
Abstract
Neural tube defects (NTDs) are the most common birth defects of the central nervous system and occur as either isolated malformations or accompanied by anomalies of other systems. The genetic basis of NTDs remains poorly understood using karyotyping, chromosomal microarray, and short-read sequencing, with only a limited number of pathogenic variants identified. Collectively, these technologies may fail to detect rare structural variants (SVs) in the genome, which may cause these birth defects. Therefore, optical genome mapping (OGM) was applied to investigate 104 NTD cases, of which 74 were isolated NTDs and 30 were NTDs with other malformations. A stepwise approach was undertaken to ascertain candidate variants using population and internal databases and performing parental studies when possible. This analysis identifies diagnostic findings in 8% of cases (8/104) and candidate findings in an additional 22% of cases (23/104). Of the candidate findings, 9% of cases (9/104) have SVs impacting genes associated with NTDs in mouse, and 13% of cases (14/104) have SVs impacting genes implicated in the neural tube development pathways. This study identifies RMND5A, HNRNPC, FOXD4, and RBBP4 as strong candidate genes associated with NTDs, and expands the phenotypic spectrum of AMER1 and TGIF1 to include NTDs. This study constitutes the first systematic investigation of SVs using OGM to elucidate the genetic determinants of NTDs. The data provide key insights into the pathogenesis of NTDs and demonstrate the contribution of SVs in the genome to these birth defects.
Neural tube defects (NTDs) are the most common birth defects of the central nervous system, with wide variation in prevalence from <1 per 1000 births in the Americas and Europe to >10 per 1000 births in Africa and China (Zaganjor et al. 2016; Dixon et al. 2019). Globally, 300,000 births are affected with NTDs each year and account for ∼88,000 deaths per year (Zaganjor et al. 2016). The embryological basis of NTDs is an abnormal neurulation process, wherein the neural plate fails to complete its transformation into the neural tube (Sadler 2005). Neurulation begins in the third week of gestation, with the formation of the neural plate, its longitudinal extension, and concomitant formation of the neural folds and neural grove, followed by fusion of the neural folds. Fusion begins in the cervical region and proceeds both cranially and caudally, with completion of the neural tube closure process by day 28. Failure of the neural tube to close in the cranial region, results in anencephaly or encephalocele, while failure anywhere from the cervical region caudally results in spina bifida. Most NTDs occur as isolated malformations, while ∼15%–25% may occur with additional unrelated malformations (Kälién et al. 1998; Stoll et al. 2011; Dean et al. 2020).
Epidemiological studies suggest a complex etiology underlying NTDs, with both environmental and genetic contributions. Increased dietary intake of folic acid has resulted in 50%–70% reduction in NTDs (MRC Vitamin Study Research Group 1991; Czeizel and Dudás 1992; Berry et al. 1999; Dean et al. 2020). However, among NTDs that remain folate-resistant, only ∼20% have been explained by either genetic or environmental factors. Environmental influences during pregnancy that may predispose to NTDs include maternal diabetes mellitus, maternal obesity, valproate and hyperthermia exposures, and maternal MTHFR C677T carrier state (Pulliam 2023). And while the risk for NTD is predicted to have a genetic basis in ∼60%–70% of cases, genetic causes have only been identified in <10% of cases. Genetic causes include numerical chromosomal aberrations in ∼5%, microdeletions or microduplications in <1%, and single-gene disorders in <2% of cases (Dean et al. 2020). Recently, exome (ES) and genome sequencing (GS) have been applied to investigate NTDs, but their diagnostic yield and ability to identify new genes have been limited (Lemay et al. 2019; Yang et al. 2022). Overall, these studies indicate that there is a higher frequency of structural changes in the genome compared to sequence alterations.
