
RAmbler resolves complex repeats in human Chromosomes 8, 19, and X
Abstract
Repetitive regions in eukaryotic genomes often contain important functional or regulatory elements. Despite significant algorithmic and technological advancements in genome sequencing and assembly over the past three decades, modern de novo assemblers still struggle to accurately reconstruct highly repetitive regions. In this work, we introduce RAmbler (Repeat Assembler), a reference-guided assembler specialized for the assembly of complex repetitive regions exclusively from Pacific Biosciences (PacBio) HiFi reads. RAmbler (1) identifies repetitive regions by detecting unusually high coverage regions after mapping HiFi reads to the draft genome assembly, (2) finds single-copy k-mers from the HiFi reads, (i.e., k-mers that are expected to occur only once in the genome), (3) uses the relative location of single-copy k-mers to barcode each HiFi read, (4) clusters HiFi reads based on their shared barcodes, (5) generates contigs by assembling the reads in each cluster, and (6) generates a consensus assembly from the overlap graph of the assembled contigs. Here, we show that RAmbler can reconstruct human centromeres and other complex repeats to a quality comparable to the manually curated Telomere-to-Telomere human genome assembly. Across more than 250 synthetic data sets, RAmbler outperforms hifiasm, LJA, HiCANU, and Verkko across various parameters such as repeat lengths, number of repeats, heterozygosity rates, and depth of sequencing.
Given the broad biological impact of obtaining the genome for a new organism, de novo genome assembly is one of the most critical problems in computational biology. Despite tremendous algorithmic progress, the problem is not yet completely solved. The assembly problem remains challenging due to the high repetitive content of eukaryotic genomes, short read length, uneven sequencing coverage, nonuniform sequencing errors, and chimeric reads. Repetitive regions (or segmental duplications) are the primary reasons for which de novo genome assemblies are often fragmented and incomplete. A large fraction of eukaryotic genomes is made of repetitive elements, including satellite DNA, minisatellites, microsatellites, and DNA/RNA transposons (Jurka et al. 2007; Mrázek et al. 2007; Treangen et al. 2009; Bustos et al. 2023). For instance, Supplemental Figure S1 illustrates the frequency of repeats present in seven plant species (Chan et al. 2015). Several studies have shown that expansions or mutations of repetitive regions are linked to a variety of human diseases, ranging from neurological diseases to cancers (for a review, see Liao et al. 2023b). Although many repeats were considered nonfunctional, they have been shown to impact gene expression, contributing to genetic disorders (Hannan 2018; Ishiura et al. 2019; Shah et al. 2023).
The third generation of sequencing technology on the market, e.g., Pacific Biosciences (PacBio) (Eid et al. 2009; Qin et al. 2012; Roberts et al. 2013; Huddleston et al. 2014; Kim et al. 2014) and Oxford Nanopore Technologies (ONT) (Clarke et al. 2009; Quick et al. 2014; Ashton et al. 2015; Loose et al. 2016), offers longer reads at a higher cost per base than the second generation, but the sequencing error rate is much higher. The introduction of PacBio HiFi sequencing at the end of 2019 has been a “game-changer” in genome assembly, because it can produce read lengths typically ranging 10–25 kb with accuracy >99.8% (Wenger et al. 2019). HiFi sequencing greatly improved human assemblies (Nurk et al. 2020; Shumate et al. 2020; Garg et al. 2021; Porubsky et al. 2021). The Telomere-to-Telomere (T2T) human genome sequencing project took advantage of PacBio HiFi and ultra-long ONT reads to close most of the repetitive gaps and achieved 99.9% completeness (Miga et al. 2020; Logsdon et al. 2021; Hoyt et al. 2022; Nurk et al. 2022; Rautiainen et al. 2023). In particular, the method developed to assemble human Chromosome 8 depended on the use of single-copy k-mers (hereafter called unikmers, also known as SUNKs in Logsdon et al. [2021] or SUNs in Sudmant et al. [2010]) to resolve repetitive regions.
The problem of reconstructing repetitive regions (segmental duplications) has been addressed several times in the literature (e.g., Chaisson et al. 2017; Vollger et al. 2019; Bzikadze and Pevzner 2020). The Segmental Duplication Assembler (SDA) by Vollger et al. (2019) is no longer maintained. More recently, Bzikadze and Pevzner (2020) proposed CentroFlye to address the problem of reconstructing human centromeres. CentroFlye is a specialized assembler that uses error-prone ONT or PacBio CLR reads. It also requires additional information such as higher-order repeats (HORs) or monomers. This limits its applicability to species for which HORs or monomers are known. In addition, CentroFlye is very demanding in terms of computational resources. While SDA, CentroFlye, and the assembler for human Chromosome 8 by Logsdon et al. (2021) are specialized tools for reconstructing segmental duplications or centromeres, they were designed for error-prone long reads. To the best of our knowledge, there is no specialized assembler for reconstructing repeats that uses PacBio HiFi reads exclusively.
In this work, we introduce RAmbler, a reference-guided assembler that takes advantage of single-copy k-mers to resolve complex repetitive regions. We show that RAmbler can resolve complex repeats in human Chromosomes 8, 19, and X from HiFi data to a quality comparable to the T2T human genome assembly. Due to the lack of specialized HiFi assemblers for repeats, we compare RAmbler against four general-purpose state-of-the-art assemblers, namely, hifiasm (Cheng et al. 2021), LJA (Bankevich et al. 2022), HiCANU (Nurk et al. 2020), and Verkko (Rautiainen et al. 2023) on more than 250 synthetic data sets and five real Homo sapiens data sets.
Results
Experimental results on Homo sapiens
We used three human genome assemblies as a reference, namely, GRCh38.p13 (hereafter called HG38), T2T-CHM13.v2.0 (T2T), and the maternal strand of HG002 (MAT002). We used PacBio HiFi reads from four different cell lines, namely, CHM13 (from the T2T project), HG002, HG00733, and HG01346 (these last three from the human pan-genome project) (Liao et al. 2023a). Supplemental Table S1 reports the accession numbers and statistics for these human data sets, while Supplemental Table S2 summarizes accession numbers and statistics for Saccharomyces cerevisiae (used in our simulation studies).
We used RAmbler to assemble some of the repetitive regions in the reference assemblies HG38, T2T, and MAT002, using various sets of HiFi reads. We used the following naming convention to identify the assemblies produced by RAmbler. Hereafter, an RAmbler assembly using reference assembly G and HiFi reads H will be denoted by RA.G.H. For example, the RAmbler assembly using the HG38 assembly and the HG00733 HiFi reads will be called RA.HG38.HG00733.
We focused on five complex repetitive regions within the human genome: the centromeres of Chromosome 8 and X, and three noncentromeric regions from Chromosome 19. While the selection of these regions was somewhat arbitrary, it was motivated by a few factors: (1) these regions were overcollapsed in the HG38 assembly (Fig. 1A), (2) Chromosomes 8, 19, and X have no unplaced contigs within the HG38 assembly, and (3) Chromosome 19 contains a few unresolved noncentromeric repeats (Supplemental Fig. S2). Our intent was to show that using newer HiFi reads, RAmbler could improve some of the over-collapsed repetitive regions, in particular in the HG38 assembly.
