
A prospective trial comparing programmable targeted long-read sequencing and short-read genome sequencing for genetic diagnosis of cerebellar ataxia
- Haloom Rafehi1,2,43,
- Liam G. Fearnley1,2,43,
- Justin Read3,4,43,
- Penny Snell3,
- Kayli C. Davies3,5,
- Liam Scott1,
- Greta Gillies3,
- Genevieve C. Thompson3,5,
- Tess A. Field3,
- Aleena Eldo3,
- Simon Bodek6,
- Ernest Butler7,
- Luke Chen8,
- John Drago9,10,
- Himanshu Goel11,
- Anna Hackett11,12,
- G. Michael Halmagyi13,14,
- Andrew Hannaford15,16,17,
- Katya Kotschet18,
- Kishore R. Kumar19,20,21,22,
- Smitha Kumble23,24,
- Matthew Lee-Archer25,
- Abhishek Malhotra26,
- Mark Paine27,
- Michael Poon28,
- Kate Pope3,
- Katrina Reardon9,29,
- Steven Ring30,
- Anne Ronan12,31,
- Matthew Silsby15,16,17,
- Renee Smyth32,
- Chloe Stutterd23,
- Mathew Wallis33,34,
- John Waterston4,
- Thomas Wellings35,
- Kirsty West36,
- Christine Wools37,38,
- Kathy H.C. Wu32,39,40,
- David J. Szmulewicz41,42,
- Martin B. Delatycki3,5,23,
- Melanie Bahlo1,2,44 and
- Paul J. Lockhart3,5,44
- 1Population Health and Immunity Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3052, Australia;
- 2Department of Medical Biology, University of Melbourne, Parkville, Victoria 3052, Australia;
- 3Bruce Lefroy Centre, Murdoch Children's Research Institute, Royal Children's Hospital, Parkville, Victoria 3052, Australia;
- 4Department of Neuroscience, Central Clinical School, Monash University, The Alfred Centre, Melbourne, Victoria 3004, Australia;
- 5Department of Paediatrics, University of Melbourne, Royal Children's Hospital, Parkville, Victoria 3052, Australia;
- 6Austin Health, Heidelberg, Victoria 3084, Australia;
- 7Monash Medical Centre, Clayton, Victoria 3168, Australia;
- 8Department of Neurology, Alfred Hospital, Melbourne, Victoria 3004, Australia;
- 9Department of Medicine, St Vincent's Hospital, University of Melbourne, Fitzroy, Victoria 3065, Australia;
- 10Florey Institute of Neuroscience and Mental Health, Parkville, Victoria 3052, Australia;
- 11Hunter Genetics, Hunter New England Health Service, Waratah, New South Wales 2298, Australia;
- 12University of Newcastle, Callaghan, New South Wales 2308, Australia;
- 13Neurology Department, Royal Prince Alfred Hospital, Camperdown, New South Wales 2050, Australia;
- 14Central Clinical School, University of Sydney, Camperdown, New South Wales 2050, Australia;
- 15Department of Neurology, Westmead Hospital, Hawkesbury Westmead, New South Wales 2145, Australia;
- 16Brain and Nerve Research Centre, Concord Clinical School, University of Sydney, Camperdown, New South Wales 2050, Australia;
- 17Department of Neurology, Concord Repatriation General Hospital, Concord, New South Wales 2139, Australia;
- 18Department of Clinical Neurosciences, St Vincent's Hospital, University of Melbourne, Fitzroy, Victoria 3065, Australia;
- 19Molecular Medicine Laboratory and Neurology Department, Concord Repatriation General Hospital, Concord, New South Wales 2139, Australia;
- 20Faculty of Medicine and Health, The University of Sydney, Camperdown, New South Wales 2050, Australia;
- 21Genomics and Inherited Disease Program, The Garvan Institute of Medical Research, Darlinghurst, New South Wales 2010, Australia;
- 22School of Medicine, University of New South Wales, Sydney, New South Wales 2052, Australia;
- 23Victorian Clinical Genetics Services, Murdoch Children's Research Institute, Royal Children's Hospital, Parkville, Victoria 3052, Australia;
- 24Department of Clinical Genetics, Austin Health, Viewbank, Victoria 3084, Australia;
- 25Department of Neurology, Launceston General Hospital, Launceston, Tasmania 7250, Australia;
- 26Department of Neuroscience, University Hospital Geelong, Geelong, Victoria 3220, Australia;
- 27Department of Neurology, Royal Brisbane and Women's Hospital, Herston, Queensland 4006, Australia;
- 28Neurology Footscray, Footscray, Victoria 3011, Australia;
- 29Department of Neurology, St Vincent's Hospital, University of Melbourne, Fitzroy, Victoria 3065, Australia;
- 30Albury Wodonga Health, West Albury, New South Wales 2640, Australia;
- 31Newcastle Medical Genetics, Lambton, New South Wales 2299, Australia;
- 32St Vincent's Clinical Genomics, St Vincent's Hospital, Darlinghurst, New South Wales 2010, Australia;
- 33Tasmanian Clinical Genetics Service, Tasmanian Health Service, Royal Hobart Hospital, Hobart, Tasmania 7001, Australia;
- 34School of Medicine and Menzies Institute for Medical Research, University of Tasmania, Hobart, Tasmania 7000, Australia;
- 35Department of Neurology, John Hunter Hospital, New Lambton Heights, New South Wales 2305, Australia;
- 36Genomic Medicine, The Royal Melbourne Hospital, Parkville, Victoria 3052, Australia;
- 37Department of Neurology, Calvary Health Care Bethlehem, Caulfield South Victoria 3162, Australia;
- 38Department of Neurology, The Royal Melbourne Hospital, Parkville, Victoria 3052, Australia;
- 39School of Medicine, University of Notre Dame, Darlinghurst, New South Wales 2010, Australia;
- 40Discipline of Genomic Medicine, Faculty of Medicine and Health, University of Sydney, Camperdown, New South Wales 2050, Australia;
