the expressed transcriptome. The distribution of transcript lengths depends on many factors, including the species and tissue used as input. Consistent with the species transcript model annotations, we observe that Drosophila has less complex and shorter transcripts than humans (Supplemental Fig. 3). A data set with reads substantially shorter than the annotated, transcriptome but a high proportion of FSM indicates capture of short transcripts, while combining a high proportion of incomplete-splice match (ISM) may indicate RNA degradation. ISM refers to a read whose junction matches an annotated transcript but is missing junctions on the 5′ end, 3′ end, or both ends. We looked at these values for an initial assessment of the quality of the Drosophila experiment.
First, we compared the number of reads and distributions of read lengths for all samples. The difference in sequencing depth (number of reads) between the two developmental stages was evident, as expected because of the additional PromethION run on the 3–8 day samples (Fig. 1A). For all samples, most reads were shorter than 1 kb, with <20% of them above the 1 kb threshold (Fig. 1B; Supplemental Figs. 4A, 5). When we evaluated all reads, 53% to 67% of the reads across samples were classified as FSM, 20% to 38% were labeled as ISM. For all samples, the proportion of NIC/NNC was <10% of the reads (Fig. 1C). For the reads that were above the 1 kb threshold, between 73% and 82% of the reads were FSM, with only 10% to 18% ISM (Fig. 1D). The decrease in proportion of ISM and increase in proportion of FSM when evaluating only the reads >1 kb suggests that the shorter reads represent incomplete transcript sequences, potentially due to mRNA degradation.
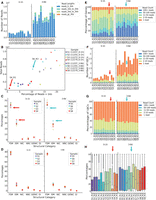
SQANTI-reads analysis of Drosophila samples. (A) Number of mapped reads by experimental group labeled with read length. (B) Percentage of mapped reads >1 kb versus percentage of reads that are FSM. Dots represent the early stage (0–1 h after eclosion) and crosses indicate the adult stage (3–8 days old). The four genotypes are indicated with four different colors. (C) Percentage of reads mapping to genes in each SQANTI3 structural category (D) Percentage of reads mapping to genes in each SQANTI3-QC structural category for reads >1 kb (E) Number of genes detected with breakdown by the number of reads mapped to each gene. (F) Number of UJCs detected with breakdown by the number of reads associated with each UJC. (G) Percentage of UJCs detected with breakdown by the number of reads associated with each UJC. (H) Distribution of the percentage of FSM reads by gene across samples.
Reads that share all internal junctions are grouped together and annotated using a string made up of the junction locations, the unique junction chain (UJC) (Nanni et al. 2024). We examined both gene-level and UJC-level metrics. We found that, despite the large sequencing depth differences between developmental stages, the number of detected genes was only slightly lower in the 0–1 h samples (Fig. 1E). However, these genes were quantified with fewer reads (80% genes with <50 reads) than the 3–8 days samples, which had between 30% and 50% of genes with more than 100 reads (Fig. 1E). When evaluating UJC, we found that, while the number of UJC mirrored the sequencing depth pattern (Fig. 1F), with 3–8 days samples showing five times more UJC than 0–1 h samples, and a larger number of FSM and ISM UJC, there were many additional UJC detected by fewer than 10 reads, and usually by a single read (Fig. 1F,G). These UJC were most frequently NIC/NNC (Supplemental Fig. 4B,C). Downstream analyses would therefore need to address whether this represents novel low-expressed transcripts or technology errors. In contrast, the percentage of FSM reads between the two time points differed by less than 1× in all replicates (Fig. 1H). These results indicate that the higher sequencing depth of the 3–8 days samples does not change the number of detected genes or annotated transcripts (FSM). The higher read depth per gene/UJC suggests that more genes and transcripts will be able to be quantitatively evaluated in the 3–8 day samples compared to the 0–1 h samples.
In the Drosophila data, we noticed two samples (Sample 4 Rep 1 [RIL 12279, 0–1 h]—red arrow; Sample 6 Rep 3 [RIL 11255, 3–8 day]—teal arrow) that had the lowest percentage of FSM and highest percentage of ISM in the 0–1 h and 3–8 day groups, respectively (Fig. 1C). To determine whether these two samples were of overall lower quality than the rest, we examined their SQANTI-reads metrics. We found that Sample 4 Rep 1 had a lower proportion of FSM across all genes (Fig. 1H) and a higher proportion of genes quantified with only one read (Fig. 1F), while Sample 6 Rep 3 had a similar gene (Fig. 1H), UJC (Fig. 1G), and % FSM in genes (Fig. 1H) than other 3–8 day samples. We concluded that RIL 12279 rep 1 0–1 h is a lower-quality sample.