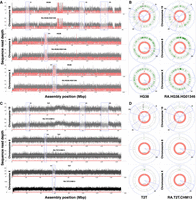
Comparing RAmbler's assemblies (RA.HG38.HG01346 and RA.T2T.CHM13) against (A,B) the GRCh38.p13 assembly (HG38) and (C,D) the T2T-CHM13.v2.0 assembly (T2T) of human Chromosomes 8, 19, and X. In all plots, blocks (i), (ii), and (iii) are noncentromeric repeats in Chromosome 19; blocks (iv) and (v) are the centromeric regions of Chromosomes 8 and X, respectively. (A,C) NucFreq plots illustrating HiFi read mapping coverage (clipped at 60×); (B,D) CRAQ Circos plots illustrating (a) assembly quality index (AQI) score (higher is better), (b) base errors (fewer is better), and (c) misjoins (fewer is better).
We carried out five experiments on human data sets. The first two experiments were aimed at demonstrating RAmbler's ability to resolve repeats without manual curation. For these two experiments, we used as inputs (1) the T2T assembly with CHM13 HiFi reads and (2) the MAT002 assembly with HG002 HiFi reads. The other three experiments focused on RAmbler's performance. For these three, we used as input the HG38 assembly and HiFi reads from (3) HG002, (4) HG00733, and (5) HG01346.
We carried out these five experiments on the centromeric regions of Chromosomes 8 and X, as well as three noncentromeric regions on Chromosome 19. We ran RAmbler, hifiasm, LJA, HiCANU, and Verkko on the subset of HiFi reads mapped to these regions. SDA was excluded because it “is no longer maintained and should not be used … assembly tools like Flye, HiCanu, and hifiasm outperform any results previously possible with SDA” (quote from https://github.com/mrvollger/SDA). CentroFlye was also excluded because we were unable to run it on our 2.8 GHz 32-core processor server with 512 GB of RAM. We tried first to reconstruct the centromere of Chromosome X using the HiFi reads and the HORs provided by the authors, but CentroFlye failed during the error-correction step. When we tried to reconstruct the same centromere using the CHM13 ONT reads from Bzikadze and Pevzner (2020), CentroFlye ran out of memory. The instructions claim that CentroFlye requires ∼800 GB of RAM to complete the assembly of Chromosome X.
The comparative results for RAmbler, hifiasm, LJA, HiCANU, and Verkko on the centromeric regions of Chromosomes 8 and X are summarized in Table 1. Columns 2, 3, and 4 indicate the input assembly, the HiFi sample, and the chromosome-level sequencing depth, respectively. Columns 5 and 6 indicate the boundaries of the regions to be reassembled. Columns 7 and 8 indicate the number of HiFi reads mapped to the target region and the total number of bases in these reads, respectively. In Column 9, we show RAmbler's estimate of the size of the repetitive region, obtained by computing the ratio between the total number of bases of the selected HiFi reads and the average chromosome-level coverage depth. While it has been reported that repetitive regions can affect HiFi sequencing coverage (Nurk et al. 2022), we determined that this coverage bias would have a small impact on RAmbler's estimation of the length of the repetitive region to be assembled. When we masked all repetitive regions from the human genome, the changes in HiFi coverage were relatively small. Supplemental Table S3 shows that on Chromosomes 8, 19, and X, the change in coverage after repeat masking was smaller than 4%. Overall, the variations in coverage were under 9% at the chromosome-level and under 3% at the genome level. The largest change was observed for Chromosome 17 (8.57% variation in coverage using the HG002 HiFi reads). Such a coverage fluctuation would mean that, in the worst case, a repetitive region with 10 copies of the monomer could be underestimated by one copy. Columns 11–15 show the assembly statistics for RAmbler, hifiasm, LJA, HiCANU, and Verkko. Observe that RAmbler consistently produced the least fragmented assemblies, and in most cases, the longest contig produced by RAmbler was the longest among all assemblers. In many cases, the total assembly size generated by RAmbler was close to the expected assembly size, although hifiasm also produced assemblies whose total size was also consistent with the expectation. Similar observations could be made for the three noncentromeric regions on Chromosome 19 (Supplemental Table S4 for T2T and MAT002; Supplemental Table S5 for HG38).
Comparison of the assemblies produced by RAmbler, hifiasm, LJA, HiCANU, and Verkko on the HiFi reads extracted around the centromeres of human Chromosomes 8 and X, for several choices of input assemblies and HiFi reads
Unlike de novo assemblers which consistently produce the same outputs for the same set of input reads, RAmbler's assembly depends on the reference used as it relies on the input assembly to identify the reads that belong to a repetitive region. Observe that in Table 1 we used the same set of HiFi reads (HG002) with two reference assemblies, namely, HG38 and MAT002. Despite the difference between HG38 and MAT002, in particular, in the centromeric regions, the analysis in Supplemental Table S6 shows that more than 85% of the selected reads were shared. As a result, the RAmbler assemblies are relatively consistent. For instance, on the centromere of Chromosome X, the length difference for the longest contigs was ∼3%. On the centromere of Chromosome 8, the length difference for the longest contigs was ∼10%.
While the assembly contiguity statistics reported in Table 1, Supplemental Tables S4 and S5 are important, they did not tell us whether these assemblies had high quality. To quantitatively assess the assembly quality, we used a two-pronged approach. First, we used the manually curated T2T assembly as the ground truth and we measured the agreement between the RAmbler assemblies and the T2T assembly using QUAST (Gurevich et al. 2013) and SyRI (Goel et al. 2019). This approach, however, can be problematic because the HiFi reads used for the RAmbler assemblies did not originate from the cell line used to generate the T2T assembly. Human centromeres are known to exhibit high variation across individuals (Altemose et al. 2022). For instance, the NucFreq coverage plots in Supplemental Figure S2 show coverage spikes in the centromeric regions of the T2T assembly when the HG01346 reads were mapped to it. As a consequence, some level of divergence would be expected in RAmbler's centromeric assembly when compared to the T2T assembly. To obtain both qualitative and quantitative assessments of RAmbler's improvements, we measured the assembly quality using CRAQ (Li et al. 2023) and NucFreq (Vollger et al. 2019). CRAQ is a tool that measures assembly quality without the need of a reference; in particular, it can detect local and global assembly errors based on the alignment information of mapped reads (long and short). CRAQ was given in input Illumina reads (which were not used in the assembly) and HiFi reads (which were used for the assembly). While we expected a reduction in coverage spikes in the repetitive regions of the NucFreq plots because those HiFi reads were also used in the assembly, we were interested in measuring the extent of this reduction. Additionally, we used NucFlag (an extension of NucFreq) to flag assembly errors or collapses along with the coverage plots.