- 41Royal Victorian Eye and Ear Hospital, East Melbourne, Victoria 3002, Australia;
- 42Bionics Institute, East Melbourne, Victoria 3002, Australia
Abstract
The cerebellar ataxias (CAs) are a heterogeneous group of disorders characterized by progressive incoordination. Seventeen repeat expansion (RE) loci have been identified as the primary genetic cause and account for >80% of genetic diagnoses. Despite this, diagnostic testing is limited and inefficient, often utilizing single gene assays. This study evaluates the effectiveness of long- and short-read sequencing as diagnostic tools for CA. We recruited 110 individuals (48 females, 62 males) with a clinical diagnosis of CA. Short-read genome sequencing (SR-GS) was performed to identify pathogenic RE and also non-RE variants in 356 genes associated with CA. Independently, long-read sequencing with adaptive sampling (LR-AS) was performed to identify pathogenic RE. SR-GS provided a genetic diagnosis for 38% of the cohort (40/110) including seven non-RE pathogenic variants. RE causes disease in 33 individuals, with the most common condition being SCA27B (n = 24). In comparison, LR-AS identified pathogenic RE in 29 individuals. RE identification for the two methods was concordant apart from four SCA27B cases not detected by LR-AS due to low read depth. For both technologies manual review of the RE alignment enhances diagnostic outcomes. Orthogonal testing for SCA27B revealed a 15% and 0% false positive rate for SR-GS and LR-AS, respectively. In conclusion, both technologies are powerful screening tools for CA. SR-GS is a mature technology currently used by diagnostic providers, requiring only minor changes in bioinformatic workflows to enable CA diagnostics. LR-AS offers considerable advantages in the context of RE detection and characterization but requires optimization before clinical implementation.
Genetic technologies are transforming healthcare by enabling genomic medicine, an emerging discipline that uses genetic data to improve clinical care and outcomes. High-throughput sequencing (HTS) is an important tool for genomic medicine, underpinning discovery, diagnostics, and understanding of disease mechanisms (Rehm 2017). Genomic medicine provides diagnostic certainty, key information for prognosis, genetic counseling, and reproductive planning and facilitates the development and delivery of improved treatments targeted to disease mechanisms (McCarthy et al. 2013; García-Foncillas et al. 2021). There is a significant treatment pipeline for personalized therapies, including gene and pharmacological therapies. For instance, treatment for spinocerebellar ataxia 27B (SCA27B) shows promising therapeutic outcomes with 4-aminopyridine (4-AP) (Wilke et al. 2023). However, genetic technologies have had limited success in extending genomic medicine's benefits to disorders caused by pathogenic nucleotide repeat expansions (REs).
RE disorders typically have significant neurological and/or neuromuscular outcomes. They occur when a segment of repetitive DNA, termed a short tandem repeat (STR), expands beyond a gene-specific threshold. STR is composed of tandem arrays of 1–12 bp sequence motifs and constitute ∼6% of the human genome (Willems et al. 2014; Mousavi et al. 2019). To date, the genetic basis of 79 RE disorders has been described (for reviews, see Cortese et al. 2024; Rajan-Babu et al. 2024), including polyglutamine disorders such as Huntington's disease, fragile X syndrome, and hereditary cerebellar ataxias (CA). RE can occur within exonic, intronic, untranslated, or intergenic regions and are associated with various pathogenic mechanisms including loss-of-function, gain-of-function, transcriptional dysregulation, protein misfolding/aggregation, and repeat-associated non-AUG (RAN) translation. Some disorders result from combinations of these mechanisms (for review, see Depienne and Mandel 2021). Collectively, RE disorders cause some of the most common genetic disorders seen by neurologists (Paulson 2018). Moreover, the RE-mediated disease burden is significantly underestimated. RE is difficult to amplify using standard molecular technologies such as polymerase chain reaction (PCR) (Schlötterer and Tautz 1992) and there is evidence suggesting that additional RE remains to be identified (Depienne and Mandel 2021). In addition, RE is often located in noncoding DNA, “hidden” from the gene, panel- and exome-based discovery approaches. Pathogenic RE embedded deep within nonpathogenic STR (Rosenbohm et al. 2022) or composed of novel motifs presents additional challenges (Dolzhenko et al. 2020). The timeline of RE identification demonstrates the issues. While the first pathogenic RE was described in 1991 (La Spada et al. 1991; Oberlé et al. 1991; Verkerk et al. 1991), ∼50% of pathogenic RE identified to date have been described in the last 10 years. The acceleration in discovery has been driven largely by the development of PCR-free short-read and long-read genomic sequencing technologies and associated bioinformatic tools (Depienne and Mandel 2021; Gall-Duncan et al. 2022; Read et al. 2023).