Altogether, this example shows that SQANTI-reads metrics can be used to compare samples and experimental conditions in a multisample experiment, detect outliers, and suggest points of attention for downstream data processing.
SQANTI-reads metrics can be used to identify systematic differences among samples
The previous example demonstrated that SQANTI-reads metrics are effective in assessing data set consistency. However, SQANTI-reads evaluates over 35 quality metrics, making it challenging to determine which features contribute to potential differences among samples. We include principal component analysis (PCA) analysis to identify which metrics are the most relevant for quality variability when there are differences among samples or between groups. The percentage of reads and UJCs in each structural category, percentage of artifact reads (RT-switching, noncanonical junctions, and intrapriming), percentage of junctions in each category, as well as length metrics, are included in the PCA.
We applied SQANTI-reads PCA analysis of quality features to investigate differences in read quality among various LRS methods used in the Long-read RNA-seq Genome Annotation Assessment Project (LRGASP) challenge (Pardo-Palacios et al. 2024b), focusing on the WTC11 data set. We evaluated triplicate transcriptome measurements of the WTC11 human cell line, analyzed using three technologies: cDNA PacBio Sequel II (cDNA PacBio), cDNA Oxford Nanopore MinION (cDNA ONT), and direct RNA Oxford Nanopore MinION (dRNA ONT). The analysis revealed that WTC11 samples clustered based on the long-read technology applied (Fig. 2A). Specifically, PC1, which explains 56% of the variance, distinguished cDNA ONT samples from those generated by the other two technologies, while PC2, accounting for 35% of the variance, highlighted differences between dRNA ONT and cDNA PacBio. To further explore these differences, we examined the loadings for each principal component. Quality features with the highest positive loadings in PC1 included the number of reads, the percentage of reads, and the proportion of UJCs in the NNC category, while features with high negative loadings included Intergenic and Genic Genomic reads. Several junction-related variables also exhibited high absolute loadings on PC1 (Fig. 2B). SQANTI-reads plots confirmed these structural category differences between cDNA ONT samples and other library preparations. cDNA ONT had both the highest proportion of NNC reads and UJCs (Fig. 2C,D) and also had the lowest proportion of intergenic reads (Fig. 2C). Other differences in sequencing throughput and junction characteristics were also confirmed (Supplemental Fig. 6).
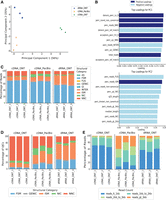
SQANTI-reads PCA analysis of LRGASP WTC11 samples. (A) PCA using SQANTI-reads quality features. The percentage of variance explained by each principal component (PC) is labeled on each axis in brackets. (B) Top 10 Loadings for PC1 and PC2. (C) Distribution of reads in SQANTI3 structural categories. (D) Distribution of UJCs in structural categories. (E) Distribution of read lengths for all mapped reads.
Upon examining the feature loadings for PC2, we found that variables with high contributions included several metrics related to read length (Fig. 2B). Consequently, we evaluated the SQANTI-reads “Lengths of All Mapped Reads” plot for this experiment. Indeed, we observed that the cDNA PacBio method produced a significantly higher proportion of reads between 1–2 kb, 2–3 kb, and >3 kb, as suggested by their negative loadings, compared to the dRNA ONT method, which predominantly generated reads shorter than 1 kb (Fig. 2E). Similarly, the percentage of reads assigned as ISM with high positive values was higher in dRNA ONT samples (Fig. 2C).
In conclusion, we showed that the SQANTI-reads PCA analysis is an effective tool for uncovering significant read quality differences between LRS methods. The technological differences explored here are based on a benchmarking experiment that has been superseded by new technology and the results here should not be interpreted as an evaluation of current technological capabilities. Nonetheless, we have shown that SQANTI-reads can be used to evaluate LRS methods and for revealing technological biases.
