
Candida albicans isolates contain frequent heterozygous structural variants and transposable elements within genes and centromeres
Abstract
The human fungal pathogen Candida albicans poses a significant burden on global health, causing high rates of mortality and antifungal drug resistance. C. albicans is a heterozygous diploid organism that reproduces asexually. Structural variants (SVs) are an important source of genomic rearrangement, particularly in species that lack sexual recombination. To comprehensively investigate SVs across clinical isolates of C. albicans, we conducted long-read sequencing and genome-wide SV analysis in three distantly related clinical isolates. Our work includes a new, comprehensive analysis of transposable element (TE) composition, location, and diversity. SVs and TEs are frequently close to coding sequences and many SVs are heterozygous, suggesting that SVs might impact gene and allele-specific expression. Most SVs are uniquely present in only one clinical isolate, indicating that SVs represent a significant source of intraspecies genetic variation. We identify multiple, distinct SVs at the centromeres of Chromosome 4 and Chromosome 5, including inversions and transposon polymorphisms. These two chromosomes are often aneuploid in drug-resistant clinical isolates and can form isochromosome structures with breakpoints near the centromere. Further screening of 100 clinical isolates confirms the widespread presence of centromeric SVs in C. albicans, often appearing in a heterozygous state, indicating that SVs are contributing to centromere evolution in C. albicans. Together, these findings highlight that SVs and TEs are common across diverse clinical isolates of C. albicans and that the centromeres of this organism are important sites of genome rearrangement.
Genomes are under constant evolutionary pressure from environmental stressors. Populations with higher amounts of standing genetic variation, including single-nucleotide polymorphisms and structural variants (SVs), adapt more quickly than those with lower standing genetic variation (Feurtey et al. 2023). SVs consist of deletions, insertions, translocations, duplications, inversions, and complex rearrangements, including chromosomal fission and fusion events. A special type of SVs are transposable elements (TEs), mobile genomic sequences that can autonomously create new copies of themselves. SVs can induce reorganization of the genome resulting in the disruption, loss, reordering, or duplication of genes (Hartmann et al. 2017; Yang et al. 2019; Fouché et al. 2023). SVs can influence gene expression, for example by the insertion or deletion of regulatory elements upstream of a gene (Butelli et al. 2012). Deleterious SVs are typically eliminated from populations by strong purifying selection (Elyashiv et al. 2010). SVs that are neutral or slightly deleterious can persist in a population for a long period of time via genetic drift (Berdan et al. 2021; Hartmann 2022). Occasionally, SVs are advantageous in a specific environment even when they are heterozygous (Lucek et al. 2019; Hämälä et al. 2021; Massonnet et al. 2022). Several examples of SVs caused by TE activity highlight how genome structure and function are strongly altered via SVs. For example, TE insertions in the promoter region of a gene encoding a major facilitator superfamily protein in a plant fungal pathogen led to increased resistance to antifungal drugs (Omrane et al. 2015, 2017). Similarly, in the same pathogen, chromosomal rearrangements likely caused by TEs led to an advantageous deletion of a gene encoding for an effector, which was likely recognized by the host (Hartmann et al. 2017). Overall, SVs encompass diverse recombination mechanisms, including TE mobilization or homologous repair following DNA-double-stranded breaks (Todd et al. 2019, Berdan et al. 2021).
The human fungal pathogen Candida albicans is an asexual diploid organism that displays significant genomic plasticity (Vande Zande et al. 2023). SVs are important drivers of genome evolution and clinical isolates contain chromosome-level polymorphisms leading to karyotype variability (Rustchenko-Bulgac 1991; Navarro-García et al. 1995; Selmecki et al. 2005). Stress increases genome plasticity in C. albicans (Forche et al. 2011, 2018). For example, antifungal drug exposure, oxidative stress, and elevated temperature increase the rate of loss of heterozygosity (Forche et al. 2011, 2018). Indeed, nearly 50% of azole drug-resistant clinical isolates have one or more aneuploidy (Selmecki et al. 2006). The most common aneuploidy across diverse isolates is a duplication of the left arm of Chr 5 in an isochromosome structure, called i(5L) (Selmecki et al. 2006; Butler et al. 2009; Hickman et al. 2013; Harrison et al. 2014; Ford et al. 2015; Ropars et al. 2018). i(5L) contains two genes involved in antifungal resistance, ERG11, encoding the azole drug target, and TAC1, encoding a transcriptional activator of drug efflux pumps (Vanden Bossche et al. 1994; White et al. 1998; Selmecki et al. 2008). Recently, we identified an isochromosome that contains two copies of the right arm of Chr 4, i(4R), that also provides a fitness benefit in the presence of azole drugs (Todd et al. 2019). These isochromosomes arise in diverse genetic backgrounds of C. albicans during adaptation to azole drugs (Selmecki et al. 2009; Todd and Selmecki 2020; Todd et al. 2023). Importantly, the breakpoints of both i(5L) and i(4R) are located near the centromere of each respective chromosome (Selmecki et al. 2006; Todd et al. 2019). These two centromeres contain long inverted repeat sequences that are likely involved in nonallelic homologous recombination resulting in the isochromosome structure (Sanyal et al. 2004; Burrack et al. 2013; Todd et al. 2019).
Centromeres faithfully segregate chromosomes during cell division. Within the fungi, centromeres range from point centromeres in Saccharomyces cerevisiae, to epigenetically defined regional centromeres that range from short (<20 kb) to long (>20 kb, Cryptococcus neoformans) (Meraldi et al. 2006). C. albicans has short (∼3–4.5 kb), regional, epigenetically marked centromeres, composed of a central core sequence surrounded by pericentromeric regions (Sanyal et al. 2004; Baum et al. 2006; Mishra et al. 2007; Ketel et al. 2009; Koren et al. 2010; Tsai et al. 2014; Chatterjee et al. 2016; Freire-Benéitez et al. 2016). The central core sequence is dominated by the presence of the centromere-specific histone H3 variant Cse4p/CENPA, while the pericentric sequence is dominated by normal histone H3 (Burrack et al. 2011). Centromeres frequently harbor repetitive sequences and complex chromatin structures that make the centromere especially susceptible to double-stranded breaks and erroneous repair (Croll et al. 2013). In many organisms, centromeres show sequence similarity to each other and contain centromere-specific repeats (Talbert et al. 2004). However, the eight centromeres of C. albicans lack conserved sequences and have likely undergone multiple rearrangements (Sanyal et al. 2004; Guin et al. 2020). Several centromeric regions in C. albicans and C. dubliniensis contain unique inverted repeats (Padmanabhan et al. 2008; Burrack et al. 2016; Todd et al. 2019). Given the repeats and erroneous repair around centromeres, we hypothesize that there is undetected variation in the centromeric regions of C. albicans that might further contribute to chromosome and genome plasticity of C. albicans.