RE disorders also challenge diagnostic service providers, impacting the implementation of genomic medicine. One issue is the relatively low incidence of some RE disorders; in many settings, it is not economically viable to provide current, RE-specific diagnostic testing for loci/conditions with low prevalence (Stevanovski et al. 2022). In addition, technological challenges exist in the molecular characterization of expanded repeat DNA. Traditional diagnostic techniques such as Southern blot, PCR sizing, and repeat-primed PCR (RP-PCR) analysis are labor-intensive, imprecise and do not scale easily to large cohorts requiring testing; generally, each individual RE requires a separate assay with specific probes or primers (Bahlo et al. 2018). Exome analysis using short-read HTS has become a mainstay diagnostic methodology for diagnosing pathogenic single nucleotide variants (SNVs), small insertions and deletions (indel), or copy number variants (CNVs) but is yet to be widely implemented in RE diagnostics (Lappalainen et al. 2019). While the technology can successfully identify RE (Tankard et al. 2018), there are limitations. Genomic DNA is fragmented and size-selection is performed to isolate inserts of ∼250–1000 bp. Therefore, larger RE may not be represented in this process. In addition, the capture probes used may be disrupted by the RE and not efficiently hybridize to the expanded allele. Moreover, like other high-throughput diagnostic technologies such as multiplex amplicon sequencing and targeted gene panel analysis, exome sequencing uses PCR amplification of the captured target sequences, which also may not work efficiently, or at all, with large and complex RE. While bioinformatic tools now available in the research space allay concerns about the effects of PCR stutter and nonunique mapping of repetitive HTS reads (Bahlo et al. 2018; Tankard et al. 2018), they are yet to be widely implemented in diagnostic pipelines. In addition, another shortcoming of exome sequencing for diagnosing pathogenic RE is that most loci are outside coding regions and therefore not captured by current “off-the-shelf” library preparation kits.
Hereditary CAs exemplify the diagnostic and healthcare challenges of RE disorders. CA is a heterogeneous group of rare, incurable, and often life-limiting disorders characterized by progressive incoordination (Jayadev and Bird 2013). There are ∼100 clinically recognized CAs, with similar numbers caused by autosomal dominant and autosomal recessive mechanisms. The prevalence of these disorders varies widely depending on genetic ancestry and geographical location, with estimates of a global average of ∼6:100,000 (Ruano et al. 2014; Rudaks et al. 2024). These debilitating disorders predominantly impact locomotion, hand coordination, speech, swallowing, and vision. Apart from the recent approval of omaveloxolone for Friedreich ataxia (FRDA) (Lee 2023), there are no disease-modifying treatments for these conditions. Treatment consists of symptom management, such as using adaptive devices and ongoing physical and occupational therapy (de Silva et al. 2019). The predominant genetic cause of CA, accounting for over 80% of diagnoses, is a pathogenic RE in one of the 17 loci associated with CA identified to date. Non-RE pathogenic variants in ∼100 other genes also contribute to disease prevalence (Beaudin et al. 2019; Rudaks et al. 2024), presenting a daunting panel of unusual variants and genes requiring examination. Diagnostic testing methodologies and outcomes for CA vary broadly. In Australia, standard clinical diagnostic testing uses single PCR/capillary array assays for between five and seven RE loci. Our center, servicing a population of ∼6 million, only tests for six RE-causing CAs (spinocerebellar ataxia [SCA]1/2/3/6/7 and FRDA) with a diagnostic rate of ∼5% (unpublished data 2015–2022). A higher diagnostic rate can be achieved by utilizing multiple testing methodologies to ensure the evaluation of multiple genetic variant types, including RE, SNV/indel, and CNV, with yields ranging between ∼30% and 60% (Hadjivassiliou et al. 2017; Kang et al. 2019). However, these comprehensive, long-term studies were performed in a research setting. Clinical service providers prefer to deploy a single frontline test with high sensitivity and specificity to maximize yield and minimize cost. Notably, the yield for CA with short-read exome sequencing, arguably the most used and cost-effective frontline diagnostic test, is only ∼25% (Rexach et al. 2019; Ngo et al. 2020).