SQANTI-reads identifies potentially under-annotated genes
Long-read data often contain a large number of sequences that cannot be exactly matched to existing annotations. In many cases, these UJCs belong to annotated genes and are identified by only a few reads, as illustrated by the SQANTI-reads analysis in Figures 1 and 2. This suggests they could be either low-expressed transcripts or technological artifacts. However, in some instances, a high proportion of reads in a gene may correspond to the same novel UJC, indicating the possibility of a previously unannotated transcript that warrants closer examination. SQANTI-reads includes a customizable decision tree to identify such cases (see Methods; Fig. 3A). Basically, the tool identifies genes with a high (R > threshold) number of reads, with novel UJC containing a large fraction (Q > threshold) of the gene's splice sites and capturing a high proportion (P > threshold) of the reads. We applied this approach to the WTC11 PacBio data using default parameters (R = 100, P = 20, and Q = 80). In addition, we classify expressed genes as well annotated or underannotated using the parameters P and R. The logic for defining expressed genes as well annotated or underannotated is described in Figure 3A. The annotation category for all expressed genes (default: number of reads in gene [R] > 100) is provided in the gene_classification.csv file.
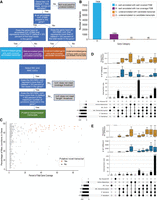
Evaluation of WTC11 PacBio samples for under-annotated genes. (A) Decision tree for classifying genes as well-annotated or under-annotated and classifying transcripts as putative novel transcripts. (B) Number of genes by their annotation status according to SQANTI-reads parameters. (C) The coverage (percent of total reads) versus length (percentage of maximum junctions) for all UJCs in under-annotated genes with well-covered candidate transcripts. UJCs that meet the thresholds for putative novel transcripts are colored in orange. (D,E) UpSet plot of putative novel transcripts with the most expressed annotated transcript in that gene for well-annotated genes with well-covered FSMs (D) and with an FSM detected but without <20% of the total reads in the gene (E). Upper and middle panels show the distribution of the number and proportion of nucleotides different between the putative novel transcript and most expressed FSM.
From the set of expressed genes, we identified 8556 well-annotated genes, 88% of which have a well-covered annotated transcript (>20% of total gene coverage) (Fig. 3B; Supplemental Fig. 7). We also identified 101 genes for which there are no reads with an FSM match to an annotated transcript. Of these, 54% have a well-covered UJC. For all expressed genes with a well-covered unannotated UJC, we identified 424 that contained most of the observed junctions in that gene (>80%), and we label these putative novel transcripts (Fig. 3C; Supplemental Fig. 7). Of these, 316 were NIC and 108 were NNC (Supplemental Fig. 7). The SQANTI-reads output for putative novel transcripts is included in the putative_underannotation.csv file (Table 1).
SQANTI-reads-specific output files
For genes with at least one putative novel transcript (R > 100, P > 20, and Q > 80) and an annotated transcript (FSM), we selected the FSM with the highest proportion of reads. We then compared the structure of the putative novel transcripts to the most expressed annotated transcript using TranD (Nanni et al. 2024). For genes with an annotated transcript that is relatively highly expressed (>20% of the reads for that gene), 103 putative candidate transcripts differed from the annotated transcript by donor/acceptor variation, suggesting a possible alternative splice site. In addition, 10 putative candidate transcripts had an extra exon, 15 a skipped exon, and 9 with both missing and skipped exons relative to the most expressed annotated UJC (Fig. 3D). For the genes where the annotated transcript represented <20% of the reads in that gene, the putative novel transcript differed from the annotated transcript by an alternative exon in 147 cases (33 extra exons, 86 skipped exons, and 43 with both an extra and skipped exon) (Fig. 3E). Details for this analysis are provided in the Supplemental Methods.
This analysis shows that SQANTI-reads can readily identify under-annotated genes and flag putative novel transcript models that contain interpretable alternative exonic patterns that deserve further attention.
SQANTI-reads metrics for donors/acceptors identify noisy splicing and potentially novel splice sites
SQANTI-reads calculates the mean, standard deviation, and coefficient of variation (CV) for all expressed annotated donors/acceptors using the absolute distances of each read donor and acceptor from the nearest annotated donors and acceptors (Fig. 4A). A CV > 0 indicates variability in the donor/acceptor, with higher CV values indicating more variability. Variability around a splice junction may be due to weak splicing (Wang and Marín 2006) or to technology errors and mapping accuracy, for example, due to junction ambiguity (Li 2018). We evaluated these metrics on the WTC11 data set for reference junctions with at least 10 reads. We found similar patterns in the variability (CV > 0) in donors and acceptors (Fig. 4B). All three technologies identify donors and acceptors with variability around the splice site (CV > 0). Donors/acceptors with CV > 0 consistently across the three technologies are highly suspicious of “noisy” splicing or a weak splice site and may warrant follow-up (Supplemental Fig. 8A,B). The SQANTI-reads output file cv.csv identifies the donors/acceptors with CV > 0, making it straightforward to follow up on particular locations with tools such as the Integrative Genomics Viewer (IGV) (Robinson et al. 2011).