The TE content of C. albicans and other yeast species is low compared to other eukaryotes (Maxwell 2020; Wells and Feschotte 2020). Sixty-three TE families or TE fragments (i.e., solo-long terminal repeats [solo-LTRs]) were previously identified in C. albicans in an earlier and more fragmented version of the reference genome (Goodwin and Poulter 2000; Goodwin et al. 2001). Most TE families have only a few copies in the current version of the reference genome, and a few of the previously reported families are not detectable in the current version. C. albicans TE families include DNA transposons and retrotransposons with LINE and LTR-retrotransposons (Wicker et al. 2007). LTR-retrotransposons contain LTR on each side of the element, which can undergo ectopic recombination, removing the majority of the retrotransposon and leaving only a solo-LTRs (Devos et al. 2002). There are 36 described solo-LTRs in C. albicans, 21 of which are not associated with an existing full-length LTR-retrotransposon (Goodwin and Poulter 2000; Goodwin et al. 2001). The presence of solo-LTRs indicates a previously active full-length element, but solo-LTRs themselves are not fully functional or active anymore. The DNA transposon Cirt2 and the LTR-retrotransposon Tca2 are expressed in C. albicans (Matthews et al. 1997; Potocki et al. 2019). However, it is not clear whether TE expression is sufficient to generate new insertions, or if new TE insertions can remain over longer periods of time. Regardless, TEs do remain in the species, and it is not clear what role TEs play in this species. Importantly, no prior study has used long reads to determine precisely how many TE copies exist, where TEs are located in different clinical isolates, and how TEs might impact the genome structure of C. albicans.
In addition, the mechanisms and sources of SVs are understudied. Conventional methods of SV characterization rely on extensive molecular experiments, which are limited to a few loci at the time, or short-read sequencing alignments that do not reliably detect most SVs (Mahmoud et al. 2019). Short-read sequencing data underestimates SVs longer than the read length and has a limited ability in determining the genomic positions, zygosity, or allele frequencies of SVs. Additionally, mapping of short-read data to the reference genome has further limited the ability to determine the precise location and structure of many SVs, especially those found multiple times in the genome, like TEs.
Here, we describe the first long-read sequencing analysis of three diverse clinical isolates of C. albicans to assess the genome-wide distribution and zygosity of SVs. We combine multiple analysis tools to improve the detection and confirmation of SVs, TEs, and TE remnants as well as the zygosity state, with a focus on the pericentromeric regions.
Results
De novo long-read genome assembly indicates the presence of large-scale C. albicans rearrangements
To conduct genome-wide analyses of SVs, we generated Oxford Nanopore Technologies long-read sequencing data for the reference isolate SC5314, along with two additional clinical isolates, L26 and P75063. These isolates are euploid and comprise two distantly related C. albicans clades (SC5314 and L26 in clade I and P75063 in clade SA) (Hirakawa et al. 2015). Well-characterized phenotypic data and Illumina short-read genome sequences are available for these isolates (Hirakawa et al. 2015). Furthermore, these isolates frequently acquire large DNA amplification events during adaptation to antifungal drug stress in vitro (Todd et al. 2019, 2023). To determine genome completeness, we conducted de novo genome assemblies. The de novo assemblies of SC5314, L26, and P75036 were fragmented into 37, 38, and 48 contigs with N50 of 1.16 Mb, 1.83 Mb, and 1.03 Mb, respectively (Supplemental Fig. S1A). The length of the largest contigs in SC5314 and L26 corresponded to the Chr 1 length described in the reference genome. We assessed the completeness of the genome assemblies using Benchmarking Universal Single-Copy Orthologs (BUSCO) (Simão et al. 2015; Manni et al. 2021). Of the 2137 Saccharomycotina BUSCO genes, 95.2%, 96.2%, and 96.0% were present in SC5315, L26, and P75036, respectively, indicating a high level of completeness (Supplemental Fig. S1B). Comparing the contigs to the reference genome showed large contiguity, with some potential chromosomal rearrangements and duplications found mostly in the subtelomeric regions (Supplemental Fig. S1C). These de novo assemblies provide a first overview of potential large-scale rearrangements and completeness.
Long-read sequencing reveals the frequency of structural variants
Next to de novo assembly, long-read sequencing provides an opportunity to detect SVs through mapping to a reference genome. We mapped the long reads to the reference genome SC5314 and identified genome-wide coverage differences based on the separate MinION runs (Supplemental Fig. S2A). After mapping the raw reads to the reference genome, we used DELLY and TELR for SV and TE detection and filtered for structures larger than 50 bp, and identified extensive SV and TE polymorphism among all three isolates (Fig. 1) (690 SVs in SC5314; 679 SVs in L26; and 864 SVs in P75063). SVs and TEs that were present on only one homolog were defined as heterozygous, and SVs and TEs that were present on both homologs were defined as homozygous (Fig. 1B). SVs were distributed throughout all chromosomes. Most SVs were insertions (INS; n = 877), followed by deletions (DEL; n = 662), translocations to another chromosome (TRA; n = 126), inversions (INV; n = 11), and duplications (DUP; n = 9) (Supplemental Table S4). Almost all SV were heterozygous in SC5314 (95.5%), and a large number of SVs were heterozygous in L26 (75.7%) and P75063 (63.2%) (Figs. 1A, 2A). To confirm that genome coverage had no impact on SV detection, we repeated SV detection on downsampled input, with reduced number of reads. Downsampling indicated that the coverage was sufficiently high for a reliable SV detection, even for L26 with its lower coverage (Supplemental Fig. S2B). Most SVs were smaller than 100 bp, with the exception of TEs that were 500 bp or larger (Supplemental Fig. S2C). Short SVs were mostly insertions or deletions. The GC content of short SVs (<100 bp) was variable and similar to the genome-wide GC content distribution (Supplemental Fig. S2D). Out of 927 short SVs, seven include homopolymers, and 87 include short tandem repeats. Additionally, short SVs are enriched at replication origins (Supplemental Fig. S2E), confirming previous reports of SC5314 (Muzzey et al. 2013).