To establish RAmbler's assembly quality, we carried out two sets of experiments. In the first set, we show that RAmbler can drastically improve the HG38 assembly in the five repetitive regions to a QUAST-based quality comparable to the T2T assembly using HG002, HG00733, or HG01346 HiFi reads. In the second set, we show that if RAmbler was given the same set of HiFi reads used for the T2T or the MAT002 assemblies, it would produce an assembly of the five repetitive regions that matches, if not exceeds, the quality of the corresponding reference assemblies.
Figure 1A and B summarizes the results of one run from the first set of experiments using HG01346 HiFi reads. Figure 1A shows the HiFi coverage. The coverage depth in Figure 1A is clipped at 60× for better visualization, whereas Supplemental Figure S3 shows full-scale plots for HG38 and the RAmbler assembly RA.HG38.HG01346. Observe that on Chromosome 19, RA.HG38.HG01346 has a more uniform coverage across the three repetitive regions compared to HG38, in particular for regions (i) and (ii). Also observe that (1) while the coverage on region (iv) of Chromosome 8 of HG38 is ∼600×, it reduces to ∼100× in RA.HG38.HG01346 indicating a partial resolution of the repeat; (2) while the coverage on region (v) of Chromosome X of HG38 is ∼800×, it reduces to ∼30× in RA.HG38.HG01346 indicating a full resolution of the repeat. From a purely coverage viewpoint, these results indicate that RA.HG38.HG01346 is significantly improved compared to HG38. Supplemental Figure S4A illustrates the NucFlag plots for the five regions analyzed. Observe that NucFlag indicates much fewer errors in the RA.HG38.HG01346 assembly compared to HG38.
Figure 1B shows the results of the CRAQ analysis (Li et al. 2023). We used HG01346 HiFi reads and HG01346 Illumina reads (∼10× coverage) to evaluate the quality of HG38 and RA.HG38.HG01346. The Circos plots (Krzywinski et al. 2009) in Figure 1B illustrate the (a) assembly quality indices (AQI) score (blue curve, higher is better, see Section “Performance metrics for real data”), (b) base errors (green bands, fewer is better), and (c) misjoins (red bands, fewer is better). Cones (i)–(v) highlight the repetitive regions. On Chromosome 19, observe that (1) the assembly quality index (AQI) score for RA.HG38.HG01346 is higher than the AQI score for HG38 on all three regions; (2) RA.HG38.HG01346's base errors are significantly better than HG38; (3) CRAQ reports four misjoins in HG38, and zero in RA.HG38.HG01346. Also observe that on the centromeres of Chromosome 8 and Chromosome X, the AQI scores for RA.HG38.HG01346 are higher than the AQI scores for HG38. On Chromosome 8, HG38 has two misjoins, and RA.HG38.HG01346 has none. RA.HG38.HG01346 also has fewer base errors. On Chromosome X, RA.HG38.HG01346 has no misjoins (HG38 has two) with fewer base errors than HG38. The NucFreq, the Circos/CRAQ, and the NucFlag plots for HiFi reads HG002 and HG00733 are shown in Supplemental Figures S5 and S6. The coverage plots and the Circos plots from both of these runs demonstrate similar results as shown in Figure 1A and B. Table 2 reports all the statistics produced by CRAQ, which clearly indicates that RA.HG38.HG002, RA.HG38.HG00733, and RA.HG38.HG01346 are improved compared to HG38.
Comparing RAmbler's assemblies (RA.HG38.HG002, RA.HG38.HG00733, and RA.HG38.HG01346) for selected regions of human Chromosomes 19, 8, and X against the GRCh38.p13 assembly (HG38) using CRAQ
Supplemental Table S7 reports the QUAST metrics for HG38, RA.HG38.HG002, RA.HG38.HG00733, and RA.HG38.HG01346, using the T2T assembly as the ground truth and for HG38 and RA.HG38.HG002, using the MAT002 assembly as the ground truth. When T2T was used as the reference, (1) the number of misassemblies on Chromosome 8 decreased from 371 in HG38 to 239–278 in the RAmbler assemblies; on Chromosome 19, they decreased from 101 in HG38 to 92–100; on Chromosome X, they decreased from 374 in HG38 to 205–250, (2) the genome fraction for Chromosome 19 increased from ∼90.7% to ∼91.5%; on Chromosome X, it increased from ∼98.6% to ∼99%; on Chromosome 8, it remained the same ∼98%. When MAT002 was used as the reference, (1) the number of misassemblies on Chromosome 8 decreased from 382 in HG38 to 205 in RA.HG38.HG002; on Chromosome 19, they reduced to 102 from 119; on Chromosome X, they decreased from 382 in HG38 to 169, (2) the genome fraction for Chromosome 8 increased from 97.5% to 98.4%; on Chromosome 19, it improved from 91.2% to 92.3%; on Chromosome X, it increased from 98.6% to 99.6%. Supplemental Figure S7 shows the synteny analysis based on SyRI (Goel et al. 2019). Observe the much stronger synteny between the three RAmbler assemblies and T2T, compared to HG38.
Figure 1C and D summarizes the results of one of the runs from the second set of experiments obtained by RAmbler using the PacBio HiFi reads that were used for the T2T project (∼32.4× coverage). Hereafter, this RAmbler assembly is called RA.T2T.CHM13. Observe in Figure 1C that T2T and RA.T2T.CHM13 have nearly identical HiFi coverage across all five regions, which is also reflected in the NucFlag analysis in Supplemental Figure S4B. Figure 1D illustrates the CRAQ assessments based on the alignment of CHM13 HiFi reads and CHM13 Illumina reads. Observe that both T2T and RA.T2T.CHM13 have similar AQI scores across all three chromosomes with no misjoins, except for three base errors introduced in the centromere of Chromosome 8 and one base error corrected in the Chromosome X by RAmbler (CRAQ numerical scores are reported in Supplemental Table S8). We also used ultra-long ONT reads (longer than 100 kb, ∼17× coverage) to further evaluate and compare these assemblies. The UL-ONT coverage and CRAQ plot in Supplemental Figure S8, and the CRAQ numerical scores in Supplemental Table S9 indicate that the RA.T2T.CHM13 has the same quality as T2T. The NucFreq plots, the Circos plots based on CRAQ (numerical scores are reported in Supplemental Table S10), and the NucFlag plots for the other run from this set of experiments are shown in Supplemental Figures S9 and S10. We used the MAT002 assembly as input assembly with the HG002 HiFi reads. The coverage plots and the Circos plots from this run demonstrate similar results as shown in Figure 1C and D. However, the NucFlag analysis reveals that the RA.MAT002.HG002 assembly had fewer errors than MAT002 in three out of the five repetitive regions. Supplemental Figure S11A indicates a perfect synteny between RA.T2T.CHM13 and T2T across Chromosomes 8, 19, and X. Similarly, Supplemental Figure S11B illustrates a perfect synteny between RA.MAT002.HG002 and MAT002 across Chromosomes 8 and 19, and a near-perfect synteny with a small inversion in Chromosome X.