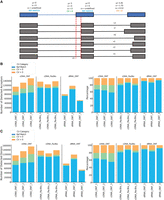
Variation in donors/acceptors. Metrics are only calculated for annotated donor/acceptors with a minimum threshold of reads (10 by default). (A) Metrics for the classification of donor/acceptor variation. When all reads align to the annotated donor/acceptor this is classified as a reference match (Ref Match). When all reads align to the same donor/acceptor location, but this is not the annotated position this is classified as CV = 0. When reads align in multiple positions in proximity to an annotated donor/acceptor this is classified as CV > 0. (B) Classification of the number (left) and percentage (right) of detected acceptors faceted by technology. (C) Classification of the number (left) and percentage (right) of detected donors faceted by technology.
A reference match junction indicates that the splice signal is strong (Wang and Marín 2006; Dent et al. 2021). Results for cDNA PacBio and dRNA ONT were similar, with both showing a higher number and proportion of reference match donors/acceptors compared to cDNA ONT (Fig. 4B,C), despite these technologies detecting similar numbers of FSM UJCs (Fig. 2D). We compared the FSM UJCs identified by the three methods (Supplemental Fig. 9A). Most of the FSM UJCs were detected by all three technologies (n = 19,690), with a similar number detected by cDNA PacBio only (n = 8110) and cDNA ONT (n = 9360) only. We hypothesized that the difference in the number of reference match donors/acceptors was potentially due to longer transcripts with more junctions being detected in cDNA PacBio compared to shorter transcripts with fewer junctions in dRNA ONT and cDNA ONT. For the FSMs detected only in one technology, we plotted the distribution of the number of junctions and confirmed that the cDNA PacBio FSM transcripts had a larger number of junctions compared to dRNA ONT and cDNA ONT (Supplemental Fig. 10). This agrees with cDNA PacBio showing longer reads than both ONT technologies in the WTC11 data set (Fig. 2E).
Splice junctions may differ from annotated sites due to the presence of novel donors/acceptors. The category CV = 0 identifies donors/acceptors with no variability but differing from the annotated donor/acceptor, representing strong candidates for bona fide alternative splice sites. We evaluated the donors and acceptors with CV = 0 in all the technologies (Supplemental Fig. 8E,F). We identified 51 donors and 34 acceptors with CV = 0 in all three technologies. Of these, 76% of donors and 65% of acceptors are within 12 nt of an annotated donor/acceptor, indicating a potential misannotation of the splice site (Supplemental Fig. 11). These donors and acceptors with CV = 0 across all technologies were detected by a minimum of 10 reads and, in some cases, could be detected with >100 reads (Supplemental Fig. 12). Supplemental Figure 13 shows an example of one of the reference donors with CV = 0 detected across all three technologies. This donor is in the H2AZ1 gene at position 99,948,814 on Chromosome 4. All the reads associated with this donor map to position 99,948,811, which is 3 nt away from the annotated donor position (Supplemental Fig. 13). Donors and acceptors with CV = 0 detected consistently across samples and/or technologies indicate potential robust detection of alternative splice sites.
Memory usage and runtime of SQANTI-reads
Computational efficiency is a critical factor for users when choosing a quality control tool. To evaluate memory usage and running time of SQANTI-reads, data sets containing five different number of reads were generated, as well as five replicates for each data set. After independently processing these data sets through SQANTI3, they were tested five times in 15 combinations to analyze memory usage and runtime for: a single sample of each size, three samples of each size, and five samples of each size.
The performance evaluation of SQANTI-reads revealed that memory consumption remained consistent at 2 GB of RAM across all tested conditions, regardless of the number of reads or the number of samples analyzed. Runtime, however, increased with both the number of reads per sample and the total number of samples processed simultaneously (Supplemental Fig. 14).