These observations highlight the need for comprehensive cytogenomic analysis beyond the detection of numerical or large copy number changes. Optical genome mapping (OGM) is a cytogenomic technique that has the ability to detect several classes of structural variants (SVs) including triploidy, aneuploidy, copy number changes (deletions and duplications), balanced and unbalanced events (insertions, inversions, and translocations), and repeat expansions and contractions, with a higher sensitivity and resolution compared to conventional methods (Broeckel et al. 2024). The strengths and limitations of OGM were previously discussed (Sahajpal et al. 2021, 2023a; Iqbal et al. 2023; Broeckel et al. 2024). In this study, OGM was applied to investigate a cohort of 104 NTD cases, with the hypothesis that the identification of rare SVs would shed additional light on the genetic basis of these birth defects.
Results
Optical genome mapping variant calls
OGM identified 6334 ± 218 (average ± SD) variants per sample. In each case, the variants were filtered against >300 control samples, resulting in 85 ± 67 variants per sample (8827 variants in total). These included 5847 deletions, 2397 insertions, 449 duplications, 75 inversions, 24 rearrangements, one translocation, 30 regions of the absence of heterozygosity (AOH), and four aneuploidies. Of these, a total of 175 variants overlapped a mouse NTD-associated gene, which included 73 deletions, 52 insertions, 17 duplications, five inversions, and 28 regions of AOH. Overall, 8827 variants were manually reviewed for potential relevance to the NTD phenotype. The relevance of SVs/genes was investigated using population databases (gnomAD and database of genomic variants), Online Mendelian Inheritance in Man, Human Gene Mutation Database, and an internal database of >20,000 chromosomal microarray analyses performed on samples submitted for clinical testing.
This analysis identified diagnostic findings in 8% of cases (8/104) and candidate findings in an additional 22% of cases (23/104). Of the candidate findings, 9% of cases (9/104) had SVs impacting genes associated with NTDs in mouse, and 13% of cases (14/104) had SVs impacting genes implicated in the neural tube development pathways (Fig. 1).
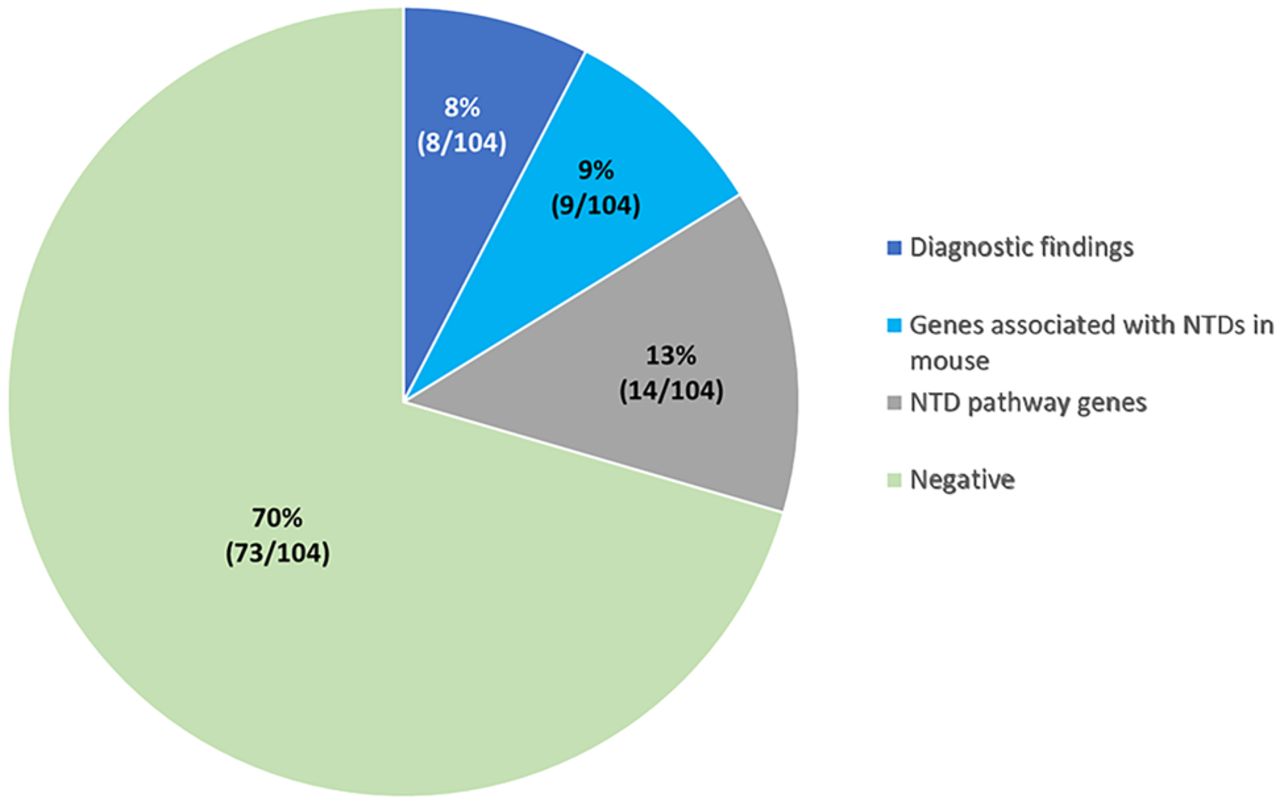
Diagnostic findings
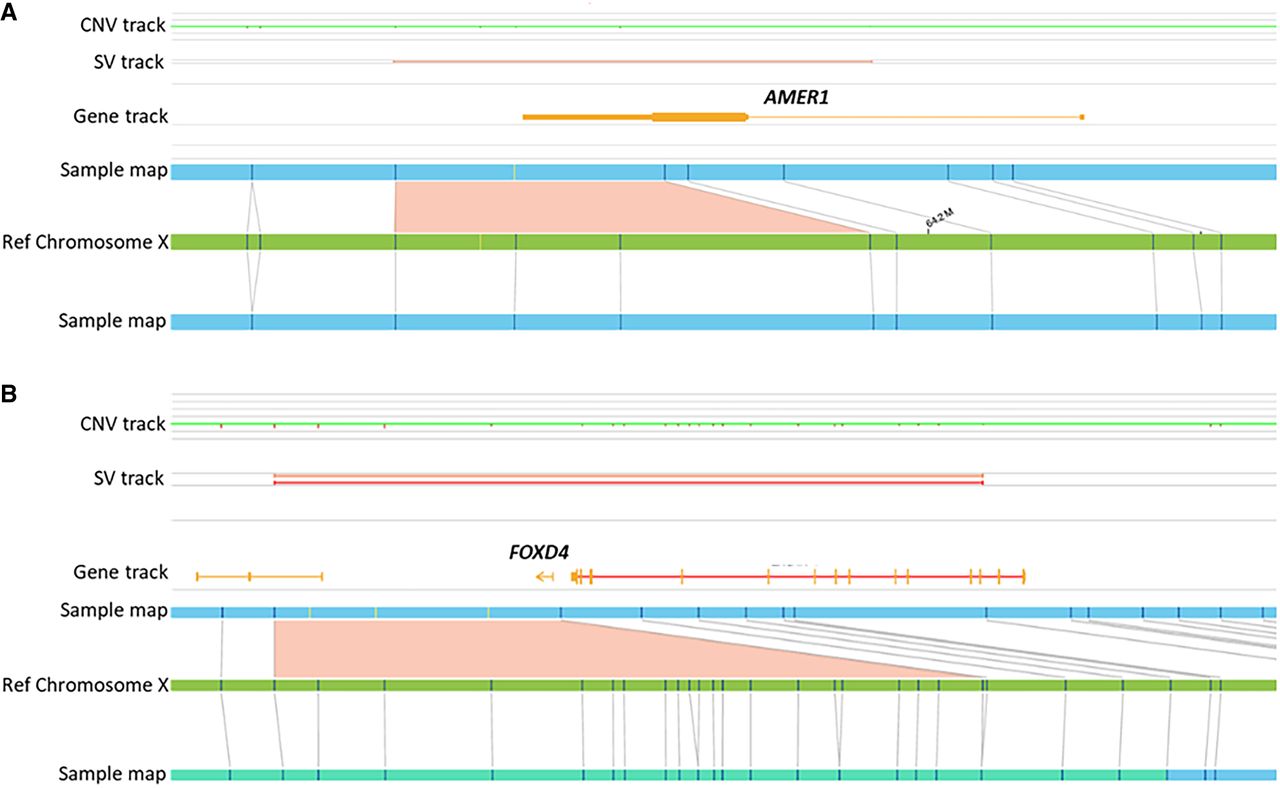
OGM identified diagnostic findings in 8% of cases (8/104). These included four cases with an aneuploidy (trisomy 18), one case with a deletion involving 15q24.1q24.2, one with an unbalanced translocation involving Chromosomes 10 and 13 that resulted in a partial gain of Chromosome 10p and partial loss of the terminal region of Chromosome 13q, one with a deletion of the promoter region of the VANGL2 gene, and one with a deletion of exon 2 of the AMER1 gene (Fig. 2A; Table 1).
Optical genome maps show deletions in two cases. (A) Partial heterozygous deletion of the AMER1 gene in a female fetus with osteopathia striata with cranial sclerosis (OS-CS) including anencephaly and cervical rachischisis (case 8). (B) Heterozygous deletion of the FOXD4 gene in a fetus with spina bifida (case 9).
List of diagnostic findings
| Case ID | NTD type | Variant | Nomenclature | Gene(s) | Region(s) | Size (bp) | Variant confirmation | Inheritance | Evidence (literature/internal database) | Evidence for association with NTDs |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Anencephaly | Trisomy | ogm[GRCh38] 18(p11.32q23)(1–80,373,285) × 3 | _ | Whole Chromosome 18 | _ | Karyotype | de novo | Dean et al. (2020) | S |
| 2 | Spina bifida | Trisomy | ogm[GRCh38] 18(p11.32q23)(1–80,373,285) × 3 | _ | Whole Chromosome 18 | _ | Karyotype | de novo | S | |
| 3 | Spina bifida | Trisomy | ogm[GRCh38] 18(p11.32q23)(1–80,373,285) × 3 | _ | Whole Chromosome 18 | _ | Karyotype | de novo | S | |
| 4 | Spina bifida | Trisomy | ogm[GRCh38] 18(p11.32q23)(1–80,373,285) × 3 | _ | Whole Chromosome 18 | _ | Karyotype | de novo | S | |
| 5 | Anencephaly | Unbalanced translocation | ogm[GRCh38] t(10;13)(p15.2;q22.2)(3,519,095;75,729,511), 13q22.2q34(75,729,511_114,352,102) × 1, 10p15.3p15.2(18,514_3,463,629) × 3 | _ | Partial loss of Chr 13 and gain of Chr 10 | 38,622,592 (Chr 13), 3,445,116 (Chr 10) | Karyotype, CMA | Paternal (balanced carrier) | Lou et al. (2000); Lurie et al. (2016) | S |
| 6 | Spina bifida | Deletion | ogm[GRCh38] 15q24.1q24.2(72,656,367_75,788,985) × 1 | _ | Partial loss of Chr 15 | 3,120,511 | CMA | de novo | El-Hattab et al. (2009) | S |
| 7 | Anencephaly | Deletion | ogm[GRCh38] 1q23.2(160,457,896_160,469,769) × 1mat | VANGL2 | Promoter | 3042 | qPCR | Maternal | MIM# 182940 | S |
| 8 | Anencephaly and cervical rachischisis (OS-CS) | Deletion | ogm[GRCh38] Xq11.2(64,180,374_64,197,924) × 1 | AMER1 | Exon 2 | 7665 | X-Array | Maternal (affected with OS-CS) | MIM# 300373; Winter et al. (1980) | S |
[i] (S) Strong, (OS-CS) osteopathia striata with cranial sclerosis.