Long-read sequencing reveals dynamic genome-wide SVs in three clinical isolates. (A) Genome-wide genomic landscape: Gene content and TE content in 5000 bp windows of the C. albicans reference genome. Distribution of SVs in SC5314, L26, and P75063. Colors indicate the SV type in comparison with the reference genome: green—(TE) transposable element; red—(DEL) deletion; blue—(INS) insertion; gray—other types of SV (inversions, duplications, translocations). The fill represents zygosity (filled symbol = homozygosity, unfilled symbol = heterozygosity). Centromeres are marked with white notches and the mating type locus with a red notch in Chr 5. (B) Schematic example of heterozygous and homozygous SV definitions.
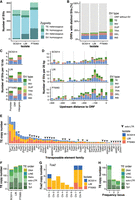
Genome-wide distribution of SVs. (A) Zygosity of SVs and TEs. The bar plot represents the copy number and zygosity for SVs and TEs in the three isolates. (B) Open reading frames (ORFs) close to SVs and TEs: Color indicates the percentage of ORFs that either overlap with at least one SV or TE, or that have at least one SV or TE in a 500 bp window upstream or downstream from the ORF. Gray indicates that no SV or TE is present. Color by SV type or TE: (TE) transposable element, (INV) inversion, (DUP) duplication, (TRA) translocation from another chromosome, (DEL) deletion, (INS) insertion. (C) SVs and TEs by distance to the ORF: location in the promoter region (500 bp upstream of the ORF), overlapping with the ORF, or intergenic. Color by SV type or TE. Corrected by the number SV per 100 kb, as the three different regions cover space of different lengths. (D) Fine-scale SV and TE distribution in the promoter region (500 bp upstream of the ORF). Color by SV type or TE. (E) Frequency of TE families between the three isolates. A triangle indicates this element is a solo-LTR. The sequences and their corresponding full-length elements can be found in Supplemental Table S1. (F) TE copy numbers in the three clinical isolates. Colors indicate the classification (Ty1 and Ty3 are superfamilies in the LTR-retrotransposons; DNA and retrotransposon are on the class level; LINE is the order of non-LTR-retrotransposons; solo-LTR are fragments of Ty1 or Ty3, and unknown retrotransposons). (G) Copy number of the Tca2 transposon on each of the C. albicans chromosomes. Colors indicate the isolate. (H) Frequency distribution of TE loci that are identical in location between the three isolates. Colors indicate the classification.
We detected and quantified TEs independently of SVs. We found more than 75% of all TEs to be heterozygous in the isolates: SC5314 (78.3%), L26 (70.9%), and P75063 (69.6%) (Figs. 1A, 2A); 20.1% (59 TEs) of all TE loci were identical in location across all three isolates, while another 19.4% (57 TEs) were only shared between the closely related isolates SC5314 and L26 (Supplemental Fig. S2F). The high number of shared TE loci between distantly related isolates was unexpected and likely indicates that most TE insertions are ancient, at least in a heterozygous state. Shared TEs are likely identical by descent, but might have diverged via mutation accumulation. Thirty-five percent of all TE loci were only detected in the more distantly related isolate P75063, indicating that some TEs might still be active (Supplemental Fig. S2F). Overall, the fact that low numbers of SVs are shared even between the closely related SC5314 and L26 indicates that SVs contribute significant sequence diversity between isolates and SV formation is ongoing.
Structural variants are close to open reading frames
We next quantified the frequency of SVs within coding sequences and promoter regions. The C. albicans reference genome is gene-dense compared to other fungal species, with a mean distance of 824 bp between annotated ORFs (Supplemental Fig. S2G). Given the short distance between ORFs, we defined the promoter region to be 500 bp upstream of the start codon. We scanned all annotated ORFs including a window 500 bp upstream and 500 bp downstream for overlaps with at least one SV or TE. Only 16.6% of promoter regions and ORFs contained SVs or TEs (Fig. 2B). SVs and TEs in the promoter region and overlapping ORFs are predominantly heterozygous and present on only one homolog (98.7% for SVs and 91.0% for TEs in SC5314; 71.2% for SVs and 83.0% for TEs in L26; 60.6% for SVs and 73.5% for TEs in P75063).
To determine the density of SVs and TEs in genomic regions, we corrected for the length of the actual chromosomal regions defined as intergenic regions, ORFs, and promoter regions. We found the highest SV and TE density in the intergenic regions, followed by the promoter region and then the ORF (Fig. 2C). Due to the high gene density in C. albicans, SVs and TEs are generally close to ORFs, especially in the 200 bp upstream region (Fig. 2D). We tested the 75 genes with a TE in the promoter or coding region for enrichment in Gene Ontology (GO) terms against a background of all C. albicans genes. We found a significant enrichment for the molecular function “3′–5′ RNA helicase activity” (P-value 0.02918). Several other genes encoding proteins involved in transport, infection, and resistance to antifungal drugs contained TEs in their promoter regions, including ISC1, NSP1, SEC18, BNI4, CDC3, DYN1, RHO2, and MRE11; however, these processes were not significantly enriched. The presence of TEs in the promoter region can have a significant impact on gene expression, resulting in up- or downregulation of the gene. Only a few TEs overlap with ORFs (4 in SC5314, 7 in L26, and 5 in P75063). One of these, FMA1, encodes a putative oxidoreductase that is associated with fluconazole resistance (Rogers and Barker 2003). FMA1 contains a heterozygous insertion of Tca7 in SC5314 and the corresponding Tca7-derived solo-LTR zeta in L26. These TEs likely disrupt the function of FMA1, but the TE copy in L26 subsequently underwent ectopic recombination. SVs and TEs are likely deleterious when located in an ORF or promoter region, but the effect could also be beneficial and generate novel functionality.