When processing a single sample, runtimes ranged from 100 sec for data sets with 10,000 reads to ∼3 min for data sets with 1,000,000 reads. For multisample runs, processing three samples of 1,000,000 reads each required ∼4 min, while processing five samples of the same size took about 5 min. These findings highlight the scalability of SQANTI-reads, with a predictable increase in runtime proportional to the data set size and sample number. Overall, SQANTI-reads demonstrated computational efficiency with minimal memory requirements, making it suitable for large-scale transcriptomic analyses.
Discussion
The increase in throughput and generalization of lrRNA-seq has led to the growing popularity of experiments containing many samples. Multiple lrRNA-seq studies have shown that, despite technological improvements, biases associated with read length and accuracy are still present (Amarasinghe et al. 2020; Delahaye and Nicolas 2021). These biases vary between the major LRS platforms and with the introduction of new instruments and chemistries. Other types of systematic biases, such as those introduced by batches utilized in large experiments, may also occur. Benchmarking studies have also revealed that transcript reconstruction methods can yield highly different results due to the varying strategies used to resolve data inaccuracies (Pardo-Palacios et al. 2024b). This highlights the need for tools that can comprehensively evaluate the characteristics of raw long-read data before making decisions on downstream analyses. Direct examination of read quality enables the researcher to evaluate the experiment for consistency and identify any outlier samples and any systematic differences in read quality between sample groups.
We have developed SQANTI-reads as a tool that enables a comprehensive assessment of reads obtained from an LRS experiment. We leverage the widely adopted SQANTI3 framework and add several metrics for the assessment of reads. SQANTI-reads is a flexible tool that can be used to evaluate the quality of lrRNA-seq multisample experiments. We present metrics that evaluate reads in terms of whether they correspond to annotated transcript models using SQANTI3 categories and subcategories. If metadata are included in the design file, alternate sample groupings can be used without needing to rebuild the classification and junction files.
For example, in Oxford Nanopore Technology (ONT), the existence of multiple platforms at different price points for different numbers of pores (Flongle, MinION, GridION, PromethION) but with the same library protocols means that, in a large experiment, samples can be initially evaluated at low cost on one of the lower-throughput platforms (Flongle MinION, GridION). If samples are of sufficient quality, they can then be run on higher-throughput platforms (PromethION). Sample multiplexing and running on multiple “lanes” is good experimental design practice (Auer and Doerge 2010). Our SQANTI-reads analysis of the Drosophila data set illustrates such a scenario. In this case, the pool for the 0–1 h samples was initially evaluated on the MinION. The resulting data from TR1 were unbalanced and had relatively short reads with a high proportion of ISM. These observations enabled adjustment of the library concentrations and run parameters, and a second MinION run resulted in fewer ISM and more balance in read numbers across libraries. A PromethION run based on the rebalanced libraries efficiently used the sequencing resources across samples. The same procedure was deployed with the 3–8 day samples, and rebalancing helped ensure the efficiency of the subsequent two PromethION runs. This example illustrates both good experimental design practices for large experiments and the utility of SQANTI-reads in assessing data quality at the early stages of data acquisition, enabling corrections that lead to a successful sequencing experiment.
Another important aspect of lrRNA-seq multisample experiments is the ability to quickly assess whether the data can address the biological questions that motivated the study. This includes, among other things, whether genes and transcripts are sufficiently quantified and if potential novel transcripts are adequately supported. While these questions may ultimately be answered after full data processing with transcript reconstruction algorithms, users may find it helpful to evaluate support directly from the raw data.
SQANTI-reads provides information on the distribution of reads across genes and UJC (a raw-data proxy for transcripts) and introduces new metrics for identifying variations in donors/acceptors, under-annotated genes, and putative novel transcripts for further evaluation. These metrics enable the researcher to quickly determine if more reads are needed and whether there are highly expressed putative novel transcripts potentially worth detailed experimentation.
The examples presented in this work demonstrate that SQANTI-reads is flexible and customizable, allowing users to explore the impact of various experimental design factors on read, UJC, and donor/acceptor properties, as well as identifying potential novel transcripts. The output from SQANTI-reads can be easily mined for additional insights and used to direct attention and resources toward interesting and novel features of lrRNA-seq experiments. We expect SQANTI-reads to become an essential tool for the QC of multisample lrRNA-seq data sets.