Supplemental Table S11 shows the runtime and the memory consumption for RAmbler to resolve the centromeres of human Chromosomes 8 and X. Currently RAmbler takes longer than hifiasm, LJA, and Verkko. RAmbler's memory consumption is reasonable. The current implementation of RAmbler is not multithreaded, so there is an opportunity to make it faster and more scalable.
Experimental results on synthetic data
Synthetic data generation and parameter optimization
We generated synthetic repetitive regions by selecting a combination of (1) repeat unit size: 10 kb, 15 kb, 20 kb, and 25 kb; (2) 2, 5, and 10 copies of the repeat unit; (3) mutation rate in each copy of the repeat P = {1/100, 1/250, 1/500, 1/1000, 1/2000}. For each combination, we generated HiFi reads using PBSim (Ono et al. 2013) on the CCS model with read coverage of 10×, 20×, 30×, and 40×. PBSim requires other parameter values to be set before generating reads, which are provided in Supplemental Table S12.
RAmbler has five main parameters (summarized in Supplemental Table S13). Based on the analysis in Section “Analysis of k-mer distribution,” we determined that k = 21 and t = 3. To find the optimal values for to and th, we conducted a grid search where to = {1, 5, 10, 15, 20} and th = {5, 10, 15, …, 50} (50 combinations).
RAmbler was tested on 135 synthetic data sets, obtained from the combinations of different choices of the repeat unit size {10, 15, 20} kb, repeat copies {2, 5, 10}, mutation rate P = {1/100, 1/250, 1/500, 1/1000, 1/2000}, and read coverage depth {20 × , 30 × , 40 × }. Supplemental Figure S12 shows the experimental results for different metrics (namely, number of contigs, number of misassembled contigs, effective genome fraction per contig, and normalized NG50), with the best choices for to and th highlighted in colored rectangles.
When considering the number of contigs (Supplemental Fig. S12A), observe that RAmbler achieved the best performance for to = {15, 20} and th = {10, 15}. Regarding the number of misassembled contigs produced by RAmbler (Supplemental Fig. S12B), the best values were to = {15, 20} and th = {10, 15, 20, 25}. In terms of effective genome fraction per contig (ζ) (Supplemental Fig. S12C), RAmbler had better results with to = {10, 15, 20} and th = {10, 15, 20}. When considering the normalized NG50 (η) (Supplemental Fig. S12D), the best outcomes were obtained with to = {15, 20} and th = 15. By combining all these metrics in the assembly score (defined later in Section “Assembly score”), we determined that the optimal values were to = 15 and th = 15 (Supplemental Fig. S13).
The value of mo (minimum overlap for the overlap graph) was set to 1000 bp in all our experiments (both on synthetic and real data). To ensure that mo = 1000 was sufficiently stringent to avoid spurious overlaps in the human genome, we carried out the following experiment. (i) We collected the centromeric regions for all human chromosomes in the T2T assembly (excluding the rDNA regions). (ii) We extracted 1 kb sequences every 100 bp in these centromeric regions. (iii) We computed an alignment between all pairs of sequences extracted in Step (ii) using minimap2 (Li 2018). Out of 2,860,045 sequences generated in Step (ii), only three had an identical full match to another sequence in the set. There was (i) a 1 kb sequence on Chromosome 16 starting at position 33,712,533 matching a 1 kb sequence starting at position 35,108,033, (ii) a 1 kb sequence on Chromosome 16 starting at position 33,713,733 matching a 1 kb sequence starting at position 35,109,233, and (iii) a 1 kb sequence on Chromosome 3 starting at position 95,760,872 matching a 1 kb sequence starting at position 95,843,772. This analysis demonstrates that spurious overlaps that are 1 kb or longer are extremely rare in the human genome, even in repetitive regions like the human centromeres. We used windows every 100 bp due to the computational cost aligning pairwise all these sequences. The minimap2 alignment file for 2.86 M sequences was 1.43 TB. The number of 1 kb sequences every 10 bp would be ∼28.6 M, and the number of 1 kb sequences every bp would be ∼286 M. Since the alignment file grows quadratically with the number of sequences, it would quickly become infeasible to process. We believe that the conclusions would still hold with 10× or 100× more data.
Comparing RAmbler, hifiasm, LJA, HiCANU, and Verkko on fixed-length repeats
We conducted an extensive performance comparison of RAmbler with other state-of-the-art HiFi assemblers, namely, hifiasm, LJA, HiCANU, and Verkko, using 36 synthetic data sets. SDA is no longer maintained and CentroFlye requires additional auxiliary information (HORs and monomers), thus they were both excluded from this comparison. We generated repetitive regions with two choices of the repeat unit size {15, 20} kb, two choices for the number of copies {5, 10}, and three values of mutation rate P = {1/250, 1/500, 1/1000}. Synthetic HiFi reads were generated using PBSim with coverage depths of {20 × , 30 × , 40 × } (see Section “Synthetic data generation and parameter optimization” for details).
Figure 2 summarizes the results in terms of four different performance metrics. In Figure 2A, we compare the number of contigs produced by the different assemblers, where a single contig would be the ideal assembly. Observe that RAmbler consistently produced the lowest number of contigs among all the assemblers. Figure 2B–D shows the results in terms of effective genome fraction per contig (ζ), normalized NG50 (η), and assembly score (numerical scores are in Supplemental Table S14), respectively. For all these metrics, the best assembly is the one that gets closer to 1.0. Observe again that RAmbler achieved higher values on all these metrics compared to the other assemblers.
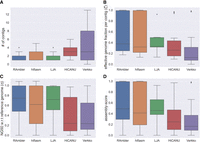
Assembly statistics for RAmbler, hifiasm, LJA, HiCANU, and Verkko for 36 different combinations of synthetic data with repetitive regions having repeat sizes {15, 20} kb, number of copies {5, 10}, mutation rate P = {1/250, 1/500, 1/1000}, and coverage depth {20× , 30× , 40× }. (A) Number of contigs, (B) effective genome fraction per contig ζ, (C) normalized NG50 η, and (D) assembly score.
Comparing RAmbler, hifiasm, LJA, HiCANU, and Verkko on variable-length repeats
To evaluate RAmbler's ability to resolve tandem repeats in the presence of variable-length repeat units, we created synthetic repetitive regions in which each repeat copy can vary up to ±5% of the length of a repeat unit. We used five copies of repeat units of {15, 20, 25} kb with mutation rates P = {1/250, 1/500, 1/1000}. Synthetic HiFi reads with a coverage depth of 30 × were generated with PBSim. Table 3 summarizes assembly score results for these nine data sets. RAmbler outperformed the other assemblers in six out of nine runs. Supplemental Figure S14 shows that RAmbler, hifiasm, and LJA produce contigs with either zero or one misassembled contig. It also shows that RAmbler, hifiasm, and LJA produce more contiguous assemblies than HiCANU and Verkko. In general, while HiCANU and Verkko rarely introduce misassemblies, they produce more fragmented assemblies than RAmbler, hifiasm, and LJA. This is reflected by the RAmbler's best assembly score in Supplemental Figure S15.