SVs overlapping genes associated with NTDs in mouse
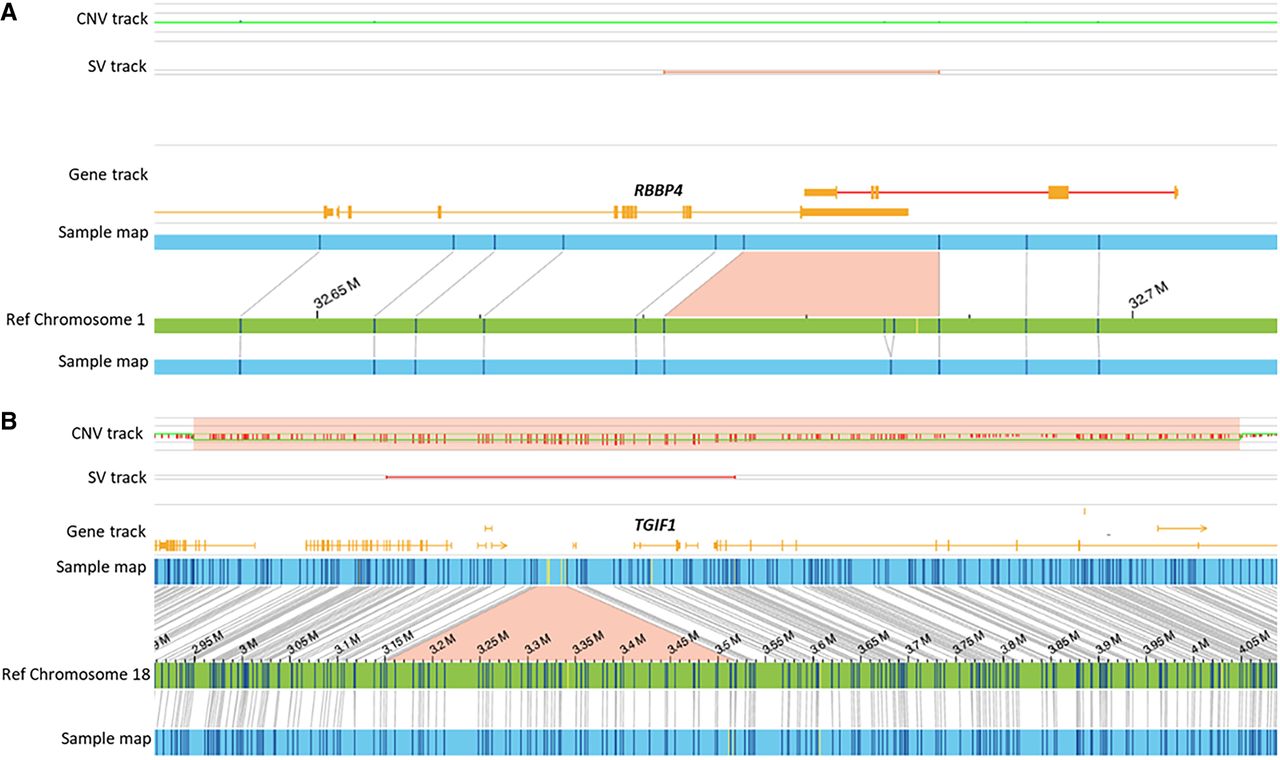
OGM identified a total of 175 variants overlapping genes associated with NTDs in mouse, of which 11 variants in nine cases were deemed relevant after manual review (Table 2). These included six deletions, three duplications, and two insertions (Figs. 3B, 4). Of these nine cases, two cases harbored multiple SVs, overlapping multiple genes associated with NTDs in mouse or rare SVs in genes of uncertain significance. Additionally, parental samples were available in three cases, of which a 9p24.3 deletion that included the FOXD4 gene was confirmed as nonmaternal.
Optical genome maps showing deletions in two cases. (A) Heterozygous deletion in the 3′ UTR of the RBBP4 in a fetus with spina bifida (case 10). (B) Paternally inherited heterozygous deletion of TGIF1 in a fetus with anencephaly (case 11).
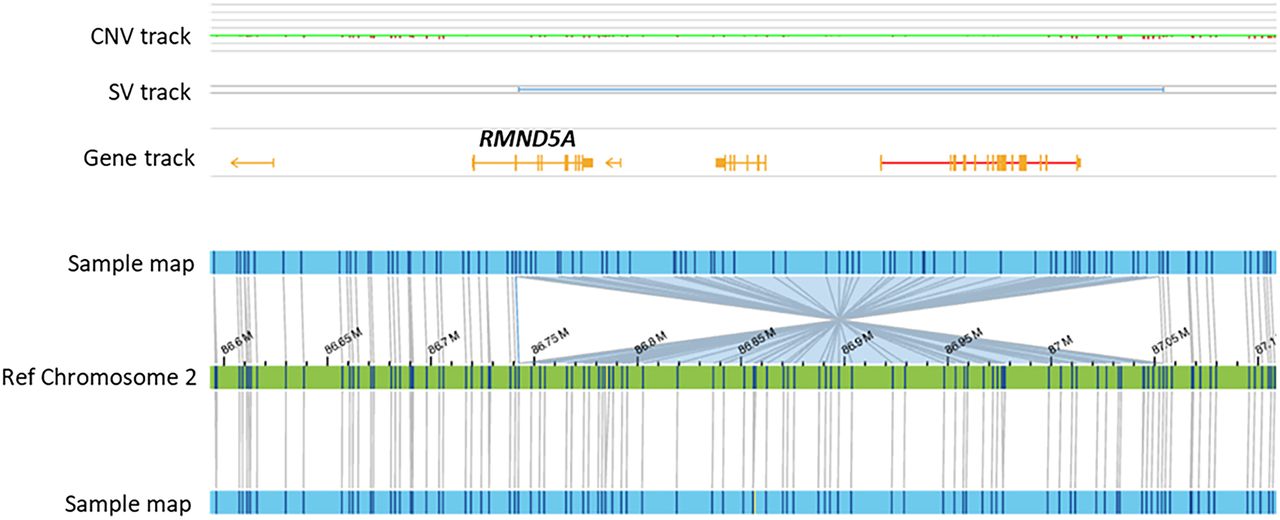
Optical genome maps showing a balanced 311 kb inversion disrupting intron 2 of the RMND5A gene (detected in cases 18 and 19).
List of rare SVs impacting genes associated with NTDs in mouse
| Case ID | Mouse NTD | NTD observed in this study | Variant | Nomenclature | Gene | Size (bp) | Region(s) | Variant confirmation | Inheritance | Evidence (literature/ internal database) | Evidence for association with NTDs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 9 | Open NTD | Spina bifida | Deletion | ogm[GRCh38] 9p24.3(82,669_173,653) × 1 | FOXD4 | 54,206 | Whole gene | qPCR | Not maternal | Neilson et al. (2012); McMohan et al. (2022) | S |
| 10 | Open NTD | Spina bifida (OEIS) | Deletion | ogm[GRCh38] 1p35.1(32,671,257_32,688,145) × 1 | RBBP4 | 4904 | 3′ UTR | qPCR | Not determined | Miao et al. (2020) | S |
| 11 | Exencephaly | Anencephaly | Deletion | ogm[GRCh38] 18p11.31(3,151,509_3,517,870) × 1 | TGIF1 | 334,545 | Whole gene | CMA | Paternal | MIM# 142946 | S |
| 12 | Exencephaly | Anencephaly | Insertion | ogm[GRCh38] ins(13)(q12.11)(20,599,264_20,606,931) | IFT88 | 2707 | Intron 11 | _ | Not determined | Pazour et al. (2000); Ohazama et al. (2009); Tian et al. (2017); Au et al. (2021) | M |
| Deletion | ogm[GRCh38] 1p34.2(42,442,313_42,468,422) × 1 | ZMYND12 | 6794 | Exons 2 and 3 | qPCR | Not determined | Wang et al. (2022) | ||||
| 13 | Anencephaly | Craniorachischisis | Deletion | ogm[GRCh38] 8q24.3(144,474,228_144,518,767) × 1 | FOXH1 | 2759 | Regulatory region | CMA* | Not determined | Roessler et al. (2008); Internal database: One anencephaly case with FOXH1 deletion | M |
| Deletion | ogm[GRCh38] 16p12.2(21,660,813_22,434,434) × 1 | 16p12.2 | 773,622 | CMA | Not determined | 16p12.2-p11.2 deletion syndrome (MIM# 613604) | |||||
| 14 | Abnormal neural tube closure | Meroacrania Type Anencephaly | Insertion | ogm[GRCh38] ins(3)(p22.3;p22.3)(32,703,720–32,716,482) | CNOT10 | 3236 | Intragenic | _ | Not determined | Internal database: One fetal demise case carries a heterozygous deletion of CNOT10 | M |
| 15 | Exencephaly | Anencephaly | Duplication | ogm[GRCh38] dup(7)(p21.1p21.1)(18,813,738_19,816,010) × 3 | TWIST1 | 1,002,272 | Whole gene | CMA | Paternal | MIM# 601622 | W |
| 16 | Open neural tube | Spina bifida | Duplication | ogm[GRCh38] 19q13.43q13.43(58,513,157_58,564,834) × 3 | TRIM28 | 51,677 | Whole gene | qPCR | Paternal | Lee and Gleeson (2020) | W |
| 17 | Exencephaly | Spina bifida | Duplication | ogm[GRCh38] 1p36.33(1,624,078_1,691,055) × 3 | MIB2 | 66,977 | Whole gene | _ | Not determined | Wu et al. (2007) | W |
[i] (S) Strong, (M) moderate, (W) weak; CMA*(CMA was indicative of the copy number loss), (OEIS) omphalocele-exstrophy-imperforate anus-spinal defects.