Methods
SQANTI-reads basics
SQANTI-reads is an adaptation of SQANTI3 designed to evaluate individual reads rather than transcript models and is available in SQANTI3 version 5.3.0. It allows for the comparison of multiple samples, providing quality control results across the entire experiment. Several new features have been introduced to address the specific needs of QC in multisample experiments, while some functionalities of SQANTI3 have been removed as they are not applicable to read-level processing. Table 2 highlights the major differences between SQANTI3 and SQANTI-reads. The input files for SQANTI-reads include: (1) a GTF file of read alignments, (2) a reference genome FASTA file, (3) a GTF file of the reference transcript model annotation, and (4) a design file containing metadata for multiple samples. The first step of SQANTI-reads involves using the SQANTI3 QC module to generate SQANTI3-like classification and junction files, with the classification file containing one row for each mapped read. Reads are classified according to the SQANTI categories (Tardaguila et al. 2018) as FSM, ISM, NIC, NNC, antisense, fusion, genic genomic, and intergenic. SQANTI3 subcategories are also included, based on 5′ and 3′ end positions relative to the annotated transcription start sites (TSS) and transcription termination sites (TTS) (Pardo-Palacios et al. 2024a). Additionally, the reverse transcriptase (RT) switching algorithm of SQANTI3 identifies reads with evidence of RT switching events, while reads with more than 60% adenines in the 20 bp downstream from the reported TTS at the genomic level are flagged as potential intrapriming events. The length of each read and the number of exons in each read are also recorded in the classification file.
Comparison between SQANTI3 and SQANTI-reads
Junction metrics
The SQANTI-reads junction file follows the same format as the SQANTI3 junction file, with each row representing a junction in a read, including the start and end positions of the junction. The distance from the junction start and end to the nearest annotated junction start and end in the reference GTF is calculated. It is important to note that the nearest annotated start and end positions may not belong to the same annotated junction. SQANTI classifies junctions as known or novel, and as canonical or noncanonical, based on the dinucleotide pairs at the junction's start and end. By default, dinucleotide combinations of GT-AG, GC-AG, and AT-AC are considered canonical, while any other combinations are classified as noncanonical, although the user can specify additional canonical sites.
SQANTI-reads introduces new metrics to evaluate the relationship between the junctions in mapped reads and the annotated donors and acceptors. In the SQANTI3 junction file, the distance from each donor/acceptor in each read to the nearest annotated donor/acceptor is recorded. In SQANTI-reads, the mean absolute distance in nucleotides from the annotated donor/acceptor site, the standard deviation, and the coefficient of variation (CV = standard deviation/mean) are calculated and included in the cv.csv file. Each detected junction is classified as: (1) Reference Match junction if the mean distance and the standard deviation to an annotated junction are both equal to 0; (2) CV = 0 junction when the mean distance is greater than 0 and the standard deviation equals 0, and (3) CV > 0 junction when the CV is greater than 0.
Unique junction chain and gene-level information
SQANTI-reads groups mapped reads based on their full junction pattern and refers to them as UJCs. Each UJC is labeled with a string that includes the chromosome and junction coordinates (Nanni et al. 2024). To enhance computational efficiency, UJC strings are encoded as an index in a hash table (JxnHash). The read count for each JxnHash is calculated and included in the ujc_counts.csv file. Additionally, the number of known canonical, known noncanonical, novel canonical, and novel noncanonical junctions within each UJC is annotated, along with the SQANTI structural category of the UJC. The number of reads within each structural category for each gene, as well as the total number of reads per gene, is stored in the summary file gene_counts.csv.
Identifying genes that may be under-annotated and transcripts that may be mis-annotated
For expressed genes, a high proportion of reads from a UJC classified as NIC/NNC may indicate the presence of a potentially novel transcript. SQANTI-reads includes a customizable pipeline to identify genes with such potential under-annotation events. The procedure identifies NIC/NNC UJCs present in genes with a minimum number of reads (R) and representing a minimum proportion (P) of reads in the gene, with default values set at 100 reads and 20%, respectively. To mitigate the risk that the NIC/NNC UJC is merely a degradation product, an additional condition is applied: the candidate UJC must include at least 80% of the gene's junctions (Q). The R, P, and Q thresholds are pipeline parameters that can be adjusted by the user. Furthermore, SQANTI-reads allows for the evaluation of under-annotated genes and novel transcripts within a specific subset of samples associated with a particular experimental factor (e.g., developmental stage or technology) using the ‐‐factor-level option.