Two technologies have recently been proposed as potential rapid and comprehensive diagnostic methods for RE disorders. Single-molecule long-read sequencing (LRS) routinely generates reads >10 kb in length and supports de novo assembly and detection of all variant types including structural variants (Amarasinghe et al. 2020). The two leading LRS providers are Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT). PacBio performs sequencing by synthesis and requires the construction of a library of circular DNA molecules. In contrast, ONT is a nanopore-based technology. The DNA sequence is determined by measuring nucleotide-specific current changes as the molecule passes through a nanopore. While both technologies have strengths and weaknesses, one recent application of ONT appears to have considerable potential for RE. Single-molecule long-read sequencing using adaptive sampling (LR-AS, ONT) enables user-defined target selection in real time (Payne et al. 2021). This technology allows simultaneous screening of multiple target loci, capturing up to ∼4% of the genome. Two recent proofs of concept pilot studies have demonstrated the method's potential utility to diagnose RE disorders by simultaneous analysis of 37 (Stevanovski et al. 2022) or 59 (Miyatake et al. 2022a) RE loci in small retrospective cohorts of 37 and 22 patients, respectively. Alternatively, short-read genome sequencing (SR-GS) is emerging as a potential replacement for exome sequencing in rare disorders. This shift is driven by recognition of the gains achievable in diagnostic rates and the availability and decreasing cost of genome sequencing. Standard diagnostic analysis pipelines previously had limited capability to assess RE loci (Ashley 2016; Turro et al. 2020); however, recent advances in bioinformatic tools and technologies mean that it is now feasible to efficiently detect both non-RE and RE pathogenic variants in SR-GS (Leitão et al. 2024). Notably, a large study recently demonstrated high sensitivity and specificity (>97%) to detect 13 pathogenic RE in a retrospective cohort of 404 patients. The prospective analysis of these 13 RE in 11,631 patients identified 81 RE with a false discovery rate of 16% (Ibañez et al. 2022).
Here, we report a prospective trial comparing diagnostic outcomes of standard clinical testing in the Australian context (five pathogenic RE) with both short-read and long-read technologies in a cohort of 110 individuals with a clinical diagnosis of CA and clearly demonstrate the substantial diagnostic gains achievable for individuals and families affected by CA.
Results
Participant recruitment and details
The study design is summarized in Figure 1. A cohort of 110 individuals with a clinical diagnosis of CA, referred for diagnostic testing, were recruited to the research program over a 2-year period (2022–2023). Individuals were excluded if there was clinical suspicion of an acquired cause of ataxia, based on the history of acute injury or illness, toxic exposure, or rapid onset. All participants were singletons, 17% had a family history of ataxia, and included 48 female/62 male individuals with adult-onset ataxia. The mean age at onset was 56 ± 14 years (range 15–77) and the mean age at testing 68 ± 13 years (range 29–88). All individuals completed diagnostic testing for five pathogenic RE in SCA1, SCA2, SCA3, SCA6, and SCA7 before research-based genetic testing. Any individuals with a positive diagnostic test result were excluded from the trial.

Overview of the study and investigations performed. (SCA) spinocerebellar ataxia, (EH5) ExpansionHunter5, (RE) repeat expansion, (LP) likely pathogenic, (P) pathogenic, (VUS) variant of uncertain significance.
RE identification with short-read genome sequencing
The primary purpose of this trial was to compare the performance of SR-GS and LR-AS for the identification of pathogenic RE. At the time of trial initiation (2022), there were 67 known pathogenic REs, 17 directly associated with CA, and the remainder with a broader range of neurogenetic, neuromuscular, and other health conditions. Therefore, all 67 REs were interrogated as part of this program. However, given the cohort composition and clinical indication for study inclusion was CA, a specific focus of the analysis was the 22 REs listed in Table 1. This included the 17 REs associated with CA and five additional pathogenic REs that cause neurological movement disorders that could possibly be a differential diagnosis for our cohort. In addition, these five REs are also commonly tested with single gene assays at our center (Victorian Clinical Genetics Services, VCGS) and more broadly. Therefore, they represent appropriate, cost-effective targets for inclusion in a diagnostic single test.
Repeat expansion target loci for CAs and clinically significant noncerebellar ataxia conditions (non-CAs), adapted from Tankard et al. (2018) and Depienne and Mandel (2021)
ExpansionHunter5 (EH5) (Dolzhenko et al. 2019) was used to genotype the 22 RE loci in the SR-GS data. A postanalysis custom k-mer filter was used to remove genotype calls where the predominant motif detected in the reads does not match the motif tested by EH5 (https://doi.org/10.5281/zenodo.11514479). All individuals with potential expansions were also manually reviewed in the Integrative Genomics Viewer (IGV) (Robinson et al. 2011) to confirm motif composition. We detected two individuals with dominant ATXN8OS expansions greater than the pathogenic threshold for SCA8 (Fig. 2A). One individual was found to have an expanded allele for NOP56; however, the estimated size (317 repeats) is lower than the pathogenic threshold (650 repeats) for SCA36 (Fig. 2A). We have previously shown that expansions in NOP56 are typically underestimated by EH5 due to their large size vastly exceeding the read length (Rafehi et al. 2020). In addition, the NOP56 STR is stable in the general population, with most alleles reported in gnomAD in the 4–10 repeats range, and only three alleles between 15 and 21 repeats. Therefore, any individual with an NOP56 expansion ≥30 repeats is further tested to confirm or exclude an SCA36 diagnosis. Manual review confirmed a GGGCCT expansion at the locus.