Indication of recent TE activity
To estimate recent TE activity, we fine-tuned our insertion and deletion polymorphism analysis, as well as the zygosity state of TEs. Within the three isolates, we detected 480 TE copies belonging to 51 TE families (out of the 63 families previously identified in C. albicans) (Goodwin and Poulter 2000; Goodwin et al. 2001; Fig. 2E). One hundred and fifty-three of the full-length TEs belonged to the superfamily Ty1, followed by unclassified retrotransposons (n = 94), Ty3 (n = 30), LINE (n = 20), and DNA transposons (n = 19) (Fig. 2F). One hundred and sixty-four TE copies were solo-LTRs, either belonging to Ty1 or Ty2 (Fig. 2E,F). Solo-LTRs are fragments that remain after ectopic recombination of the two LTRs of a retrotransposon (Supplemental Fig. S5D). The highest number of TEs was detected in the isolate P75063 (n = 173), followed by L26 (n = 164) and SC5314 (n = 143). P75063 also had the highest number of solo-LTRs (n = 78) compared to SC5314 (n = 45) and L26 (n = 41). L26 had an increased number of Tca2 retrotransposons (n = 34) compared to SC5314 which belongs to the same clade (n = 6) and P75063 (n = 5). Copies of Tca2 were present on every chromosome in L26, while Tca2 copies were restricted to Chr 1, Chr 7, and Chr R in SC5314 and P75063 (Fig. 2G). The corresponding solo-LTRs gamma from Tca2 were not detected in any isolate. Because new TE insertions are perfect copies and mutations only slowly accumulate over time, a high sequence similarity between TE copies indicates recent activity. To determine sequence similarity, we conducted multiple sequence alignments with all Tca2 copies from the three isolates. Overall, all Tca2 copies showed high sequence similarity of more than 99.5%, ranging between 0 and 14 single-nucleotide polymorphisms compared to the Tca2 consensus sequence (Supplemental Fig. S3A,B). Our data indicate recent activity of Tca2 in the species, yet it remains unclear why and how Tca2 increased copy numbers in L26 and not in the other two isolates.
We next tested if any TE copies were identical in location and likely ancestral. TEs were clustered into 294 individual loci, of which 167 were only present in one isolate, 68 were shared between two isolates, and 59 were shared by all three isolates (Fig. 2H). DNA transposon loci generally were present in all three isolates. We found the distance to the closest ORF to be highly variable between TE families with a range from 0 to 5282 bp (Supplemental Fig. S3C). The same TE families almost always had a similar range of distance to the closest ORF. Yet, while copies of Tca2 were located close to genes in SC5314 and P75063 (range 0–667 bp, mean 103 bp), the copies in L26 showed a significantly higher distance (range 0–942 bp, mean 221 bp). This indicates a relaxed selection on new TE copies inserted into gene-poor niches. Once activated, several new TE insertions may emerge. Increases of copy numbers of single TE families indicate ongoing and autonomous TE activity, yet at a much smaller scale compared to other fungal species where bursts can create hundreds to thousands of new copies (Badet et al. 2020; González-Sayer et al. 2022).
Heterozygous inverted centromere orientation and evidence of transposon activity at CEN4 and CEN5
We next focused on SVs and TEs that are in the pericentromeric regions. We detected SVs and TEs in six of the eight pericentromeric regions, although only three (Chr 2, Chr 4, and Chr 5) contained TEs or TE fragments in or very close to the central core region (Supplemental Fig. S4A). Chr 2 had a heterozygous solo-LTR epsilon in the central core region in all three isolates. Chr 4 had a Cirt2 DNA transposon in the pericentromeric region, which was homozygous in SC5314 and L26, but heterozygous in P75063. This Cirt2 insertion corresponds to the annotated ORF orf19.3830, and is located only 51 bp from the central core region of CEN4 (Freire-Benéitez et al. 2016). Chr 5 had a heterozygous solo-LTR psi in the central core region in both SC5314 and P75063 that was not detected in L26. To validate SV and TE calls in the pericentromeric regions, we generated ribbon plots from the long reads mapped to the reference genome for these regions. Chr 4 and Chr 5 were unique among all other chromosomes in that their pericentromeric regions contained additional, larger SVs including large inversions covering the central core regions of CEN4 and CEN5 (Supplemental Fig. S4B).
The pericentromere of Chr 4 contained multiple SVs between the isolates. All SC5314 reads mapped to the CEN4 region in the reference orientation and showed no indication of SVs (Fig. 3A). In L26, only half of the reads mapped to CEN4 in the reference orientation and the other half contain an inversion of the CEN4 sequence (Fig. 3B). In P75063, all reads mapped to the CEN4 reference orientation, however, approximately half of the reads contained a 523 bp insertion upstream and a 1824 bp deletion downstream from the CEN4 sequence compared to the reference genome (Fig. 3C; Supplemental Fig. S5A). The same long reads carrying the insertion were also carrying the deletion, indicating that both SVs were phased on the same homolog. When inspecting the sequences, we found that both the insertion and the deletion correspond to TEs described in the literature in C. albicans (Goodwin and Poulter 2000; Goodwin et al. 2001). The insertion polymorphism belongs to episemon. Episemon is a solo-LTR with no described full-length retrotransposon in C. albicans, and episemon only had one copy in SC5314 and L26 in the pericentromeric region of Chr 5, yet 4 additional copies in different genomic locations in P75063. The deletion contains the annotated orf19.3820 and corresponds to the full-length DNA transposon Cirt2. Cirt2 belongs to the superfamily Tc1-Mariner and contains a transposase, 46 bp terminal inverted repeats, and generally contains “TA” target site duplications upon insertion (Supplemental Fig. S5B). We detected Cirt2 to be heterozygous present in both SC5314 and L26. A potential explanation for the absence of Cirt2 in one allele could be mobility, with the element excising and inserting elsewhere in the genome. Indeed, our TE screen identified another copy of Cirt2 in the right arm of Chr 4 and contained the annotated orf19.2866 (Supplemental Fig. S5C). This second copy shared a 99.89% sequence identity with the CEN4 proximal copy. Long-read analysis showed that the second copy was present in a homozygous state in SC5314 and P75063 and a heterozygous state in L26.