Multisample processing
SQANTI-reads processes multiple samples to generate classification and junction files when a design file (e.g., Supplemental File 2) is provided to the sqanti-reads.py command. If individual samples have already been preprocessed with SQANTI3, SQANTI-reads can be run in ‐‐fast mode, where the design file links the individual classification and junction files to sample IDs for the calculation of SQANTI-reads metrics, summaries, and a series of visualizations. If preprocessing has not been done, SQANTI-reads is run in ‐‐simple mode, where SQANTI3 is run on each sample, followed by the calculation of SQANTI3 metrics and summaries. The output also includes a summary for each sample, reporting the mean, median, upper quartile, and lower quartile of mapped read length, as well as the number and proportion of reads that are shorter than 1 kb, between 1 and 2 kb, between 2 and 3 kb, and >3 kb in length, all of which are included in the length_summary.csv file.
Drosophila melanogaster data
A total of 24 female D. melanogaster abdomen samples corresponding to two developmental stages (0–1 h and 3–8 days posthatching), four genotypes (dmel 11037, 11255, 12272, and 12279), and three replicates (2 time points × 4 genotypes × 3 replicates = 24 samples) were sequenced using ONT. For each sample, mRNA was isolated (DynaBeads mRNA direct kit) from a pool of ∼20 abdomens. ONT libraries were constructed using the ONT PCR-cDNA Barcoding Kit (SQK-PCB109) starting with poly(A) mRNA according to the manufacturer's protocol. Libraries were pooled to a total of 100 fmol and run on a MinION Mk1c with real-time basecalling and demultiplexing (Guppy v6.1.5, MinKNOW v22.05.8). Read length and quality were evaluated with all samples passing the pycoQC metrics (v2.5.2, Leger and Leonardi 2019). Based on the MinION read counts, libraries were repooled prior to obtaining additional sequencing data on the ONT PromethION (Guppy v5.1.13, MinKNOW v23.04.5) at the University of Florida Interdisciplinary Center for Biotechnology Research (ICBR). TRs are defined as the same library run on different ONT flow cells (MinION or PromethION). TRs 1–3 were run on the MinION and TRs 4–6 were run on the PromethION. Detailed metadata for these samples and TRs are provided in Supplemental Table 1.
Dorado basecalled reads were generated from the FAST5 files by converting to pod5 formats (pod5 v 0.3.6) prior to basecalling by Dorado (v 0.5.2) (https://github.com/nanoporetech/dorado) using options ‐‐recursive ‐‐device “cuda:0,1” ‐‐kit-name SQK-PCB109 ‐‐trim none. Reads were demultiplexed using the demux mode of Dorado (v 0.5.2) with options ‐‐no-classify ‐‐emit-fastq.
Both Guppy and Dorado basecalled reads were processed using pychopper (v 2.7.1). Re-oriented FASTQ files were aligned to D. melanogaster 6.50 using minimap2 (v 2.17) (Li 2018) and the resulting SAM files converted to GTF format using SAMtools (v 1.10) (Li et al. 2009) and BEDTools (v 2.29.2) (Quinlan and Hall 2010).
The resulting 67 GTF files ((5 samples × 2 TRs) + (19 samples × 3 TRs) = 67) were used as input into SQANTI-reads along with the D. melanogaster 6.50 FASTA and GTF reference files (https://ftp.flybase.net/releases/FB2023_01/dmel_r6.50/, (Öztürk-Çolak et al. 2024)) and a design file (Supplemental File 3). The design file included all experimental factors for evaluation, including time, genotype, sequencing platform (MinION and PromethION), and basecaller (Dorado vs. Guppy).
Human cell line WTC11
We used publicly available lrRNA-seq data from the Long-read RNA-seq Genome Annotation Assessment Project (Pardo-Palacios et al. 2024b) to illustrate the utility of SQANTI-reads. Specifically, we used triplicate measurements of the transcriptome of the WTC11 human cell line that were profiled by cDNA PacBio Sequel II, cDNA Oxford Nanopore MinION, and direct RNA Oxford Nanopore MinION methods. Data were downloaded from the ENCODE website (https://www.encodeproject.org/search/?type=Experiment&internal_tags=LRGASP). Accession numbers for these samples are provided in Supplemental Table 2. The FASTQ files were preprocessed by LRGASP researchers as described in Pardo-Palacios et al. (2024b). We used the GTF files of read alignments, GENCODE's GRCh38.p13 reference genome GTF and FASTA for release 38 (https://www.gencodegenes.org/human/release_38.html), and a design file (Supplemental File 4) to run SQANTI-reads on the WTC11 samples. The SQANTI-reads output is provided in Supplemental File 5.