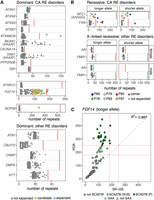
Targeted RE screening in SR-GS identifies potential RE diagnoses. STR genotypes were determined for the 22 loci associated with CA (Table 1) with EH5. (A) Genotypes are shown for the longer allele in dominant RE disorders that cause ataxia (top 3 plots, loci separated based on maximum allele size) and those that cause other disorders (bottom). Blue circles indicate individuals with an expansion in the respective allele that is larger than the pathogenic threshold (solid red line), while yellow circles are individuals who exceed an EH5-specific threshold (dashed red line) and are candidates for further investigation. Benign AAAAT motifs do not have a threshold. (B) Genotypes for the shorter and longer alleles are shown for autosomal recessive RE ataxia disorders (top) and X-linked recessive disorders other than ataxia, split by sex (bottom). Individuals who are heterozygous carriers for an allele expanded beyond the pathogenic threshold (solid red line) or an allele larger than the EH5-specific threshold (dashed red line) are shown as brown squares. For FMR1, the blue dashed line indicates the threshold for FXTAS while the solid red line is the threshold for FXS. Individuals who carry two alleles expanded beyond either the pathogenic or EH5-specific threshold are shown as colored circles. (C) Concordance plot showing a comparison of the FGF14 STR EH5 genotypes from SR-GS compared to PCR sizing (longer allele only). The pathogenic (≥250 repeats) and VUS (≥180 repeats) thresholds are shown as dashed red lines. A 1:1 correlation is shown as a dashed blue line. The R2 is a Pearson's correlation. Dark green circles are individuals with a confirmed SCA27B diagnosis and those in light green are SCA27B VUS. Circles indicate GAA motifs and squares are non-GAA motifs, determined by observation in IGV.
FGF14 STR size is also known to be underestimated by RE calling in SR-GS data. Our previous work identified high concordance between PCR and SR-GS sizing for STR up to ∼(GAA)100; however, larger RE is typically underestimated by RE genotyping tools such as EH5 (Rafehi et al. 2023). In contrast to NOP56, in which outliers are easy to detect, the FGF14 GAA STR is highly unstable and there is significant variation in the length in the general population. As a result, it is not possible to distinguish pathogenic expansions (≥250) from alleles of ∼100–249 using EH5. Based on previous experience with this locus, we prioritize any individual with an EH5 genotype call of ≥90 repeats as candidates for SCA27B. Using this approach, we identified 37 individuals suspected to have SCA27B (Fig. 2A). Manual review confirmed that all 37 REs were composed of pure (GAA)n.
Expanded sequences were also detected for the benign reference motif (AAAAT) in BEAN1 and DAB1 (Fig. 2A), but neither of the pathogenic motifs (TGGAA) nor (ATTTC) were identified in the short-read data. Although expansions of the reference motif alone are not pathogenic for these two loci, we cannot exclude the possibility that these individuals have a pathogenic motif embedded deep within the reference motif as these cannot be detected with SR-GS (Rosenbohm et al. 2022).
Analysis of recessive loci also identified potential pathogenic RE in FXN and RFC1. One individual has two expanded alleles in FXN, consistent with a diagnosis of FRDA (Fig. 2B). We also identified four individuals with a single expanded FXN allele (Supplemental Table S1). We tested for the possibility of a second pathogenic small variant or CNV on the other FXN allele utilizing research tools seqr and CXGo; however, no candidate variants were identified, suggesting these individuals are heterozygous carriers. Testing for the nonreference pathogenic AAGGG motif in RFC1 did not identify any individuals with an RE greater than the pathogenic threshold (>250 repeats) (Dominik et al. 2023). However, we have previously shown that AAGGG RE sizing of RFC1 is underestimated in SR-GS (Rafehi et al. 2019). Therefore, any individual with an AAGGG allele ≥30 repeats is considered likely to carry a pathogenic RE in RFC1. Using this threshold, we identified five individuals with pathogenic biallelic RE in RFC1, consistent with the clinical presentation of CA, neuropathy, and vestibular areflexia syndrome (CANVAS) in these individuals (Table 2). In addition, we identified three individuals with a heterozygous pathogenic AAGGG RFC1 allele (Supplemental Table S1) but failed to identify a second pathogenic variant on the other allele, suggesting they are carriers but that this is not the cause of their presentation.