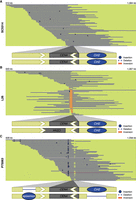
SVs in the pericentromere of Chr 4 in clinical isolates. Representative long reads aligned to CEN4 in the C. albicans reference genome for (A) SC5314, (B) L26, and (C) P75063. Binding of the centromere-specific histone H3 variant Cse4p/CENPA delineates the central core sequence of the centromere and is indicated with a gray box (Sanyal et al. 2004; Ketel et al. 2009). Blue lines indicate reads in the same orientation as the reference genome, while red reads indicate an inverted orientation compared to the reference genome. Insertions and deletions compared to the reference genome are denoted with blue dots or a thinner blue line, respectively. Insertions that are not shared by more than one reads are considered to be sequencing errors. The schematic at the bottom represents a model of the CEN4 structure (lengths not to scale). Blue circles indicate the presence of a TE. White arrows indicate inverted repeat sequences identified previously (Selmecki et al. 2006; Todd et al. 2019).
The pericentromere of Chr 5 contained a similar set of SVs in the three isolates. For SC5314, half the reads mapped to the CEN5 reference sequence while the other half reads contained two insertions compared to the reference genome (Fig. 4A; Supplemental Fig. S6A). The first 523 bp insertion is upstream of CEN5 and the second 470 bp insertion is located within the Cse4p binding site in CEN5. We found that both insertions were solo-LTRs belonging to two different retrotransposon TE families. The CEN5-adjacent solo-LTR belonged to the episemon family with no described full-length TE family in C. albicans. The CEN5 episemon is the only episemon locus that is shared between all three isolates. The second insertion located in CEN5 belonged to the psi family, the solo-LTR of the Tca9 TE family (Selmecki et al. 2006). Genome-wide, we detected 3, 2, and 7 loci of psi in SC5314, L26, and P75063, with the CEN5 psi copy as the only shared copy. In each isolate, we found three copies of the full-length element Tca9, yet none of the sequences overlapped with the pericentromeric or central core region. Both solo-LTRs are heterozygous and were phased on the same long reads in all three isolates (Supplemental Fig. S6B). All reads from L26 mapped to CEN5 in an inverted orientation, and half of the reads had the same insertions as SC5314 that belonged to episemon and psi (Fig. 4B). In P75063, the reads mapped to CEN5 had either the two insertions episemon and psi or had an inversion (Fig. 4C). Overall, short solo-LTRs seem to be persistent in the central core and pericentromeric regions, yet only in heterozygous form, while the larger DNA transposon Cirt2 was able to persist in a homozygous form.
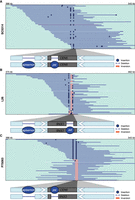
SVs in the pericentromere of Chr 5 in clinical isolates. Representative long reads aligned to CEN5 in the C. albicans reference genome for (A) SC5314, (B) L26, and (C) P75063. Binding of the centromere-specific histone H3 variant Cse4p/CENPA delineates the central core sequence of the centromere and is indicated with a gray box (Sanyal et al. 2004; Ketel et al. 2009). Blue lines indicate reads in the same orientation as the reference genome, while red reads indicate an inverted orientation compared to the reference genome. Insertions and deletions compared to the reference genome are denoted with blue dots or a thinner blue line, respectively. Insertions that are not shared by more than one reads are considered to be sequencing errors. The schematic at the bottom represents a model of the CEN5 structure (lengths not to scale). Blue circles indicate the presence of a TE. White arrows indicate inverted repeat sequences identified previously (Selmecki et al. 2006; Todd et al. 2019).
Structural variants in the pericentromeric region are frequent in clinical isolates
To determine the frequency of SVs across diverse clinical isolates, we designed PCR primers that captured the orientation and TE presence in the pericentromeric regions of CEN4 and CEN5 (Fig. 5A,E; Supplemental Table S3). Then, we screened 100 C. albicans clinical isolates from different patients, body sites, geographic regions, and hospitals for the CEN4 and CEN5 SVs (Supplemental Table S2). For CEN4, we confirmed the heterozygous inversion in L26 (Fig. 5B). This heterozygous inversion was shared with 41% of the clinical isolates, while the other isolates were homozygous for the reference orientation. No instance of homozygous inversion was detected. We confirmed the homozygous presence of the pericentromeric Cirt2 in SC5314 and L26, while the heterozygous presence in P75063 could not be confirmed, only the absence was confirmed. In the 100 clinical isolates, 41% had Cirt2 homozygous and 9% heterozygous, while the element was absent in 50% of isolates. We confirmed the homozygous presence of the Chr 4 right arm copy of Cirt2 in SC5314 and P75063 while the heterozygous presence in L26 could not be confirmed, again only the absence was confirmed (Fig. 5D). In the 100 clinical isolates, 11% had the right arm copy of Cirt2 homozygous state and 7% heterozygous state, while the element was not present in 82% of isolates. We did not find a correlation between the presence or absence of polymorphism of the CEN4 proximal and subtelomeric Cirt2 copies in the 100 isolates. Likely these are independent copies (Supplemental Fig. S7A,B).
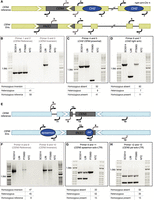
Confirmation of centromere inversions and TE polymorphisms. Diagnostic PCR of CEN4 and CEN5 and representative strains. The table below each EtBr-stained DNA gel indicates the PCR results from 100 compiled clinical isolates. A full visualization of the 100 clinical isolates is available in Supplemental Figure S7. (A) Schematic of the primers for CEN4. Cse4p/CENPA binding delineates the central core sequence of the centromere and is indicated with a gray box (Sanyal et al. 2004; Ketel et al. 2009). (B) CEN4 inversion is indicated by primer pair 1 and 2 for a reference orientation, and primer pair 3 and 2 for an inversion. (C) CEN4 pericentromeric Cirt2 (orf19.3820) presence is indicated by the primer pair 4 and 5. (D) Right arm Chr 4 Cirt2 (orf19.2866) presence is indicated by the primer pair 6 and 7. (E) Schematic of the primers for CEN5. (F) CEN5 inversion is indicated by primer pair 9 and 8 for a reference orientation, and primer pair 8 and 10 for an inversion. (G) CEN5 solo-LTR episemon presence is indicated by the primer pair 9 and 11. (H) CEN5 solo-LTR psi presence is indicated by primer pair 12 and 13. All primers are described in Supplemental Table S3.