SVs overlapping genes implicated in NTD pathways
On manual review of rare variants, 15 variants in 14 cases were found to be candidates relevant to the NTD phenotype (Table 3). Of these 15 variants, there were six deletions, six duplications, two inversions, and one complex rearrangement. Of note, two cases carried an inversion disrupting the RMND5A gene (Fig. 4). Two cases (one with OEIS and the other with anencephaly) carried a duplication of the HNRNPC gene, the upregulation of which has been previously associated with NTDs (Niggl et al. 2023). Parental samples were available in one case, and the duplication was found to be de novo (Fig. 5A). In one case with anencephaly, deletion of exons 2 and 3 of the PFKFB4 gene, which has been shown to be involved in neural tube development (Pegoraro et al. 2015), was found to be a de novo change. In another case with anencephaly, an Xp22.2 duplication that included the HCCS and AMELX genes was identified to be maternally inherited. Loss-of-function (LoF) variants in the HCCS gene are associated with XLD linear skin defects with multiple congenital anomalies; 1 and LoF variants in the AMELX gene are associated with XLD amelogenesis imperfecta, type 1E. For this case, X-Chromosome inactivation (XCI) was randomly skewed in the mother, while the proband was highly skewed, presumably expressing the X carrying the duplication (Supplemental Fig. S1).
Optical genome maps show duplication in two cases. (A) de novo 5′-UTR region duplication of HNRNPC in a fetus with anencephaly (case 20). (B) A heterozygous duplication of HNRNPC in a fetus with spina bifida (OEIS; case 21).

List of rare SVs impacting genes implicated in NTD pathways
| Case ID | NTD type | Variant | Nomenclature | Gene(s) | Region(s) | Size (bp) | Variant confirmation | Inheritance | Evidence (literature/ internal database) | Evidence for association with NTDs |
|---|---|---|---|---|---|---|---|---|---|---|
| 18 | Anencephaly | Inversion | ogm[GRCh38] inv(2)(p11.2p11.2)(87,054,207_86,742,515) | RMND5A | Gene disruption | 311,692 | _ | Not maternal | Vogel et al. (2012); Pfirrmann et al. (2015) | S |
| 19 | Encephalocele (Meckel syndrome) | Inversion | ogm[GRCh38] inv(2)(p11.2p11.2)(87,054,207_86,742,515) | RMND5A | Gene disruption | 311,693 | _ | Not determined | S | |
| 20 | Anencephaly | Duplication | ogm[GRCh38] 14q11.2(21,253,761_21,311,033) × 3 | HNRNPC | Exon 1 and 2 (both noncoding) | 57,272 | qPCR | de novo | MIM # 620688; Liu et al. (2020); Niggl et al. (2023) | S |
| 21 | Spina bifida (OEIS) | Duplication | ogm[GRCh38] 14q11.2q11.2(20,990,605_21,390,099) × 3 | HNRNPC | Whole gene | 399,494 | qPCR | Not determined | S | |
| 22 | Anencephaly | Deletion | ogm[GRCh38] 3p21.31(48,540,706_48,558,562) × 1 | PFKFB4 | Exon 2 and 3 deletion | 3260 | qPCR | de novo | Pegoraro et al. (2015) | M |
| 23 | Encephalocele | Deletion | ogm[GRCh38] 19p13.2(9,370,139_9,529,559) × 1 | ZNF266 | Whole gene | 147,320 | qPCR | Not determined | Internal database: Two cases with similar deletion: 1 with MCA; 2 in fetal demise | W |
| 24 | Spina bifida (OEIS) | Deletion | ogm[GRCh38] 15q25.3(85,036,822_85,102,540) × 1 | PDE8A | Exons 2–10 | 50,956 | qPCR | Not determined | Internal database: same deletion in a case of fetal demise | W |
| Deletion | ogm[GRCh38] 18p11.21(10,982,344_11,061,530) × 1 | PIEZO2 | Intron 2 | 72,238 | qPCR | Not determined | (MIM# 248700, 114300, 108145) | W | ||
| 25 | Anencephaly | Deletion | ogm[GRCh38] 5p13.2(34,009,171_34,049,829) × 1 | C1QTNF3 | Whole gene | 30,311 | qPCR | Not determined | MIM# 612045 | W |
| 26 | Anencephaly | Deletion | ogm[GRCh38] 5q35.1(173,140,602_173,149,664) × 1 | BNIP1 | Exon 1 and 2 | 3853 | _ | MIM# 603291 | W | |
| 27 | Anencephaly | Duplication | ogm[GRCh38] Xp22.2(11,017,889_11,437,394) × 3 | HCCS, AMELX | Whole genes | 419,505 | qPCR | Maternal | HCCS: MIM# 309801, AMELX: MIM# 301200 | W |
| 28 | Anencephaly | Rearrangement | ogm[GRCh38] dup(1)(p36.13p36.13) | SDHB, MFAP2 | Multiple genes | 378,106 | _ | Not determined | MIM# 185470 | W |
| 29 | Anencephaly | Duplication | ogm[GRCh38] 8p21.2(23,701,105_24,412,415) × 2∼3 | NKX2-6 | Whole gene | 711,311 | qPCR | Not determined | MIM #217095 | W |
| 30 | Anencephaly | Duplication | ogm[GRCh38] dup(10)(q22.3q22.3) | KCNMA1 | Exon 1 and 2 | 371,692 | _ | Not determined | MIM #618729 | W |
| 31 | Anencephaly | Duplication | ogm[GRCh38] 1q21.1(145,497,462_145,815,386) × 3 | _ | Partial locus-mosaic | 317,924 | qPCR | Not determined | Partial duplication of 1q21.1 locus | W |
[i] (S) Strong, (M) moderate, (W) weak, (OEIS) omphalocele-exstrophy-imperforate anus-spinal defects.
Discussion
SVs contribute substantially to the genomic variation found in individuals and have been implicated in evolutionary processes, human health, and disease (Eichler 2019; Levy-Sakin et al. 2019). However, this contribution is underestimated, primarily due to the limitation of the available technologies used to assess SVs in the human population and rare disorders, including NTDs. To address this gap, OGM was applied to investigate this cohort of NTD cases, which led to a diagnostic yield comparable to combined conventional methods, expansion of the phenotypic spectrum associated with two genes, and identification of five strong candidate genes associated with NTDs.
The spectrum of SVs constituting the diagnostic findings in this study indicates that OGM is a comprehensive cytogenomic method with the ability to detect the different classes of SVs in a single method and analysis (Sahajpal et al. 2021, 2023a; Iqbal et al. 2023; Broeckel et al. 2024). The frequency of numerical chromosomal aberrations (∼4%) and microdeletions/duplications (∼2%) was consistent with the frequency reported in the literature (Dean et al. 2020). Trisomy 18 has been reported to be the most common aneuploidy found in NTDs, and is the only trisomy found in this study (cases 1–4), implicating potential overexpression of gene(s) on Chromosome 18 with NTDs. Of the two cases with microdeletions/duplications, anencephaly in the fetus with unbalanced t(10;13) might be attributed to the distal 13q monosomy (case 5). Distal 13q monosomy has long been associated with NTDs, especially cranial NTDs relative to spinal defects, with 13q33-34 considered to be the critical region sufficient to cause NTDs (Lou et al. 2000). The region deleted in this case extended from 13q22.2 to q34, which is associated with a more penetrant NTD phenotype. The proximal 13q22 region is hypothesized to harbor regulatory elements that regulate the NTD-associated genes in the 13q33-q34 region (Lurie et al. 2016). The 15q24.1q24.2 deletion case of spina bifida, appears to be the second deletion case with a spinal NTD (case 6). The region includes ARID3B, CSK, and PTPN9, which are constrained genes and are implicated in neural tube development/NTDs in mouse (Lee and Gleeson 2020). Previously, a 5-month-old female affected with myelomeningocele, hydrocephalus, tetralogy of Fallot, clubfeet, short stature, and facial dysmorphism was reported to carry the 15q24 deletion (72.130–76.080 Mb, hg18) (El-Hattab et al. 2009). Deletion of this region is associated with 15q24 microdeletion syndrome, which is characterized by developmental delay, intellectual disability, and characteristic facial features. While congenital malformations are rarely reported, these include structural brain anomalies, cardiovascular malformations, congenital diaphragmatic hernia, intestinal atresia, and imperforate anus (Magoulas and El-Hattab 2012). Together, these two cases demonstrate the variability of 15q24 microdeletion syndrome and suggest the phenotypic spectrum should be expanded to include spinal NTDs.