Repeat expansion variant findings
Overall, this analysis identified 46 individuals with potentially pathogenic RE causing their clinical presentation, including heterozygous RE in ATXN8/ATXN8OS (2×), NOP56 (1×), and FGF14 (37×), and biallelic RE in FXN (1×) and RFC1 (5×).
Molecular validation of RE identified by SR-GS
We and others have shown that SR-GS has high sensitivity and specificity for the identification and sizing of pathogenic RE when the DNA repeat length is similar or smaller than the standard SR-GS read length of 150 bp (Dolzhenko et al. 2017; Tankard et al. 2018). In contrast, while SR-GS demonstrates high specificity/sensitivity for larger pathogenic RE, such as those causing SCA36 and myotonic dystrophy 2 (DM2), it significantly underestimates the size of the pathogenic allele (Day et al. 2003; Rafehi et al. 2020). We previously demonstrated that size estimates of the RE in FGF14 that cause SCA27B are unreliable when the expansion is greater than ∼(GAA)100 (Rafehi et al. 2023), suggesting SR-GS may have poor specificity and/or sensitivity for this RE. Therefore, we performed orthogonal molecular testing of all individuals with potential pathogenic RE identified by SR-GS (FXN via clinical diagnostic test; NOP56 and RFC1 via RP-PCR; ATXN8/ATXN8OS and FGF14 via RP-PCR and flanking PCR/capillary array sizing). This analysis confirmed the SR-GS results for the nine individuals with pathogenic RE in ATXN8/ATXN8OS, FXN, NOP56, and RFC1 (Table 2). Of the 37 individuals with a potential pathogenic RE in FGF14 only 24 were confirmed to be expanded above the current pathogenic threshold of (GAA) ≥ 250 (Pellerin et al. 2023; Rafehi et al. 2023). We subsequently used long-range PCR to also size the FGF14 locus in the remaining 73 individuals in the cohort, which SR-GS suggested were nonpathogenic. This analysis demonstrated significant divergence in the size of the larger allele estimated by EH5 compared to PCR (Fig. 2C). These results demonstrate that SR-GS has a sensitivity of 100% and specificity of 85% to identify pathogenic RE in FGF14 when utilizing an EH5 estimate of 90 repeats as the pathogenic threshold (Supplemental Table S2). In addition, the PCR analysis identified nine individuals with an FGF14 GAA allele >179 but <250 repeats; these were classified as variants of uncertain significance (VUSs) (Supplemental Table S3; Mohren et al. 2024). Overall, while SR-GS identified 46 individuals with a potentially pathogenic RE (42% of the cohort), the actual diagnostic rate achieved was 33/110 (30%), the discrepancy being the result of 13 false positive FGF14 diagnoses (i.e., a GAA repeat <250 repeats).
Identification of non-RE pathogenic variants with SR-GS
In addition to RE, a variety of other pathogenic variants can cause CA (Beaudin et al. 2019; Rudaks et al. 2024). Therefore, we analyzed the SR-GS data for SNV/indel, CNV, and mitochondrial DNA (mtDNA) variants. No CNV or mtDNA variants were identified but an additional seven patients were solved by identification of likely pathogenic/pathogenic (LP/P) variants in ANO10 (autosomal recessive spinocerebellar ataxia 10; SCAR10), CACNA1G (SCA42), HEXA (Tay Sachs disease), PNPT1 (SCA25), SPG7 (spastic paraplegia 7; SPG7), STUB1 (SCA48), and TTBK2 (SCA11) (Table 3). This analysis also identified an additional nine individuals with suspicious variants that do not meet ACMG guidelines for classification as LP/P (Richards et al. 2015) and were, therefore, classified as VUS (Supplemental Table S4). The overall diagnostic yield achieved by SR-GS was 38% (33× RE, 7× non-RE), with a false positive and negative rate of 12% and 0%, respectively, for FGF14 RE (SCA27B).
Single nucleotide and small insertion or deletion variant findings
RE identification with long-read adaptive sequencing
Targeted sequencing is an effective tool that provides many advantages for genomic medicine, prioritizing relevant candidate genes for phenotype and significantly reducing per test cost and analysis burden. However, multiplex PCR-based enrichment and sequencing technologies have several limitations in the context of characterizing pathogenic RE, including PCR stutter affecting both RE size and composition and an inability to amplify large pathogenic RE. Alternative physical enrichment technologies that do not require PCR amplification have been developed, including CRISPR–Cas9 enrichment. However, these enrichment approaches are bespoke, difficult to multiplex, and are limited to ∼50 targets per library preparation. Recently, the development of LR-AS has provided a mechanism to couple multiplex targeting with LRS at a reasonable cost. Therefore, LR-AS was performed independently to SR-GS, targeting 334 genomic regions (111 Mb/∼3.59% of the GRCh38 [hg38] reference genome), which included 67 genes with a pathogenic RE and other non-RE genes associated with CA (Supplemental Table S5). Each sample was analyzed using a single MinION flow cell. Summary statistics are presented in Figure 3 and detailed individual sample/locus results are described in Supplemental Table S5. The on-target mean read length was 4727 bp (95% CI 2728, 6014) compared to 480 bp (95% CI 444, 493) for off-target sequence, confirming rapid and efficient rejection of nontargeted regions of the genome (Fig. 3A). The on-target mean sequencing depth was 22.1 (95% CI 12.7, 31.0) compared to 3.0 (95% CI 1.7, 4.1) for off-target regions (Fig. 3B), resulting in a mean 8.2-fold (95% CI 5.3-fold, 10.4-fold) target enrichment (Fig. 3C). Consistent with the SR-GS analysis, we focused analysis on the 22 REs listed in Table 1. The distribution of read counts across all samples at these 22 loci (Fig. 3D,E) ranged from a mean of 16.8 reads (AR) to 28.2 reads (C9orf72) across the 110 individuals in the cohort.