Computational efficiency evaluation
SQANTI-reads can operate in two modes: slow and fast. The slow mode uses SQANTI3 to generate the standard classification file, which serves as the input for further analysis. In contrast, the fast mode begins with preprocessed samples that have already been run through SQANTI3. This mode generates a UJC and hash dictionary for each sample to facilitate comparisons and calculate summary statistics.
To evaluate the performance of SQANTI-reads, we generated 25 data sets representing GTF files with varying numbers of reads. To do so, we used the shuf command line tool, with the -n flag, to extract a random set of 10,000, 50,000, 100,000, 500,000, and 1,000,000 reads from the ENCFF003QZT sample of the LRGASP study (Pardo-Palacios et al. 2024b). For each number of reads, five different random subsets of reads were extracted, representing five replicates. These data sets were first processed independently using SQANTI3 to produce the necessary classification files required for fast-mode analysis, as the SQANTI3 performance does not reflect the actual runtime or memory usage of SQANTI-reads itself.
Following preprocessing, the data sets were tested under 15 experimental conditions designed to evaluate runtime and memory usage across different scales. The conditions included processing (1) a single sample of each data set size, (2) three samples of each size, and (3) five samples of each size. All experiments were conducted on a Linux High-Performance Computing Cluster requesting 10 GB of RAM and a single CPU.
We measured runtime as the wall-clock time from the start to the completion of SQANTI-reads execution and memory usage using sacct command from Slurm workload manager. These metrics were recorded for each experimental condition to assess scalability and resource efficiency under varying data set sizes and sample counts.
Software availability
SQANTI-reads is available as Supplemental Code and at GitHub where it is integrated into SQANTI3 from version 5.3.0 (https://github.com/ConesaLab/SQANTI3). Instructions for running SQANTI-reads are provided on the SQANTI3 wiki (https://github.com/ConesaLab/SQANTI3/wiki/Running-SQANTI%E2%80%90reads).
Code for generating subsampled data sets to evaluate computational efficiency, along with scripts for analysis, is available on GitHub (https://github.com/ConesaLab/SQANTI_reads_ComputationalEfficiency_plots) and as Supplemental Code.
Data access
Drosophila long-read data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under the accession number PRJNA1134728. The WTC11 long-read data from the LRGASP project are available from the ENCODE Portal (https://www.encodeproject.org/) and accession numbers are provided in Supplemental Table 2.
Competing interest statement
A.C. has received in-kind funding from Pacific Biosciences for library preparation and sequencing. A.C. collaborates with Oxford Nanopore in the Marie Skłodowska-Curie Actions Doctoral Network project LongTREC (grant agreement no. 101072892).
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (1R21HG011280-01), the Spanish Ministry of Science Innovation and Universities (MCIU) (PID2020-119537RB-I00 and PID2023-152976NB-I00), the European Union's programme Horizon Europe under the Marie Skłodowska-Curie Actions postdoctoral fellowship to C.M. (101149931). This work is also supported in part by a grant from the National Cancer Institute (NCI P01 CA214091), the National Institute of General Medical Sciences (NIGMS GM137430), the University of Florida Department of Molecular Genetics and Microbiology, the University of Florida Genetics Institute, the University of Florida Cancer Center, and the University of Florida Research Computing Center (www.rc.ufl.edu) and the Latin American and Caribbean Scholars award to N.K. Part of the computations were performed on the high-performance computing cluster Garnatxa at the Institute for Integrative Systems Biology (I2SysBio). I2SysBio is a joint research center formed by the University of Valencia (UV) and the Spanish National Research Council (CSIC). We acknowledge Ashley Myrick for help with some of the initial CV plots and Knife Bankole for the initial coding of the junction hash. Alison Morse prepared all samples and libraries for the Drosophila experiment, ran all initial QC analyses, and recalled all the bases for ONT data. We acknowledge Rolf Renne for his support.
Author contributions: N.K. conducted research, developed software implementations, created figures, and drafted the manuscript. C.M. developed software implementations, integrated new methods into SQANTI3 GitHub, corrected figures, and helped with data analysis. L.M. conceived and supervised the study and drafted the manuscript. A.C. supervised the study, contributed to conceptualizations, and drafted the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280021.124.
- Received September 12, 2024.
- Accepted February 1, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