Assembly score results for nine different data sets of synthetic HiFi reads with coverage 30×, based on a repetitive region with a variable-length (up to ±5% per copy) repeat unit of length {15, 20, 25} kb, and P = {1/250, 1/500, 1/1000}
Comparing RAmbler, hifiasm, LJA, HiCANU, and Verkko on repetitive regions with copy number variation
Note that RAmbler was not designed to produce a haplotype-resolved assembly. An important question is what assembly would RAmbler produce in case there are copy number variations between the two haplotypes. To address this question, we carried out several experiments on synthetic diploid genomes, as follows. (1) We created a synthetic repetitive region where the primary haplotype (hap1) contained either 5 or 10 copies of a repeat unit. Each repeat unit was {5, 10, 15, 20, 50, 100} kb long, with a mutation rate of {1/250, 1/500, 1/1000}. (2) We produced the secondary haplotype (hap2) as follows. When the hap1 had 5 copies, hap2 had {3, 4, 5} copies. If hap1 had 10 copies, hap2 had {6, 8, 10} copies. (3) We added a 50 kb sequence upstream and downstream from the repetitive region on both hap1 and hap2. (4) We used PBSim to generate 30 ×-coverage HiFi reads from these 2 × 6 × 3 × 3 = 108 synthetic diploid genomes (hap1 + hap2). (5) We assembled the synthetic HiFi reads using RAmbler, hifiasm, LJA, HiCANU, and Verkko.
The goal of these experiments was to evaluate the quality of the assemblies produced by RAmbler, hifiasm, LJA, HiCANU, and Verkko on these synthetic diploid genomes using various metrics. We recorded the total number of contigs, the overall assembly size, the number of resolved repeat copies, and the number of contigs with haplotype switching. On these synthetic HiFi data sets with minimal divergence between haplotypes, LJA, HiCANU, and Verkko are expected to produce phased assemblies, but not fully haplotype-resolved assemblies due to the limited length of HiFi reads. They typically require either long-range sequencing data (e.g., ultra-long ONT or Hi-C reads) or phasing data (e.g., maternal and paternal reads) for full haplotype resolution. In HiFi-only mode, LJA, HiCANU, and Verkko generate a single FASTA file containing both haplotypes. hifiasm instead, attempts to resolve haplotypes even in HiFi-only mode by joining phased assembly blocks to achieve greater contiguity. It produces separate FASTA files for primary and alternate assemblies. Thus, the assemblies produced by LJA, HiCANU, and Verkko are expected to have a smaller number of haplotype switches, but more fragmented than hifiasm. Since LJA, HiCANU, and Verkko are expected to resolve repeat copies from both haplotypes, their total assembly size is expected to be close to the sum of the two haplotype lengths. Instead, since hifiasm assigns phased blocks to either the primary or the alternate assembly, it is expected to incur more haplotype switch errors, and generate pseudohaplotypes that capture hap1 as primary and hap2 as alternate (or vice versa). RAmbler also prioritizes long contig generation and outputs a single FASTA file representing the primary pseudohaplotype. This assembly is expected to capture hap1, which contains the larger repeat copy count, possibly including some haplotype switches.
We used BLAST (McGinnis and Madden 2004) to align all the repeat units in hap1 and hap2 to each target assembly. We defined a repeat unit R to be resolved by an assembly A if the BLAST output indicated a perfect identity (i.e., 99.99% or higher) of R in A. Any alignment with less than perfect identity was disregarded. We also recorded whether any contig of a target assembly contained a mix of repeat units from both haplotypes, which indicated switching errors.
The experimental results are summarized in Supplemental Tables S15–S18. We omitted Verkko from the tables because it failed to produce any output in most cases. Verkko failed 92 runs out of 108 for unknown reasons (the logs were uninformative). Supplemental Tables S15 and S16 report the number of contigs and the total assembly size. Supplemental Tables S17 and S18 report the number of resolved repeat copies and the number of contigs containing haplotype switching errors. In Supplemental Tables S17 and S18, red numbers indicate incorrect repeat copy counts (neither matching either haplotype copy counts nor their sum), blue numbers indicate copy counts that match the sum of copies for both haplotypes (hap1 + hap2), and gray cells indicate assemblies with haplotype switches.
Supplemental Tables S15 and S17 show the experimental results when hap1 had 5 copies and hap2 had 3 − 5 copies. For these data sets, RAmbler produced an assembly containing 5 copies of the repeat unit in 47 cases out of 54 (87%) with eight haplotype switches. hifiasm produced the correct number of copies for hap1 in 48 cases out of 54 (89%), and the correct number of copies for hap2 in 46 cases out of 54 (85%) with 7 and one haplotype switches, respectively. RAmbler, however, always produced a single contig, while hifiasm produced two to three contigs in some cases (the average number of contigs was 1.09 for both primary and alternate assembly). LJA resolved hap1 + hap2 repeat copies in 19 cases out of 54 (35%), and sometimes only hap1 repeat copies in two cases out of 54 (4%). LJA produced more fragmented assemblies (the average number of contigs was 3.65), with a total of seven haplotype switching errors. HiCANU resolved hap1 + hap2 repeat copies in 10 cases out of 54 (19%), and sometimes only hap1 repeat copies in nine cases out of 54 (17%). HiCANU produced more fragmented assemblies (the average number of contigs was 7.49), with a total of 12 haplotype switching errors. Overall, LJA and HiCANU performed worse than RAmbler and hifiasm on these data sets, producing assemblies that were more fragmented and contained less repeat copies.
Supplemental Tables S16 and S18 summarize the cases where hap1 had 10 copies and hap2 had 6, 8, or 10 copies. RAmbler resolved 10 copies of the repeat unit in 32 cases out of 54 (59%) with 14 haplotype switching errors. The average number of contigs over all RAmbler's assemblies was 1.26. hifiasm resolved 10 repeat copies in 40 cases out of 54 (74%) with 17 haplotype switching errors. The average number of contigs over all hifiasm's assemblies was 1.06 for the primary, and 1.07 for the alternate. LJA's assemblies matched either hap1 or hap1 + hap2 in 19 cases out of 54 (35%) with 18 haplotype switching errors. The average number of contigs over all LJA's assemblies was 7.52. HiCANU's assemblies matched either hap1 or hap1 + hap2 in 16 cases out of 54 (30%) with 21 haplotype switching errors. The average number of contigs over all HiCANU's assemblies was 13.81. LJA and HiCANU, again, performed worse than RAmbler and hifiasm on these data sets.
In summary, RAmbler captured the larger repeat count between the two haplotypes in most cases. It frequently produced a single contig with a low number of haplotype switching errors, relying exclusively on HiFi reads.
Discussion
We introduced RAmbler, a reference-guided genome assembler aimed at resolving complex repetitive regions. To the best of our knowledge, there is no other specialized assembler for resolving complex repeats that uses HiFi reads exclusively. Both SDA and CentroFlye expect as input error-prone PacBio CLR or ONT long reads. SDA is no longer maintained and CentroFlye requires very high computational resources and additional information about the centromeres of interest.