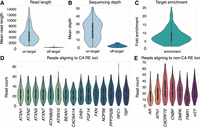
Performance metrics for adaptive sequencing in a targeted panel of RE loci. (A) Mean read length in target regions significantly exceeds that of off-target regions. (B) Sequencing depth in on- versus off-target regions. (C) Genome-wide enrichment of on- versus off-target regions in individual experiments. (D) Number of reads aligning to each of the targeted CA repeat loci. (E) Number of reads aligning to each targeted clinically significant non-CA locus.
Given that clinical testing excluded individuals with SCA1/2/3/6 and 7 from the cohort, we first tested if LR-AS was effective in identifying the corresponding pathogenic RE by analyzing individuals with known pathogenic RE status. Affected individuals, not part of the cohort, with known pathogenic RE in ATXN1, ATXN2, ATXN3, CACNA1A, and ATXN7 were subject to LR-AS and genotypes were compared to allele sizes determined by diagnostic testing (flanking PCR and capillary array analysis). LR-AS identified two alleles, one pathogenic and one nonpathogenic, for ATXN1, ATXN2, ATXN3, and ATXN7. The calculated allele sizes were concordant with those obtained by diagnostic testing (Supplemental Table S6). However, Straglr only identified a single allele (14 repeats) for CACNA1A, compared to the diagnostic result of two alleles of 23 and 12 repeats. Visual inspection in IGV showed four spanning reads with 22–23 repeats, and 10 spanning reads with 12 repeats. It is not clear why Straglr was unable to call two alleles; it may be that the relatively low read depth and allelic bias (4 reads vs. 10 reads) confounded the analysis. However, ATXN7 was called successfully despite lower read depth and similar allelic bias. Alternatively, the small relative difference in size between pathogenic and nonpathogenic alleles may have contributed to only a single allele being identified. Analysis of the LR-AS data using the alternate algorithm vamos, which can interrogate both motif size and composition (Ren et al. 2023) similarly failed to call the pathogenic RE in the SCA6-positive sample (Supplemental Table S6). While previous studies have suggested that LR-AS can effectively identify pathogenic RE in these five genes, they also highlighted the utility of manual inspection/visual confirmation of bioinformatic allele size estimations (Miyatake et al. 2022a; Stevanovski et al. 2022). This is a limitation that also impacts SR-GS as described above.
We subsequently extended the LR-AS analysis to the trial cohort, identifying individuals with potentially pathogenic RE by determining if their allele size estimates exceeded thresholds in a process similar to that used for SR-GS (Methods; Table 1). Overall, before any manual review, Straglr identified RE that supported 30 potential diagnoses. For the dominant CA disorders, Straglr identified heterozygous pathogenic RE in ATXN8OS in two individuals (Fig. 4). One RE was estimated to be 124 repeats, similar to the size estimated by orthogonal PCR analysis (134 repeats), while the second RE was estimated to be 768 repeats (Table 2), which is larger than can be determined by PCR. Heterozygous pathogenic REs in FGF14 (≥250 GAA) were identified in 20 individuals and showed very high concordance with the PCR sizing results (R2 = 0.997) (Fig. 5A; Table 2). Expanded sequences were also detected for the benign reference motif (AAAAT) in BEAN1 and DAB1 (Fig. 4A) but not the pathogenic motifs (TGGAA) or (ATTTC). Manual review of the aligned reads did not provide any evidence of pathogenic motifs embedded deep within the expanded sequence, which would have been missed with SR-GS (Supplemental Fig. S1).