For CEN5, we confirmed the CEN5 reference orientation in SC5314, the homozygous inversion orientation in L26, and both orientations in P75063 (Fig. 5F). We confirmed the heterozygous presence of the solo-LTR episemon in SC5314 and L26 and homozygous presence in P75063 (Fig. 5G). In the 100 clinical isolates, 33% lacked an episemon solo-LTR, while 52% had a heterozygous and 15% a homozygous presence. In the 100 clinical isolates, 47% had a homozygous inverted orientation, 8% a homozygous reference orientation, and 45% a heterozygous orientation. Finally, we confirmed the heterozygous presence of the psi solo-LTR (Fig. 5H). In the 100 clinical isolates, 35% had a heterozygous presence for psi, 15% had a homozygous presence, and 50% showed an absence of psi. The CEN5 proximal mating type locus was heterozygous in 83% of the isolates, while 6% of the loci were homozygous for MTLa/a and 11% homozygous for MTLα/α (Supplemental Fig. S7C). The mating type locus and the pericentromeric SVs do not correlate and likely recombine independently (Supplemental Fig. S7D). Both SVs and TEs appeared to persist in a heterozygous state in the population. One limitation of PCR was that heterozygous TEs were difficult to capture either because one PCR product preferentially amplified better than the other (typically, the smaller PCR product Fig. 5C,D, P75063 and L26, respectively) or that PCR was detecting active TE insertion/deletion events within the overnight cultures. Therefore, for the additional clinical isolates we reported simply whether a deleted TE allele amplified or not. By focusing on six SVs in two chromosomes, we were able to detect a high variability in presence, absence, and zygosity. These six SVs seem to persist over longer evolutionary time-scales, but can also easily be lost in either one or both copies. Overall, we detected several SVs and TEs with a potential influence on chromosome stability and gene expression. Our long-read sequencing and molecular validation support that SVs and TEs are frequently associated with pericentromeres in diverse C. albicans isolates. However, future experiments are needed to address the effect of SVs on C. albicans genomic plasticity and evolvability.
Discussion
SVs, and TEs in particular, consist of a spectrum of genomic rearrangements, including insertions, deletions, duplications, inversions, and translocations. SVs play a crucial role in driving genome evolution, especially in asexual organisms like C. albicans. Due to challenges with SV detection based on short reads, they are understudied in many species. Long-read sequencing offers an exciting opportunity to detect SVs and their impact on evolution with better reliability. In this study, we provide the first analysis of SVs and TEs in three clinical isolates of C. albicans using long-read sequencing data. We detected several SVs and TEs with a potential influence on chromosome stability and gene expression. Our specific focus on centromeres offers a unique perspective on the impact of SVs on chromosomal instability, given their important role in chromosome segregation and genome stability. We were able to detect a high variability in presence, absence, and zygosity at centromere regions. Centromere SVs seem to persist over longer evolutionary time-scales, but can also easily be lost in either one or both copies. More experiments are needed to address the effect of SVs on C. albicans genomic plasticity and evolvability as a fungal pathogen.
Long-read sequencing improves our understanding of SV dynamics and heterozygosity
The use of long-read sequencing technologies allowed us to detect a larger number of high-quality SVs and TEs than previously described. We frequently found individual SVs present in only one or two isolates, and approximately half of all SVs were heterozygous. In contrast, we found a large number of TEs identical in location across all three isolates, yet almost always in the heterozygous state. In other species, heterozygous SVs and TEs can be deleterious, as they hinder sexual recombination (Homolka et al. 2007), and consequently, most TE loci are strongly selected against, and remain at low frequencies in the species (Stritt et al. 2018; Oggenfuss et al. 2021). This may not hold true in C. albicans, as this species lacks a sexual cycle, and heterozygous SVs might not have a negative influence, but rather increase the genomic plasticity and increase the potential for rapid adaptation. The heterozygous presence of SVs across the genome could maintain a high level of genetic variation in the absence of meiosis. Notably, C. albicans copy number breakpoints colocalize with SVs and repetitive regions, further suggesting that the presence of heterozygous SVs could lead to chromosomal rearrangements (Todd et al. 2019). SVs and TEs in particular might remain over longer periods of time by genetic drift. Given the consistent presence of many TE loci, TEs cannot always be considered to be SVs in C. albicans. Instead, these TEs might provide transcription factor binding sites and induce chromosomal rearrangements. Finally, some SVs and TEs might have a positive impact depending on the environment. For example, FMA1 is associated with fluconazole resistance in C. albicans and contains a heterozygous TE insertion in both SC5314 and L26. Future molecular analyses are needed to determine the effect of TE insertions on drug susceptibility.
Despite their low abundance, TEs impact the genome evolution of C. albicans
Previous studies show TEs cover only ∼0.8% of the C. albicans genome, considerably less than in most other eukaryotic species, but similar to other yeast species (Maxwell 2020; Wells and Feschotte 2020). We found many TE loci are shared between the three C. albicans isolates, and they might be present in other isolates or even fixed in the species. Fixation of TEs would suggest genetic drift or even a beneficial impact. We detected a significant enrichment of ORFs with “3′–5′ RNA helicase activity” that have a TE insertion in the promoter or the ORF region. Helicases play an important role in the unwinding and binding of DNA and were shown to be involved in DNA repair (Croteau et al. 2014). Having TE insertions in the promoter or coding region might influence the expression of helicases, which could in term change the way the species responds to stress. We detected several ORFs with a function in transport that contain a TE in the promoter region. Such TE insertions could drastically change the expression of transporter genes, potentially leading to a faster efflux of antifungal drugs and increased resistance as observed in a fungal plant pathogen (Omrane et al. 2015, 2017). Most detected TEs were heterozygous. Remaining in a heterozygous state might be a strategy for TEs to escape defense mechanisms. Additionally, the heterozygosity of many SVs and TEs upstream of ORFs might cause allele-specific expression differences that can be adaptive in changing environments.