RAmbler leverages unikmers to detect overlaps and locally assemble the HiFi reads. By taking advantage of shared unikmers, RAmbler can select safe and informative overlaps that are difficult to identify by traditional assemblers. Traditional HiFi assemblers rely on highly accurate (but not necessarily perfect) overlaps to build a string/overlap graph, which is a method that works very well for nonrepetitive regions of the genome. For instance, hifiasm carries out an all-pairs sequence alignment of the HiFi reads before building the string graph. Highly repetitive regions generate an overwhelming number of prefix–suffix overlaps, which are difficult to process. In contrast, the use of unikmers allows RAmbler to detect informative overlaps without the need of sifting through a very large number of sequence alignments. The use of unikmers also allows RAmbler to eliminate the need of the error-correction step to compensate for rare sequencing errors in HiFi reads, as it is done in hifiasm.
The extensive set of experiments on human Chromosomes 8, 19, and X across centromeric and noncentromeric complex repetitive regions demonstrated RAmbler's ability to achieve T2T-level assembly quality using PacBio HiFi reads exclusively, without manual intervention. Our comparative experimental results on more than 250 synthetic data sets, and on real data for H. sapiens, indicated that RAmbler outperformed hifiasm, LJA, HiCANU, and Verkko on reconstructing these repetitive regions in the majority of cases. RAmbler generated assemblies with the fewest contigs, achieving higher completeness, contiguity, and accuracy in them. Our analysis of synthetic diploid genomes with haplotype-dependent copy number illustrated that RAmbler can produce less fragmented assemblies with fewer haplotype switching errors compared to LJA, HiCANU, and Verkko by relying exclusively on HiFi reads. hifiasm performed comparably to RAmbler on synthetic data sets with fixed-length repeat units and copy number variation between the haplotypes, but its performance declined with variable-length repeats and further deteriorated on real data sets. LJA also performed well, producing assemblies with a low number of contigs, second only to RAmbler on synthetic data. However, LJA performed poorly on contiguity when the region to be assembled was longer than 1 Mb on real data. HiCANU and Verkko generated assemblies with a large number of contigs. Although HiCANU and Verkko rarely produced misassembled contigs, they suffered from poor contiguity, creating many small contigs and inflating the total assembly size. Additionally, there were instances where HiCANU failed to complete the assembly on real data sets. Similarly, Verkko failed to generate an assembly in two separate instances of H. sapiens data set and the majority of cases of synthetic diploid genomes.
RAmbler still has some limitations. In simulations, its performance drops significantly when the mutation rate P is smaller than 1/1000 (see Fig. 3A). In this case, the individual copies inside the repetitive regions are almost identical, thus there are not enough unikmers to resolve them. This observation is supported by Figure 3B, which illustrates that the number of unikmers decreases with P. However, the mutation rate in eukaryotic genomes is generally higher than one SNP over a 1000 bp (Risch 2000; International Human Genome Sequencing Consortium 2001; Orr and Chanock 2008). The current implementation of RAmbler is not multithreaded which penalizes its runtime. This is an opportunity to improve RAmbler's runtime. In addition, RAmbler generates pseudohaplotype assemblies from HiFi data, while LJA, HiCANU, and Verkko produce phased assemblies and hifiasm always attempts to produce haplotype-resolved assemblies. HiCANU, hifiasm, and Verkko support trio-based enhanced phasing. hifiasm and Verkko can use Hi-C reads to further improve haplotype resolution. Currently, our tool lacks these features, which we plan to add in future versions of RAmbler.

Relationship among RAmbler's performance, mutation rate P, and number of true unikmers. (A) Performance of RAmbler for several choices of the mutation rate P over 27 different combinations of repetitive regions with repeat sizes {10, 15, 20} kb, number of copies {2, 5, 10}, coverage depth = {20×,30×,40×}. (B) Number of true unikmers as a function of mutation rate and repeat unit's sizes (five copies).
Methods
Problem formulation
We assume that (1) the genome G contains n repetitive regions {R1, …, Ri, …, Rn}, (2) each repetitive region Ri is composed of ti tandem copies of a string αi, (3) each tandem copy has sufficient variations that allows it to be distinguished from another copy; we assume that each
copy contains SNPs with probability P (e.g., P = 1/100), and its length can increase or decrease by at most L%. Given a set T of HiFi reads and the draft genome G, the objective is to produce a set {F1, …, Fi, …, Fn} of n assemblies, where each Fi is as “similar as possible” to Ri. In particular, if the assembly Fi contains 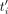 copies of the repeat unit, we want
copies of the repeat unit, we want 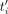 as close as ti as possible (see Supplemental Fig. S16). For synthetic data, we measure the quality of the assembly Fi by comparing it to Ri using QUAST (Gurevich et al. 2013), i.e., we report the fraction of Ri covered by Fi (ideally 100%), the number of misassembled contigs in Fi (ideally zero), and the number of contigs in Fi (ideally one). When the ground truth is unavailable (i.e., for real data sets), a qualitative assessment of the assembly's
accuracy can be obtained by aligning Fi with the corresponding repeat unit αi. The alignment, visualized as a dot plot, can provide a qualitative measure on how well the repeat units are assembled.
as close as ti as possible (see Supplemental Fig. S16). For synthetic data, we measure the quality of the assembly Fi by comparing it to Ri using QUAST (Gurevich et al. 2013), i.e., we report the fraction of Ri covered by Fi (ideally 100%), the number of misassembled contigs in Fi (ideally zero), and the number of contigs in Fi (ideally one). When the ground truth is unavailable (i.e., for real data sets), a qualitative assessment of the assembly's
accuracy can be obtained by aligning Fi with the corresponding repeat unit αi. The alignment, visualized as a dot plot, can provide a qualitative measure on how well the repeat units are assembled.
Repeat identification
To identify the repetitive regions {R1, …, Rn}, we map the HiFi reads T against the draft genome assembly G. Since unresolved tandem repeats are collapsed in the draft assembly, they can be identified by a spike in mapping coverage. For instance, Figure 4 shows the mapping coverage of an unresolved tandem repeat in Chromosome XII of S. cerevisiae which is known to contain ∼150 tandemly repeated copies of a 9.1 kb rDNA unit (Johnston et al. 1997; Kim et al. 2006). This region is the only unresolved nontelomeric gap in the current S. cerevisiae assembly.
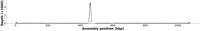
PacBio HiFi mapping coverage for Chromosome XII in S. cerevisiae illustrated using NucFreq; the coverage spike indicates the presence of a repetitive region which is known to contain ∼150 tandemly repeated copies of a 9.1 kb rDNA unit.
Analysis of k-mer distribution
Recall that we assume that the copies in the repetitive region are not identical to each other. If all the copies were identical, the problem of resolving repeats would be impossible, unless one can produce reads so long that they span the entire repetitive region. We rely on the presence of the SNPs to distinguish and partition the HiFi reads that belong to different repeat copies within a repetitive region. When distinct SNPs are present among the different copies, we expect those copies to have their own SNP signatures.