The maternally inherited deletion of VANGL2 in an anencephalic fetus (case 7), demonstrates the incomplete penetrance associated with this gene. VANGL2, associated with autosomal dominant NTDs (MIM # 182940) has an estimated penetrance of 75% (Fineman et al. 1982). The incomplete penetrance suggests the role of other modifiers, genetic, or environmental. However, none was identified in this case. Additional comprehensive genomic/epigenomic analysis in a cohort of cases with pathogenic VANGL2 variants would be needed to identify candidate modifiers, if any.
The anencephalic fetus with cervical rachischisis and multiple abnormalities (bilateral cleft lip and palate and intestinal malrotation) (case 8) carried a partial deletion of the AMER1 (Supplemental File 1), which is associated with X-linked dominant (XLD) osteopathia striata congenita with cranial sclerosis (OS-CS) (MIM# 300373). To date, one case with spina bifida occulta affecting the third through the fifth lumbar vertebrae and the first sacral vertebra was reported in one female in a kindred with four females in three generations affected with OS-CS (Winter et al. 1980). A single case of holoprosencephaly was reported in a family with five affected females in three generations with OS-CS (Savarirayan et al. 1997). However, neither of these cases had a molecular diagnosis; thus, this represents the first reported case of anencephaly and cervical rachischisis in a female with OS-CS associated with an AMER1 deletion. Collectively, these findings expand the phenotypic spectrum of AMER1 to include NTDs.
SVs overlapping genes associated with NTDs in mouse
Over 400 genes have been observed to cause NTDs in mouse models (Harris and Juriloff 2010; Lee and Gleeson 2020); however, only a limited number of these genes have subsequently been associated with NTDs in humans. In this cohort, nine human homologs of genes associated with NTDs in mouse have been identified in association with NTDs. Six of these genes are being proposed as human NTD-associated genes for the first time, with strong or moderate evidence for pathogenicity.
Four cases carried deletions of genes associated with NTDs in mice (FOXD4, RBBP4, TGIF1, and FOXH1). FOXD4 (case 9, Fig. 2B; Table 1), is a forkhead transcription factor, that regulates the transition from pluripotent embryonic stem cells (ESCs) to neuroectodermal stem cell, and is necessary for neuronal differentiation (Neilson et al. 2012). McMohan et al. (2022) observed craniofacial malformations and neural tube closure defects in mouse models with loss of FOXD4 function. FOXD4 was found to be essential in the anterior mesendoderm and later in the anterior neuroectoderm for rostral neural tube closure and neural crest specification during head development (McMohan et al. 2022). Loss of RBBP4 (case 10, Fig. 3A) results in a defective inner cell mass, severe apoptosis, hyperacetylated histones, and ultimately lethality in mice. Experiments in zebrafish show that RBBP4 is required for neural crest development and persistent neurogenesis in the midbrain (Miao et al. 2020). RBBP4 is highly conserved (pLI of 1) and the functional evidence suggests that LoF variants in this gene might result in severe birth defects, which could be lethal in humans. Loss of TGIF1 (case 11, Fig. 3B) causes exencephaly in the mouse and has been associated with holoprosencephaly type 4 in humans (MIM #142946). Odent et al. 1998 demonstrated autosomal dominant inheritance with incomplete penetrance was the mode of inheritance associated with this gene. Although human NTDs have not been previously associated with TGIF1, the observed phenotype in mouse and anencephaly in case 13, expands the phenotypic spectrum associated with TGIF1 to include NTDs.
A combinatorial effect of IFT88 and ZMYND12 variants was suspected in case 12. Loss of IFT88 is known to cause exencephaly in mice (Pazour et al. 2000; Ohazama et al. 2009; Tian et al. 2017), and has been implicated as a risk factor for myelomeningocele in humans (Au et al. 2021); while ZMYND12 has been proposed as a candidate gene for ciliary transition zone defects (Wang et al. 2022). The craniorachischisis fetus (case 13), carried FOXH1 and 16p12.2 microdeletion. The regulatory region of FOXH1 was assessed using the ENCODE data set, suggesting the deleted region included cis-regulatory elements (cCREs), also supported by H3K27 acetylation marks. Additionally, one anencephaly case in our internal database carried a heterozygous deletion of FOXH1. Functional analysis of the human NODAL-signaling pathway has linked FOXH1 with congenital heart defects and holoprosencephaly (Roessler et al. 2008), missense variants in zebrafish cause tetralogy of Fallot and holoprosencephaly, while loss of FOXH1 in mice results in anencephaly. Although the human phenotype associated with FOXH1 remains to be elucidated, the mouse phenotype and the two cases in this series suggest an association with cranial NTDs.
One anencephalic fetus (case 14), with insertion in CNOT10 was also deemed novel. Although the functional consequence of the insertion was not determined, an additional case with a heterozygous deletion of CNOT10 was identified in a case of fetal demise. CNOT10 is a core subunit of the evolutionarily conserved CCR4-NOT complex, which functions in the cytoplasm as the major mRNA deadenylase in both constitutive mRNA turnover and regulated mRNA decay pathways. CNOT10 is highly conserved (pLI of 1) and given the association of other CNOT genes with severe recognizable syndromes (CNOT1 MIM# 618500, 619033; CNOT2 MIM# 618608; CNOT3 MIM# 618672), the two cases in this cohort suggest that LoF variants in CNOT10 could be lethal. However, additional cases are required to further delineate the phenotype associated with CNOT10 variants. Three additional cases carried rare duplications of TWIST1 (case 15), TRIM28 (case 16), and MIB2 (case 17). Although these were rare SVs, whether gain-of-function (GoF) would result in NTDs is questionable as mouse associations have been for LoF variants. Nevertheless, the rare duplication of these genes associated with NTDs in mouse warrants further analysis and is reported for the purpose of future discovery and research.
SVs overlapping genes implicated in NTD pathways
The unbiased genome-wide SV analysis led to the identification of 15 additional rare variants in 14 cases. Most notable finding was in two cranial NTD cases (cases 18 and 19), that carried a balanced inversion disrupting RMND5A. The inversion was mediated by sequence homology at both breakpoints, which might explain the occurrence of the same SV in two unrelated cases. RMND5A (pLI of 1) is highly conserved and encodes a ubiquitin ligase, which is highly expressed in the neural ectoderm, and suppression of its function during embryonic development in model organisms results in brain malformations, specifically in dorsal regions of the pros- and mesencephalon, the eyes, and ciliated cells of the skin (Pfirrmann et al. 2015). An 80.65 kb gain of Chromosome 2p11.2 that partially overlapped the RMND5A gene (exons 3–9) has been previously reported in a male child born at 33 weeks gestation with a giant occipitoparietal meningoencephalocele (Vogel et al. 2012). The breakpoint of the duplication within the RMND5A gene overlaps with the inversion breakpoint detected in the two cases in this study. Although the orientation of the duplication was not investigated by the authors, an inverted duplication cannot be ruled out given the sequence homology-mediated inversion detected in this cohort. Nevertheless, the conserved role of the RMND5A gene, and these three cases collectively highlight RMND5A as a new gene associated with cranial NTDs.