Despite the low TE coverage, some C. albicans TE families are still actively expressed, even though the mechanisms that regulate and maintain their low copy numbers remain poorly understood in the species (Holton et al. 2001; Zhu et al. 2014; Potocki et al. 2019). We detected an increased copy number and a distribution to all chromosomes of the retrotransposon Tca2 in L26, indicating that this TE might still be active. The proliferation of Tca2 seems to be recent, and must have happened after the separation from SC5314, as SC5314 only contains six copies. We did not detect any copies of the solo-LTR gamma, indicating that no ectopic recombination has occurred. The lack of solo-LTRs supports the idea that the burst of Tca2 in L26 was recent. Finally, the high sequence similarity of Tca2 copies indicates that not enough time has passed to accumulate mutations. The genome-wide distribution of the Tca2 copies suggests that the Tca2 family is able to jump to new chromosomes, possibly via extrachromosomal intermediates (Holton et al. 2001). Therefore, our analysis suggests that TEs are likely still able to be active in C. albicans, although smaller in magnitude than non-yeast fungal species like the plant pathogens Zymoseptoria tritici or Pseudocercospora ulei (Badet et al. 2020; González-Sayer et al. 2022).
We detected a large number of solo-LTRs, with an increased number of solo-LTRs in P75063 compared to the other two isolates. Solo-LTRs indicate a previous insertion of the corresponding full-length retrotransposon, followed by ectopic recombination leading to the deletion of the intermediate region and one LTR, resulting in the presence of a solo-LTR (Devos et al. 2002). Similarly, ectopic recombination of two TE copies belonging to the same family will lead to the deletion of the region between the copies and one TE copy. Ectopic recombination is potentially a defense mechanism against TE proliferation and increased genome sizes. Bird genomes that are generally compact and gene-rich show a higher number of solo-LTRs as opposed to large and repeat-rich salamander genomes with low levels of solo-LTRs (Frahry et al. 2015; Ji and DeWoody 2016). C. albicans might use ectopic recombination of TEs as a similar strategy to birds to keep the TE content low and the genome size small. Solo-LTRs are not functional transposons, as they lack coding sequences needed for autonomous replication, and are not able to create new copies (Ma and Bennetzen 2004). However, solo-LTRs might still influence the expression of nearby genes, for example by providing transcription factor binding sites (Butelli et al. 2012). We detected several solo-LTRs of high interest that were present in all isolates in the centromeric region. The solo-LTR episemon was heterozygous in all three isolates in CEN5. Outside the pericentromeric region, only one additional copy of episemon was detected in P75063. However, no full-length copy of this TE is known in the literature or was detected in this study. Episemon does not share sequence similarity with any other known TE in C. albicans. This indicates that the full-length element of episemon likely is lost from at least the reference genome, maybe even from the species as a whole. As solo-LTRs cannot actively create new copies, episemon copies are likely ancient, yet remained in specific locations including the centromeres over a long period of time.
SVs and TEs in the pericentromeric region provide plasticity and adaptation potential
One observation arising from our analyses was the frequent occurrence of SVs within centromeres CEN4 and CEN5. Plasticity was not only observed between centromere regions but also between different clinical isolates and between homologous chromosomes of the same isolate. We detected inversions within centromeres that are likely caused by nonallelic homologous recombination between inverted repeat sequences, and that are likely reversible (Delprat et al. 2009). We used our PCR assay as an approximation tool to estimate the general frequency of these centromere-associated features in 100 C. albicans clinical isolates. The presence of a frequently homozygous Cirt2 TE near CEN4 in multiple isolates suggests a neutral or even beneficial impact of Cirt2. TEs in other species are covered by facultative histone marks, which silence TEs under normal conditions, and allow expression under stress conditions (Fouché et al. 2020). Self-regulation to keep TE copy numbers low has recently been hypothesized to be a survival strategy for certain TE families in eukaryotic genomes (Stritt et al. 2021). However, whether this strategy also applies to C. albicans and other yeast species, remains an open question. In some isolates, Cirt2 is still actively expressed (MacCallum et al. 2009), and indeed, we detected a nearly identical copy of Cirt2 in the right arm of Chr 4. We found the Cse4p binding site in CEN4 to be disrupted by the inversion event. Isolates carrying the inversion likely exhibit differences in Cse4p binding that are undetectable in our current data set. As we found no strains in either the clinical isolates screen or in directed evolution experiments with a CEN4 homozygous inversion orientation, we suggest that a homozygous CEN4 inversion may have a strong fitness impact, leading to purifying selection against isolates carrying inversions in both homologs.
Our findings support the high centromeric plasticity previously described in C. albicans (Ketel et al. 2009; Burrack et al. 2016; Barra and Fachinetti 2018). SVs are potentially contributing to genomic instability and altering recombination frequencies at centromeres. Frequent breakpoints in the pericentromeric region are also observed in tumor cells in humans and sometimes resolve into isochromosomes (Shih et al. 2023). The evolutionary and adaptive trajectories of centromere inversions and insertions of TEs remain poorly understood. Recombination events that occur at the centromeres, including the formation of isochromosomes, have been shown to be sufficient to confer resistance to antifungal drug stress (Selmecki et al. 2006, 2008; Todd et al. 2019). In particular, adaptive isochromosomes i(5L) and i(4R) have been observed in clinical isolates after exposure to fluconazole (Todd and Selmecki 2020; Todd et al. 2023). Previous work has identified that fluconazole destabilizes the centromeres in C. albicans through the depletion of the centromere-specific H3 histone variant, Cse4p/CENPA (Brimacombe et al. 2019). We detected most SVs around centromeres to be heterozygous, potentially allowing fast adaptation to antifungal drug conditions, while maintaining the homolog that is adapted to environments without antifungal drugs. We suggest that frequent heterozygous SVs are important in maintaining high-standing genetic variation in C. albicans. Our findings highlight the dynamics of SVs and TEs across diverse clinical isolates of C. albicans, with a special focus on the diverse pericentromeric regions. Despite their low copy numbers, TEs play an important role in genome rearrangement and centromere diversity.
Methods
Culture conditions and high molecular weight gDNA extraction
The reference isolate SC5314 (clade I) and two additional clinical isolates L26 (clade I) and P75063 (clade SA) were analyzed (Supplemental Table S2; clade information from Hirakawa et al. [2015]). SC5314 originated from disseminated candidiasis, L26 was isolated from a vaginal sample of a vaginitis patient from Iowa, and P75063 was isolated from a bloodstream infection in France (http://www.candidagenome.org/Strains.shtml) (Wu et al. 2007). Previously, these isolates were whole genome sequenced using Illumina short-read sequencing (Hirakawa et al. 2015). High molecular weight gDNA was extracted using the Oxford Nanopore Technologies protocol “High Molecular Weight gDNA Extracted from Yeast” (see Supplemental Methods).