Each SNP is likely to induce a unique (or single-copy) k-mer, i.e., a k-mer that occurs a number of times approximately equal to the expected sequencing coverage. We call these k-mers, unikmers. Unikmers (called SUNKs in Logsdon et al. [2021] or SUNs in Sudmant et al. [2010]) were crucial to resolve the assembly of human Chromosome 8, but to the best of our knowledge, they have not been used in any other assembly method. Please note that unikmers are NOT k-mers that appear only once in the reads: those k-mers correspond to sequencing errors.
One of the contributions of our study is to provide a method to identify unikmers from the reads, and analyze its accuracy and precision. RAmbler finds unikmers by selecting all k-mers in the HiFi reads that have a number of occurrences within the interval [μ − tσ, μ + tσ], where μ is the average sequencing depth of the HiFi reads, σ is the standard deviation of the sequencing depth, k and t are the user-defined parameters.
We investigate how to choose k and t in the following analysis. (1) We determine the set of true unikmers in the S. cerevisiae genome to serve as the ground truth. (2) We compute the k-mer distribution for a set of real HiFi reads (obtained from the NCBI Sequence Read Archive [SRA; https://www.ncbi.nlm.nih.gov/sra] under accession number SRR13577847) and ONT reads (SRA accession SRR18365585) for S. cerevisiae using Jellyfish (Marçais and Kingsford 2011). The left column of Figure 5 shows the k-mer distribution for HiFi reads for k = 17 (Fig. 5A1), k = 21 (Fig. 5B1), and k = 25 (Fig. 5C1). Odd integers in the range [17, 25] are typical choices for k to estimate genome size (see, e.g., Vurture et al. 2017; Ranallo-Benavidez et al. 2020) or the construction of the de Bruijn graphs for eukaryotic genomes (see, e.g., Zerbino and Birney 2008). Observe that the distributions are almost identical, which indicate that any of these k-mer choices would be appropriate. (3) We compute the average sequencing depth μ and the standard deviation of the sequencing depth σ from the k-mer distribution. (4) The k-mers in the HiFi reads that have a number of occurrences within the interval [μ − tσ, μ + tσ] for t = 0, 1, 2, 3, 4, 5 are compared against the true unikmers: true positive, false positive, true negative, and false negative are recorded.
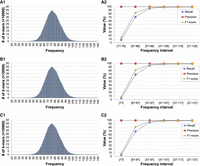
PacBio HiFi k-mer distribution for k = 17 (A1), k = 21 (B1), and k = 25 (C1) for S. cerevisiae; precision, recall, and F1-score for unikmers when k = 17 (A2), k = 21 (B2), and k = 25 (C2) for longer and longer intervals centered at the average sequencing coverage.
The results of this analysis (precision, recall, and F1-score) for HiFi reads are shown in the right column of Figure 5. The x-axis represents the choice of t, i.e., longer and longer intervals centered around the mean (the first interval is for t = 0, the second is for t = 1, etc.). Figure 5A2 shows the results for k = 17, Figure 5B2 illustrates the results for k = 21, and Figure 5C2 shows the results for k = 25. Observe that in all cases precision and recall are very close to 100% as soon as t = 3. For instance, there are 11,137,337 21-mers that occur [47 − 107] times in the HiFi reads, i.e., at most t = 3 standard deviations away from the average coverage. Of those, 11,058,290 are truly unikmers which correspond to a precision of 99.29%; only 79,047 are false positives (0.71% of the total). For t = 3, this method recalls 97.84% of the unikmers in the genome. Almost identical results can be obtained from k = 17 or k = 25. This analysis indicates that selecting k-mers that have a number of occurrences in the interval μ ± 3σ in HiFi reads can recover almost 98% of the true single-copy k-mers in the genome with a false positive rate <1%. The same analyses carried out on ONT reads show that precision, recall, and F1-score for ONT reads are slightly lower than those obtained from PacBio HiFi reads, likely due to the higher rate of sequencing errors in ONT reads (see Supplemental Fig. S17).
Based on this analysis, we used k = 21 and t = 3 for all the experiments (unless otherwise noted).
RAmbler's algorithm
The algorithm used in RAmbler is illustrated in Figure 6. It comprises of six major steps, the first two of which are data preprocessing.
-
Determine the reads corresponding to repetitive regions. As mentioned in Section “Repeat identification,” RAmbler identifies repetitive regions by mapping all HiFi reads T against the draft genome. RAmbler generates the plot of the read coverage across the genome using NucFreq (Vollger et al. 2019). Unresolved repetitive regions produce distinctive peaks in the coverage plot as illustrated in Figure 4 and Supplemental Figure S2. Then, RAmbler selects the reads that map to the repetitive regions, as well as the reads extending 50 kb upstream and downstream from the peak, as shown in Figure 6, step A. We call the set of HiFi reads extracted in this step Tr, where r identifies the repetitive region.
-
Determine unikmers. RAmbler uses Jellyfish on the entire set of HiFi reads T to obtain the count distribution of 21-mers genome-wide. RAmbler calculates the mean μ and standard deviation σ of the distribution by excluding 21-mers that appear less than 5 times since these are most likely induced by sequencing errors. RAmbler then selects the 21-mers that fall within the interval [μ − 3σ, μ + 3σ]. According to our analysis in Section “Analysis of k-mer distribution,” these 21-mers are true unikmers with high probability (Fig. 6, step B).
-
Barcode reads. RAmbler uses the set of unikmers to barcode the HiFi reads Tr (Fig. 6, step C). RAmbler searches for exact occurrences of the unikmers in the reads Tr or their reverse complement. The set of unikmers present in a read and their location is the barcode of that read. For each read, RAmbler stores pairs (u, j), where u is a unikmer and j is the location within the read.
-
Cluster the barcoded reads. RAmbler compares the barcode of all pairs of reads to identify shared unikmers. This pairwise comparison allows RAmber to determine which reads are overlapping. Two reads are overlapping if they share at least th unikmers, and the set of relative distances between the shared unikmers match within a tolerance up to to base pairs. Overlaps are stored in the barcode graph: each node in the barcode graph represents a read; nodes in the graph are connected by an edge if the corresponding reads are overlapping according to the criteria described above. Once the graph is completed, RAmbler identifies clusters of reads by determining the connected components of the barcode graph (Fig. 6, step D).
-
Assemble read clusters and build overlap graph. RAmbler carries out individual local assemblies for each set of clustered reads using a standard HiFi assembler (hifiasm in this case). Each read cluster is assembled in one or more contigs. RAmbler then uses minimap2 (Li 2018) to align assembled contigs to each other. Any contig that is fully contained within another longer contig is removed. RAmbler constructs an overlap graph based on the overlap information provided by minimap2: each node in the overlap graph represents a contig; nodes are connected by edges if they have a suffix–prefix overlap (Fig. 6, step E). It is worth noting there could be multiple suffix–prefix overlaps between a pair of contigs. RAmbler retains the overlap with the highest percentage of identity as long as the overlap is at least mo = 1000 bp. Furthermore, these suffix–prefix overlaps can occur between the positive or negative strands, resulting in three types of edges. Each edge is labeled by a pair (t, l), where t ∈ { + , − , *}, and l is the length of the suffix–prefix overlap (Supplemental Fig. S18). Given an edge (u, v) in the overlap graph, its type t is (1) “+” when there is an overlap between contig u and contig v, (2) “−” when there is an overlap between the reverse complement of contig u and contig v, and (3) “*” when there is an overlap between contig u and the reverse complement of contig v.