Also, notable were cases 20 and 21 that carried a duplication of HNRNPC (Fig. 5A,B). Dual 5′-UTR variants were constructed using synthetic biology techniques and showed increased transcription and translation of the gene (Balzer Le et al. 2020); thus, the functional consequence of both variants, the 5′-UTR duplication and whole-gene duplication, is expected to be GoF. HNRNPC belongs to the ubiquitous HNRNP family that plays various roles in mRNA processing and is a key regulator of gene expression. Seven HNRNP family members, including HNRNPC, have been associated with neurodevelopmental disorders. LoF and splicing variants in the HNRNPC gene (pLI of 1) were recently associated with neurodevelopmental disorders. Although no GoF variants were reported, functional studies demonstrated that either reduced or enhanced levels of HNRNPC affected neuronal migration (Niggl et al. 2023). HNRNPC along with two additional RNA-binding proteins were found to be overexpressed in folic acid deficient (FAD) H9 human ESCs into neural crest cell (H9-NCCs) models in vitro and in the FAD mouse model in vivo. An upregulation of these RNA-binding proteins was suggested to contribute to folate deficiency-induced neural crest cell dysfunction (Liu et al. 2020). Whether the NTDs in these cases resulted from HNRNPC duplication alone or gene–environment interaction is unknown; however, the evidence implicates HNRNPC in the development of NTDs.
Another gene identified as a candidate is PFKFB4, which is a glycolysis regulator and has been shown to regulate dorsal ectoderm global patterning in gastrulating frog embryos. PFKFB4 is required for dorsal ectoderm progenitors to proceed toward neural and nonneural ectoderm, neural crest, or placodes (Pegoraro et al. 2015). The identification of a de novo deletion in an NTD fetus, and the early embryonic function warrants further analysis of this gene to be associated with NTDs.
Four cases harbored deletions of ZNF266, PDE8A, C1QTNF3, and BNIP1 genes, and although these represent rare variants, additional cases are needed to determine the impact of these variants in the human phenotype. The last five cases are all fetuses with anencephaly that carry a duplication of a gene that is associated with a syndrome with either LoF or missense variants, but not duplication variants. These include duplication of HCCS and AMELX in a female fetus with anencephaly (case 27), which are associated with XLD linear skin defects with multiple congenital anomalies and XLD amelogenesis imperfecta, type 1E, respectively; rearrangement on Chromosome 1 that includes SDHB, which is associated with AD paraganglioma (MIM# 606864) (case 28); duplication of Chromosome 8p including the NKX2-6 gene, associated with conotruncal heart malformations (MIM# 217095) (case 29); duplication of KCNMA1, associated with Liang-Wang syndrome, AD (MIM# 618729) (with LoF) (case 30); and a partial duplication of Chromosome1q21.1 region (case 31) that is associated with dysmorphic features, developmental delay, and brain anomalies. However, additional cases and studies are needed to confirm pathogenicity and association with the NTD phenotype. This study constitutes the first systematic assessment of SVs using OGM to identify the genomic determinants in NTDs. This study identified RMND5A, HNRNPC, FOXD4, and RBBP4 to be strong candidates associated with NTDs, and expanded the phenotypic spectrum of AMER1 and TGIF1 to include NTDs. The data provide key insights into the pathogenesis of NTDs and demonstrate the contribution of SVs in the genome to these birth defects.
The results need to be validated in future studies with larger sample size. A statistical SV burden analysis was not performed for this cohort because of the lack of a comparable control data set. A statistical analysis would be performed in the future once a comparable control data set is established on the OGM platform. Sequence variants, small SVs (<500 bp; limit of resolution of OGM), short and long noncoding RNA, polygenic, and epigenetic modifications were not investigated in this study. Additionally, parental samples were not available for several cases, and all SVs were not confirmed by orthogonal methods. Expression analysis and functional studies were beyond the scope of this study.
Methods
Cohort
Samples for this study were obtained through the South Carolina anatomical gift act, and the protocol was reviewed and approved by the Self Regional Healthcare Institutional Review Board (IRB #Pro00057750). A total of 104 NTD cases were included in the study, of which 45 were spina bifida, 43 anencephaly, six encephalocele, and 10 craniorachischisis. Of these 104 cases, 74 presented as isolated defects and 30 with additional malformations. The families/mothers were interviewed under the South Carolina Neural Tube Defects Program, and clinical history was documented under the surveillance and prevention activities conducted by the Greenwood Genetic Center since 1992. The demographic and clinical parameters of females carrying fetuses with NTDs included in this study are listed in Table 4.
Demographic and clinicopathological findings in NTD cases and mothers
| Characteristics | Groups | Number (n) |
|---|---|---|
| NTD | Isolated | 74 |
| Complex | 30 | |
| NTD type | Anencephaly | 43 |
| Spina bifida | 45 | |
| Encephalocele | 6 | |
| Craniorachischisis | 10 | |
| Ethnicity | Caucasian | 69 |
| African American | 19 | |
| Hispanic | 6 | |
| Maternal age | Mean ± SD (years) | 26 ± 6 (range 14–41) |
| Maternal diabetes | Yes | 2 |
| No | 88 | |
| Maternal obesity | Yes | 10 |
| No | 79 | |
| Folic acid supplementation | Yes | 15 |
| No | 45 |
[i] Information not available: Ethnicity (n = 10), maternal age (n = 3), maternal diabetes (n = 14), maternal obesity (n = 15), and folic acid supplementation (n = 44).
Optical genome mapping
OGM was performed following the manufacturer's protocol (Bionano Genomics, San Diego, CA, USA) as described previously (Sahajpal et al. 2023b). Briefly, ultra-high-molecular-weight DNA was isolated using a nanodisc-based isolation method, which was then labeled with direct labeling (DL)-green fluorophores at a specific six-base sequence motif (CTTAAG) using an enzymatic reaction. The DNA backbone was stained blue and was loaded onto a specialized chip wherein the DNA molecules were linearized with the help of electrophoresis and the ultra-long labeled and stained DNA molecules were imaged at a single molecule resolution on the Saphyr instrument. Analytical quality control (QC) targets were set to achieve >70× effective coverage of the genome, >60% mapping rate, 13–17 label density (labels per 100 kb), and >220 kb N50 (of molecules >150 kb).
OGM variant calling and data analysis
The images of labeled and stained single DNA molecules were converted into digital barcodes from which a de novo genome assembly was constructed using the de novo genome assembly pipeline (Bionano Access (v.1.7)/Solve (v.3.7)) for all samples. The assembled de novo genomes were compared to the reference human genome (GRCh38) for variant calling (deletions, duplications, insertions, inversions, translocations, repeat expansions, and contractions) based on the differences in the alignment of labels between the sample and the reference assembly. Additionally, a coverage-based algorithm enabled the detection of large CNVs and aneuploidies. For data analysis, variants were filtered using the following criteria: (1) The manufacturer's recommended confidence scores were applied: insertion: 0, deletion: 0, inversion: 0.01, duplication: − 1, translocation: 0, and copy number: 0.99 (low stringency, filter set to 0); (2) the GRCh38 SV mask filter that hides any SVs in difficult-to-map regions was turned on for analysis; (3) to narrow the number of variants to be analyzed, polymorphic variants that were present in >1% of an internal OGM control database (n > 300) were removed; (4) The relevance of SVs was ascertained using a stepwise approach utilizing population databases and an internal CMA database (>20,000 chromosomal microarray analyses performed on samples submitted for clinical testing, primarily consisting of individuals affected with rare disorders). Briefly, an unbiased genome-wide SV analysis was performed, and relevance of each variant was ascertained manually, primarily based on frequency in population database (Database of Genomic Variants) and evidence for association/involvement in the NTD/development process based on literature reports. This approach segregated variants into (a) diagnostic findings; (b) SVs overlapping genes associated with NTDs in mouse; and (c) SVs overlapping genes involved in neural tube development pathways. The internal CMA database was reviewed to check the frequency of CNVs in these shortlisted regions/genes, and the phenotypes were compared to rule in/out the SVs for potential association with NTDs.
Confirmation of candidate SVs using orthogonal methods
Quantitative PCR
Quantitative PCR (qPCR) performed on genomic DNA (gDNA) was used to confirm candidate copy number variants. Primer sequences were designed using NCBI and Primer3 software. For each candidate and four controls, PCR amplicons were generated in triplicate using the 7500 real-time thermocycler (Thermo Fisher Scientific, San Francisco, USA). Relative quantitation of genomic dosage was performed using the ΔΔCt method to calculate the copy number of each candidate normalized to the qPCR product of a reference gene in comparison to the control DNA (Livak and Schmittgen 2001).