Genomic landscape of the C. albicans reference genome
Genome characteristics are not uniform along the chromosomes. The C. albicans reference genome SC5314 A21 (from now on called C. albicans reference genome) A21-s02-m09-r08 obtained on October 7, 2015, from the Candida Genome Database website (CGD) (http://www.candidagenome.org/download/sequence/C_albicans_SC5314/Assembly21/archive/C_albicans_SC5314_version_A21-s02-m09-r08_chromosomes.fasta.gz) (Van het Hoog et al. 2007) was used as a basis for a genome-wide study of niche characteristics on gene density, TE density, and GC content per 5000 kb window (see Supplemental Methods; Goodwin and Poulter 2000; Rice et al. 2000; Goodwin et al. 2001; Li and Durbin 2009; Li et al. 2009; Quinlan and Hall 2010).
Long-read sequencing alignment
Long-read sequencing was conducted using Oxford Nanopore MinION with R9.4.1 flow cells. Raw reads were basecalled using Guppy version 5.0.11 with the super high accuracy configuration (Wick et al. 2019). Basecalled reads were aligned to the C. albicans reference genome using minimap2 version 2.24 and the parameter -ax map-ont (Li 2018). Reads were sorted and indexed, duplicated reads were removed, and the remaining reads were reindexed using SAMtools. Reads were visualized with the Integrative Genomics Viewer (IGV) version 2.4.16 and Ribbon using “Position of primary alignment in SAM/BAM entry” as read sorting (Thorvaldsdóttir et al. 2013; Nattestad et al. 2021) (https://genomeribbon.com/).
To gain a general overview on sequence similarity between the genomes, simple de novo assemblies were conducted using Canu, quality control was conducted with BUSCO and Quast, and a visual comparison was conducted with MUMmer (see Supplemental Methods; Kurtz et al. 2004; Simão et al. 2015; Koren et al. 2017; Mikheenko et al. 2018; Manni et al. 2021).
Genome-wide structural variant and transposable element calling and genomic landscape
To detect SVs, DELLY version 1.1.6 was used (see Supplemental Methods; Li 2011a; Rausch et al. 2012). SV types are inversions (INV), duplication (DUP), translocations between chromosomes (TRA), deletions (DEL), and insertions (INS) relative to the reference genome. TE presence or absence of loci in the reference genome was confirmed based on raw reads and BLASTN, and de novo TEs were detected using the TELR version 1.0 pipeline (see Supplemental Methods; Camacho et al. 2009; Han et al. 2022). For each annotated ORF, the number of SVs and TEs in a 500 bp window upstream and downstream from the ORF region was extracted using the BEDTools window with the parameters -r 0 -l 500 and -r 500 -l 0, respectively. The closest SVs and TEs upstream were detected with BEDTools closest and the parameters -iu D b. Each SV and TE overlap with an annotated gene was detected with BEDTools intersect and the parameter -wao. The distance between genes was estimated with BEDTools closest and the parameters -iu -io -D a. To determine if any gene functions were enriched among the genes containing TEs or containing TEs in their 500 bp upstream promoter regions, we performed GO analysis using the Candida Genome Database Gene Ontology Term Finder with species C. albicans (http://www.candidagenome.org/cgi-bin/GO/goTermFinder) (Ashburner et al. 2000; The Gene Ontology Consortium 2000). To test if sequencing depth had an impact on SV and TE detection, a downsampling analysis was performed. The number of raw reads was reduced in steps of 10% and SV and TE detection tools were run again on the reduced sets of reads as described above.
Sequence comparison for the Cirt2 and Tca2 transposable element alleles
The sequence of the position of the 1551 bp orf19.3820 in the pericentromeric region of Chr 4 was extracted from C. albicans reference genome with SAMtools faidx with adding 1000 bp on each side. BLASTN was used to compare the region with known TE sequences in C. albicans (Goodwin and Poulter 2000; Goodwin et al. 2001). orf19.3820 showed a 99.95% sequence similarity on the 1822 bp consensus sequence of the DNA transposon family Cirt2. Both copies of the 46 bp terminal inverted repeats were detected. Cirt2 belongs to the TE superfamily Tc1-Mariner or pogo and class of DNA transposons. We blasted the sequence against NCBI to correctly position the transposase.
For the Tca2 TE family copies that underwent amplification in L26, phylogenetic analysis was conducted as described in Oggenfuss and Croll (2023) (see Supplemental Methods; Waterhouse et al. 2009; Li 2011b; Katoh and Standley 2013; Stamatakis 2014; Wickham 2016; Yu et al. 2017, 2018; Wang et al. 2020; Yu 2020; R Core Team 2023).
PCR screen for centromeric structural variants
To validate the presence of SVs, PCR was performed on SC5314, L26, P75063, and an additional 100 C. albicans clinical isolates from diverse patients, body sites, and geographic regions (see Supplemental Table S2; Supplemental Methods).
Data access
All raw sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA967712 for FASTQ (SC5314: SRR24449796; L26: SRR24449795; P75063: SRR24449794), FAST5 (SC5314: SRR29423996, L26: SRR29423995, P75063: SRR29423994). This Whole Genome Shotgun project has been submitted to DDBJ/ENA/GenBank under the following accession numbers SC5314: JBIBQR000000000, L26: JBIBQQ000000000, P75063: JBIBQP000000000.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Judy Berman (Tel Aviv University) for in-depth discussions regarding the orientation of CEN5, and Steven Cavaliri for providing many Candida albicans isolates. We thank Laura Burrack (Gustavus College), Pétra Vande Zande, Xin Zhou, Dalton Piotter, Dana Davis, and Nancy Scott (University of Minnesota) and the anonymous reviewers for their helpful comments on the manuscript. This work was supported by the Swiss National Science Foundation (P500PB_206850) to U.O., the National Institutes of Health (R01AI143689), and Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases Award (#1020388) to A.S. The University of Minnesota Supercomputing Institute (MSI) contributed computational resources to this project.
Author contributions: A.S., R.T.T., and U.O. contributed to the overall study design. R.T.T., A.G., A.B., B.K., and A.S. performed the experiments. U.O. and N.S. analyzed WGS data. The manuscript was written primarily by U.O. and A.S. with contributions from the other authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279301.124.
- Received March 9, 2024.
- Accepted October 21, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