-
Generate consensus assembly. At this stage, RAmbler needs an estimate of the size of the repetitive region. The size is obtained by computing the ratio between the total number of bases in the reads Tr (extracted in step A) and the average coverage depth.
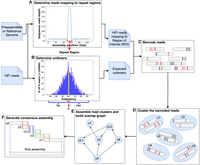
The algorithmic pipeline used in RAmbler.
To compute the final assembly, RAmbler first determines whether the overlap graph is acyclic. If it is acyclic, RAmbler enumerates all possible paths using DFS and generates a set of candidate assemblies. When computing the sequence consensus for suffix–prefix overlaps, if the suffix and the prefix do not match, RAmbler arbitrarily picks the base either from the suffix or the prefix (Fig. 6, step F). Among all assemblies, RAmbler selects the one that best matches the estimated length of the repetitive region.
When the overlap graph has cycles, RAmbler partitions the graph into three components: an acyclic precycle subgraph, the cycle itself, and a postcycle subgraph (which could be cyclic), as shown in Supplemental Figure S19. RAmbler repeats this process iteratively on the postcycle subgraph until no cycles remain. Once the graph is completely decomposed in a set of acyclic subgraphs, RAmbler generates an assembly for each subgraph as described in the previous paragraph. RAmbler then enumerates all possible combinations of partial assemblies and selects the combination such that the sum of the individual assembly's length best matches the estimated length of the repetitive region.
A summary of RAmbler's main parameters k, t, th, to, and mo with their default values is shown in Supplemental Table S13. The optimization of these parameters is discussed in Section “Synthetic data generation and parameter optimization.”
Performance metrics for real data
We used CRAQ (Clipping information for Revealing Assembly Quality) to assess the assemblies produced by RAmbler (Li et al. 2023). CRAQ utilizes both NGS short reads and long reads for identifying and classifying errors in a given draft assembly. It reports assembly errors at different scales by transforming error counts into corresponding AQIs that reflect assembly quality at both regional and structural levels. CRAQ can distinguish between assembly errors and heterozygous loci based on the ratio of mapping coverage and the effective number of clipped reads; (1) Clip-based Regional Errors (CREs): If a region with clipped NGS reads is spanned by long reads with only SNP cluster features, and (2) Clip-based Structural Errors (CSEs): if the mapped long reads around a region with errors exhibit clipping features, i.e., the NGS reads simultaneously show clipping or no coverage.
Assembly quality index (AQI)
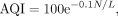 (1)
where N represents the cumulative normalized CRE or CSE count, and L indicates the total length of the assembly in the mega-base unit. Observe that a perfect assembly will yield an AQI score
of 100. To avoid excessive impacts of specific regions enriched in errors (e.g., pericentromeric regions) on the overall AQI
values, error counts were normalized within a sliding window of 0.0001*(total assembly size).
(1)
where N represents the cumulative normalized CRE or CSE count, and L indicates the total length of the assembly in the mega-base unit. Observe that a perfect assembly will yield an AQI score
of 100. To avoid excessive impacts of specific regions enriched in errors (e.g., pericentromeric regions) on the overall AQI
values, error counts were normalized within a sliding window of 0.0001*(total assembly size).
Normalized error count in a window (Nw)
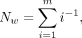 (2)
where m is the actual number of CRE/CSEs in the block. R-AQI and S-AQI can be calculated separately for CREs and CSEs.
(2)
where m is the actual number of CRE/CSEs in the block. R-AQI and S-AQI can be calculated separately for CREs and CSEs.
Performance metrics for synthetic data
Metrics such as NG50, genome fraction, number of contigs, and number of misassemblies have been traditionally employed to evaluate the quality of an assembly on synthetic data when the reference genome is known. However, each of these metrics alone does not fully capture all the desired qualities of a “good assembly.” To address this shortcoming, we introduce here a new metric called the assembly score that summarizes in one number the quality of an assembly in terms of accuracy, contiguity, and completeness. The assembly score is based on two preliminary metrics, as explained next.
Effective genome fraction per contig (ζ)
Consider an assembly that consists of a single contig but contains one misassembly. To correct the misassembly, the contig
needs to be broken, resulting in the creation of an additional contig. Based on this observation we define the effective number of contigs as the sum of the number of contigs in the assembly and the number of misassemblies. We propose to calculate the effective
genome fraction per contig as follows: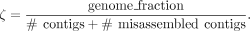 (3)
(3)
As said, metric ζ takes into account the number of misassemblies and penalizes the score accordingly. An assembly that covers 100% of the reference genome (without any misassemblies) would yield ζ = 1.0. By considering the effective genome fraction per contig, we can assess the assembly quality while accounting for the presence of misassemblies, thereby providing a more comprehensive evaluation.
Normalized NG50 (η)
While NG50 is an essential metric for evaluating the contiguity of an assembly, it depends on the size of the reference genome,
making it challenging to use it to compare an assembler's performance across genomes of different lengths. To address this
limitation, we normalize NG50 by the size of the reference genome, yielding a metric called η defined as follows: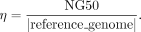 (4)
(4)
By normalizing NG50 with respect to the reference genome size, η is constrained within the range of [0, 1], with a perfect assembly achieving η = 1. Observe that it is possible that η may exceed 1 when the assembly is overinflated, i.e., longer than the actual genome. In general, a higher value of η indicates a better assembly quality, as long as it is smaller than 1.
Assembly score
The assembly score is defined by computing the harmonic mean of ζ and η, as follows: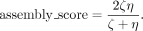 (5)
(5)
Observe that while ζ is always within [0, 1], η can exceed 1, which can result in an assembly score greater than 1. Nevertheless, an assembly score closer to 1 indicates a higher quality assembly. This score enables a holistic assessment of the assembly's quality, taking into account accuracy, contiguity, and completeness, which are all equally important.
Software availability
RAmbler is available from GitHub (https://github.com/ucrbioinfo/rambler) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This project was supported in part by National Science Foundation (NSF) Information and Intelligence Systems (IIS) #2444456, NSF Chemical, Bioengineering, Environmental, and Transport Systems (CBET) #2225878, and National Institutes of Health (NIH) 1-R01-AI169543-01 to S.L. The authors thank the anonymous reviewers who helped to improve the quality of this manuscript.
Author contributions: S.C., G.L., and S.L. conceptualized and designed the study; S.C. wrote and tested the code; G.L. provided guidance on the experimental design on the human genome; S.C. carried out the experiments and generated figures and tables; S.C. and S.L. wrote the manuscript; all authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279308.124.
-
Freely available online through the Genome Research Open Access option.
- Received March 13, 2024.
- Accepted February 6, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