Chromosomal microarray
gDNA was analyzed using the CytoScan HD assay following the manufacturer's protocol (Thermo Fisher Scientific) (Uddin et al. 2015). Briefly, gDNA was digested by the Nsp1 restriction enzyme and amplified to produce amplicons in the 200–1100 bp range, which were then purified and digested with DNase I to produce 25–125 bp fragments. The fragments were end-labeled with a modified biotinylated base and hybridized overnight (16–18 h) to the array. After incubation, the array was washed and stained with a streptavidin-coupled dye and a biotinylated anti-streptavidin antibody. Finally, the array was scanned with the GeneChip Scanner, to generate cell files that included the intensity probe signals. The data were analyzed using the Chromosome Analysis Suite (ChAs 3.0) software, where the signal for the sample was compared with a reference. Differences in signal between the sample and reference were expressed as a log2 ratio representing relative intensity for each marker. A discrete copy number value was determined from the relative intensity data (Uddin et al. 2015).
Karyotyping and fluorescent in situ hybridization
Karyotype analysis was performed by standard G-banded analysis with 20 metaphases evaluated per case. Fluorescent in situ hybridization was performed using targeted probes with 200 interphase cells analyzed per probe (Cui et al. 2016).
XCI assay
XCI assay was performed using the androgen receptor (AR) locus (Caylor 2023). Briefly, the XCI pattern was determined by PCR analysis of a polymorphic trinucleotide repeat in the first exon of the AR gene with or without digestion with the methylation-sensitive enzyme HpaII (Pegoraro et al. 1994). Samples with a skewing ratio below 80% were classified as “Randomly inactivated,” 80%–90% as “Moderately skewed,” and 91%–100% as “Highly skewed.” Cutoffs were set based on the XCI skewing distribution in healthy individuals (Amos-Landgraf et al. 2006; Shvetsova et al. 2019).
Data access
The raw OGM data generated in this study has been submitted to BioStudies (https://www.ebi.ac.uk/biostudies/studies/S-BSST1664) under accession number S-BSST1664.
Competing interest statement
R.E.S. is a clinical consultant of Bionano Genomics (San Diego, CA, USA). A.C. and A.H. are salaried employees at Bionano Genomics (San Diego, CA, USA).
Acknowledgments
Partial funding for this research was provided by Bionano Genomics (San Diego, CA, USA), and the South Carolina Department of Disabilities and Special Needs (SC, USA). We would like to thank the patients and their families for their participation in this study.
Author contributions: R.E.S. designed the study. R.E.S. and J.D. selected samples for the study. N.S.S. performed data analysis. N.S.S., B.H., and R.E.S. wrote the manuscript. N.S.S., J.D., B.H., T.F., C.S., A.H., B.R.D., A.C., M.J.F., and R.E.S. reviewed the article.
Footnotes
[5] Supplementary material [Supplemental material is available for this article.]
[6] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279318.124.
[7] Freely available online through the Genome Research Open Access option.
References
- ↵Amos-Landgraf JM, Cottle A, Plenge RM, Friez M, Schwartz CE, Longshore J, Willard HF. 2006. X chromosome-inactivation patterns of 1,005 phenotypically unaffected females. Am J Hum Genet 79: 493–499. 10.1086/507565
- ↵Au KS, Hebert L, Hillman P, Baker C, Brown MR, Kim D-K, Soldano K, Garrett M, Ashley-Koch A, Lee S, 2021. Human myelomeningocele risk and ultra-rare deleterious variants in genes associated with cilium, WNT-signaling, ECM, cytoskeleton and cell migration. Sci Rep 11: 3639. 10.1038/s41598-021-83058-7
- ↵Balzer Le S, Onsager I, Lorentzen JA, Lale R. 2020. Dual UTR-A novel 5′ untranslated region design for synthetic biology applications. Synth Biol (Oxf) 5: ysaa006. 10.1093/synbio/ysaa006
- ↵Berry RJ, Li Z, Erickson JK, Li S, Moore CA, Wang H, Mulinare J, Zhao P, Wong LY, Gindler J, 1999. Prevention of neural tube defects with folic acid in China. China-U.S. collaborative project for neural tube defect prevention. N Engl J Med 341: 1485–1490. 10.1056/NEJM199911113412001
- ↵Broeckel U, Iqbal MA, Levy B, Sahajpal N, Nagy PL, Scharer G, Rodriguez V, Bossler A, Stence A, Skinner C, 2024. Detection of constitutional structural variants by optical genome mapping: a multisite study of postnatal samples. J Mol Diagn 26: 213–226. 10.1016/j.jmoldx.2023.12.003
- ↵Caylor RC. 2023. Nonrandom X chromosome inactivation detection. Curr Protoc 3: e748. 10.1002/cpz1.748
- ↵Cui C, Shu W, Li P. 2016. Fluorescence in situ hybridization: cell-based genetic diagnostic and research applications. Front Cell Dev Biol 4: 89. 10.3389/fcell.2016.00089
- ↵Czeizel AE, Dudás I. 1992. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med 327: 1832–1835. 10.1056/NEJM199212243272602
- ↵Dean JH, Pauly R, Stevenson RE. 2020. Neural tube defects and associated anomalies before and after folic acid fortification. J Pediatr 226: 186–194.e4. 10.1016/j.jpeds.2020.07.002
- ↵Dixon M, Kancherla V, Magana T, Mulugeta A, Oakley GPJr. 2019. High potential for reducing folic acid-preventable spina bifida and anencephaly, and related stillbirth and child mortality, in Ethiopia. Birth Defects Res 111: 1513–1519. 10.1002/bdr2.1584
- ↵Eichler EE. 2019. Genetic variation, comparative genomics, and the diagnosis of disease. N Engl J Med 381: 64–74. 10.1056/NEJMra1809315
- ↵El-Hattab AW, Smolarek TA, Walker ME, Schorry EK, Immken LL, Patel G, Abbott MA, Lanpher BC, Ou Z, Kang SH, 2009. Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping. Hum Genet 126: 589–602. 10.1007/s00439-009-0706-x
- ↵Fineman RM, Jorde LB, Martin RA, Hasstedt SJ, Wing SD, Walker ML. 1982. Spinal dysraphia as an autosomal dominant defect in four families. Am J Med Genet 12: 457–464. 10.1002/ajmg.1320120409
- ↵Harris MJ, Juriloff DM. 2010. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res A Clin Mol Teratol 88: 653–669. 10.1002/bdra.20676
- ↵Iqbal MA, Broeckel U, Levy B, Skinner S, Sahajpal NS, Rodriguez V, Stence A, Awayda K, Scharer G, Skinner C, 2023. Multisite assessment of optical genome mapping for analysis of structural variants in constitutional postnatal cases. J Mol Diagn 25: 175–188. 10.1016/j.jmoldx.2022.12.005
- ↵Kälién B, Robert E, Harris J. 1998. Associated malformations in infants and fetuses with upper or lower neural tube defects. Teratology 57: 56–63. 10.1002/(SICI)1096-9926(199802)57:2<56::AID-TERA3>3.0.CO;2-4
- ↵Lee S, Gleeson JG. 2020. Closing in on mechanisms of open neural tube defects. Trends Neurosci 43: 519–532. 10.1016/j.tins.2020.04.009
- ↵Lemay P, De Marco P, Traverso M, Merello E, Dionne-Laporte A, Spiegelman D, Henrion É, Diallo O, Audibert F, Michaud JL, 2019. Whole exome sequencing identifies novel predisposing genes in neural tube defects. Mol Genet Genomic Med 7: e00467. 10.1002/mgg3.467
- ↵Levy-Sakin M, Pastor S, Mostovoy Y, Li L, Leung AKY, McCaffrey J, Young E, Lam ET, Hastie AR, Wong KHY, 2019. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat Commun 10: 1025. 10.1038/s41467-019-08992-7
- ↵Liu W, Wang K, Lv X, Wang Q, Li X, Yang Z, Liu X, Yan L, Fu X, Xiao R. 2020. Up-regulation of RNA binding proteins contributes to folate deficiency-induced neural crest cells dysfunction. Int J Biol Sci 16: 85–98. 10.7150/ijbs.33976
- ↵Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25: 402–408. 10.1006/meth.2001.1262
- ↵Lou J, Balkin N, Stewart JF, Sarwark JF, Charrow J, Nye JS. 2000. Neural tube defects and the 13q deletion syndrome: evidence for a critical region in 13q33-34. Am J Med Genet 91: 227–230. 10.1002/(sici)1096-8628(20000320)91:3<227::aid-ajmg14>3.0.co;2-i
- ↵Lurie IW, Novikova IV, Tarletskaya OA, Lazarevich AA, Gromyko OA. 2016. Distal 13q monosomy and neural tube defects. Genet Couns 27: 177–186.
- ↵Magoulas PL, El-Hattab AW. 2012. Chromosome 15q24 microdeletion syndrome. Orphanet J Rare Dis 7: 2. 10.1186/1750-1172-7-2
- ↵McMohan R, Sibbritt T, Aryamanesh N, Masamsetti VP, Tam PPL. 2022. Loss of FOXD4 impacts neurulation and cranial neural crest specification during early head development. Front Cell Dev Biol 9: 777652. 10.3389/fcell.2021.777652
- ↵Miao X, Sun T, Barletta H, Mager J, Cui W. 2020. Loss of RBBP4 results in defective inner cell mass, severe apoptosis, hyperacetylated histones and preimplantation lethality in mice. Biol Reprod 103: 13–23. 10.1093/biolre/ioaa046
- ↵MRC Vitamin Study Research Group. 1991. Prevention of neural tube defects: results of the MRC vitamin study. Lancet 338: 132–137.
- ↵Neilson KM, Klein SL, Mhaske P, Mood K, Daar IO, Moody SA. 2012. Specific domains of FoxD4/5 activate and repress neural transcription factor genes to control the progression of immature neural ectoderm to differentiating neural plate. Dev Biol 365: 363–375. 10.1016/j.ydbio.2012.03.004
- ↵Niggl E, Bouman A, Briere LC, Hoogenboezem RM, Wallaard I, Park J, Admard J, Wilke M, Harris-Mostert EDRO, Elgersma M, 2023. HNRNPC haploinsufficiency affects alternative splicing of intellectual disability-associated genes and causes a neurodevelopmental disorder. Am J Hum Genet 110: 1414–1435. 10.1016/j.ajhg.2023.07.005
- ↵Odent S, Marec BL, Munnich A, Merre ML, Bonaïti-Pellié C. 1998. Segregation analysis in nonsyndromic holoprosencephaly. Am J Med Genet 77: 139–143. 10.1002/(SICI)1096-8628(19980501)77:2<139::AID-AJMG6>3.0.CO;2-N
- ↵Ohazama A, Haycraft CJ, Seppala M, Blackburn J, Ghafoor S, Cobourne M, Martinelli DC, Fan C-M, Peterkova R, Lesot H, 2009. Primary cilia regulate Shh activity in the control of molar tooth number. Development 136: 897–903. 10.1242/dev.027979
- ↵Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG. 2000. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Bio 151: 709–718. 10.1083/jcb.151.3.709
- ↵Pegoraro E, Schimke RN, Arahata K, Hayashi Y, Stern H, Marks H, Glasberg MR, Carroll JE, Taber JW, Wessel HB, 1994. Detection of new paternal dystrophin gene mutations in isolated cases of dystrophinopathy in females. Am J Hum Genet 54: 989–1003.
- ↵Pegoraro C, Figueiredo AL, Maczkowiak F, Pouponnot C, Eychène A, Monsoro-Burq AH. 2015. PFKFB4 controls embryonic patterning via Akt signalling independently of glycolysis. Nat Commun 6: 5953. 10.1038/ncomms6953
- ↵Pfirrmann T, Villavicencio-Lorini P, Subudhi AK, Menssen R, Wolf DH, Hollemann T. 2015. RMND5 from Xenopus laevis is an E3 ubiquitin-ligase and functions in early embryonic forebrain development. PLoS One 10: e0120342. 10.1371/journal.pone.0120342
- ↵Pulliam L. 2023. “Risk factors for neural tube defects: an overview of the literature and an investigation within South Carolina.” Doctoral dissertation, Clemson University.
- ↵Roessler E, Ouspenskaia MV, Karkera JD, Vélez JI, Kantipong A, Lacbawan F, Bowers P, Belmont JW, Towbin JA, Goldmuntz E, 2008. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am J Hum Genet 83: 18–29. 10.1016/j.ajhg.2008.05.012
- ↵Sadler TW. 2005. Embryology of neural tube development. Am J Med Genet C Semin Med Genet 135C: 2–8. 10.1002/ajmg.c.30049
- ↵Sahajpal NS, Barseghyan H, Kolhe R, Hastie A, Chaubey A. 2021. Optical genome mapping as a next-generation cytogenomic tool for detection of structural and copy number variations for prenatal genomic analyses. Genes (Basel) 12: 398. 10.3390/genes12030398
- ↵Sahajpal NS, Mondal AK, Fee T, Hilton B, Layman L, Hastie AR, Chaubey A, DuPont BR, Kolhe R. 2023a. Clinical validation and diagnostic utility of optical genome mapping in prenatal diagnostic testing. J Mol Diagn 25: 234–246. 10.1016/j.jmoldx.2023.01.006
- ↵Sahajpal NS, Mondal AK, Hastie A, Chaubey A, Kolhe R. 2023b. Optical genome mapping for oncology applications. Curr Protoc 3: e910. 10.1002/cpz1.910
- ↵Savarirayan R, Nance J, Morris L, Haan E, Couper R. 1997. Osteopathia striata with cranial sclerosis: highly variable phenotypic expression within a family. Clin Genet 52: 199–205. 10.1111/j.1399-0004.1997.tb02547.x
- ↵Shvetsova E, Sofronova A, Monajemi R, Gagalova K, Draisma HHM, White SJ, Santen GWE, Chuva de Sousa Lopes SM, Heijmans BT, van Meurs J, 2019. Skewed X-inactivation is common in the general female population. Eur J Hum Genet 27: 455–465. 10.1038/s41431-018-0291-3
- ↵Stoll C, Dott B, Alembik Y, Roth MP. 2011. Associated malformations among infants with neural tube defects. Am J Med Genet A 155A: 565–568. 10.1002/ajmg.a.33886
- ↵Tian H, Feng J, Li J, Ho T-V, Yuan Y, Liu Y, Brindopke F, Figueiredo JC, Magee WIII, Sanchez-Lara PA, 2017. Intraflagellar transport 88 (IFT88) is crucial for craniofacial development in mice and is a candidate gene for human cleft lip and palate. Hum Mol Genet 26: 860–872. 10.1093/hmg/ddx002
- ↵Uddin M, Thiruvahindrapuram B, Walker S, Wang Z, Hu P, Lamoureux S, Wei J, MacDonald JF, Pellecchia G, Lu C, 2015. A high-resolution copy-number variation resource for clinical and population genetics. Genet Med 17: 747–752. 10.1038/gim.2014.178
- ↵Vogel TW, Manjila S, Cohen AR. 2012. Novel neurodevelopmental disorder in the case of a giant occipitoparietal meningoencephalocele. J Neurosurg Pediatr 10: 25–29. 10.3171/2012.3.PEDS11559
- ↵Wang J, Thomas HR, Thompson RG, Waldrep SC, Fogerty J, Song P, Li Z, Ma Y, Santra P, Hoover JD, 2022. Variable phenotypes and penetrance between and within different zebrafish ciliary transition zone mutants. Dis Model Mech 15: dmm049568. 10.1242/dmm.049568
- ↵Winter RM, Md'A C, Meire HB, Mitchell N. 1980. Osteopathia striata with cranial sclerosis: highly variable expression within a family including cleft palate in two neonatal cases. Clin Genet 18: 462–474. 10.1111/j.1399-0004.1980.tb01795.x
- ↵Wu JI, Rajendra R, Barsi JC, Durfee L, Benito E, Gao G, Kuruvilla M, Hrdličková R, Liss AS, Artzt K. 2007. Targeted disruption of Mib2 causes exencephaly with a variable penetrance. Genesis 45: 722–727. 10.1002/dvg.20349
- ↵Yang Y, Zhao S, Sun G, Chen F, Zhang T, Song J, Yang W, Wang L, Zhan N, Yang X, 2022. Genomic architecture of fetal central nervous system anomalies using whole-genome sequencing. NPJ Genom Med 7: 31. 10.1038/s41525-022-00301-4
- ↵Zaganjor I, Sekkarie A, Tsang BL, Williams J, Razzaghi H, Mulinare J, Sniezek JE, Cannon MJ, Rosenthal J. 2016. Describing the prevalence of neural tube defects worldwide: a systematic literature review. PLoS One 11: e0151586. 10.1371/journal.pone.0151586