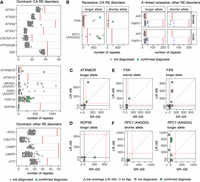
Targeted RE screening panel in LR-AS and comparison to SR-GS. STR genotypes were determined for short-listed loci with Straglr. Green points indicate individuals with a confirmed clinical diagnosis for the respective locus. (A) Genotypes are shown for the longer allele in dominant RE disorders that cause ataxia (top 2 plots, loci separated based on maximum allele size) and those that cause other disorders (bottom). (B) Genotypes for the shorter and longer alleles are shown for autosomal recessive RE ataxia disorders (left) and X-linked recessive disorders other than ataxia, split by sex (right). Comparison of allele genotyping with SR-GS and LR-GS is shown for loci with a confirmed diagnosis for the longer allele in dominant disorders. (C) ATXN8OS (SCA8), (D) NOP56 (SCA36), and for both alleles in recessive disorders for (E). FXN (FRDA) and (F). RFC1 (CANVAS, AAGGG motif only). LR-AS with low coverage (≤7 reads) are shown as triangles, those with no coverage issues flagged (>7 reads) are shown as circles. Green circles indicate individuals with an expansion in the respective allele that is larger than the pathogenic threshold (solid or dashed red line). Benign AAAAT motifs do not have a threshold. For FMR1, the blue dashed line indicates the threshold for FXTAS while the solid red line is the threshold for FXS.
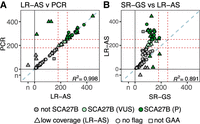
Comparison of FGF14 STR sizing between LR-AS, SR-GS, and PCR identifies the strengths of LR-AS. Comparison of FGF14 STR sizing is shown as concordance plots for (A). LR-AS compared to PCR and (B). LR-AS compared to SR-GS. The R2 is a Pearson's correlation. Dark green circles are individuals with a confirmed SCA27B diagnosis (P) and those in light green are SCA27B VUS. Triangles indicate low coverage (≤7 reads) on LR-AS, squares indicate non-GAA repeat motifs, and circles samples with coverage >7 reads and GAA motifs. The pathogenic (≥250 repeats) and VUS (≥180 repeats) thresholds are shown as dashed red lines. A 1:1 correlation is shown as a dashed blue line. The R2 is a Pearson's correlation. Individuals with no genotype call from either LR-AS, SR-GS, or PCR are indicated separately to the numbered axis and are labeled nc (i.e., no genotype call).
Analysis of recessive loci also identified expanded RE in FXN and RFC1. Pathogenic FXN RE of 947 and 296 repeats were identified in one individual, confirming a molecular diagnosis of FRDA. Three other individuals were identified with a pathogenic GAA RE in FXN, with estimated sizes of 629, 570, and 160 repeats, respectively (Fig. 4B). However, the second allele for each individual was in the normal range (32, 21, and 8 repeats, respectively), identifying these individuals as heterozygous carriers (Supplemental Table S1). Pathogenic RE in RFC1 was identified by Straglr in six individuals. Five of these were called “homozygous” REs for the pathogenic AAGGG, i.e., only a single allele size was reported. The estimated allele sizes of 872, 794, 945, 395, and 1109 repeats exceed the current pathogenic threshold of >250 repeats (Dominik et al. 2023). Three of these were confirmed as biallelic expansions by visual review of aligned reads (Table 2), while the other two (395 and 1109 repeats) were shown to be heterozygous for a single pathogenic AAGGG RE. In both cases, Straglr did not identify (call) the second nonpathogenic allele. On visual inspection in IGV, the samples appeared to be heterozygous in motif as well as size, with both samples having one pathogenic AAGGG allele, and a second AAAAG allele. Subsequent flanking PCR and gel electrophoresis confirmed the presence of a nonpathogenic AAAAG allele in both P010 and P005 (Supplemental Fig. S2A,B). In addition, one individual was called “homozygous” RE for the pathogenic AAAGG, with an estimated allele size of 640 repeats, exceeding the current pathogenic threshold of >500 repeats for this motif (Dominik et al. 2023). However, manual review determined that the motif is actually AAGGG. Additionally, Straglr did not call the second nonpathogenic 130 repeat AAAGG allele (Supplemental Fig. S2C); therefore, this individual is also a heterozygous carrier.
Comparison of testing methods
Independent analysis of the cohort by LR-AS, SR-GS, and for some loci orthogonal PCR methodologies enabled us to compare and contrast the results across the different platforms. Allele sizing was broadly concordant between LR-AS and SR-GS data up to an RE size of ∼450 bp (Fig. 4; Supplemental Fig. S3). This observation is consistent with previously published work performing PCR validation of long-read RE sizing methods (Ibañez et al. 2022). Expansions of more than 450 bp were consistently sized by LR-AS as being larger than SR-GS due to the longer read length afforded by the ONT platform. There was a very good correlation between LR-AS and orthogonal sizing methodologies (Fig. 4; Table 2), indeed LR-AS with read lengths >5 kb exceeds the sizing capabilities of PCR/capillary array technology (limited to <2 kb).
Straglr did not size the single sample with a pathogenic NOP56 expansion (P026) due to a lack of any reads completely spanning the expanded allele. However, manual review clearly identified reads demonstrating the presence of a pathogenic heterozygous expansion at this locus (Supplemental Fig. S4A). Similarly, two samples with pathogenic biallelic RFC1 expansions (P069, P097, independently identified by SR-GS and PCR testing) were not identified by Straglr due to low read depth; in each case, only a single read spanned the entire RE. However, manual review confirmed the majority of reads at this locus encoded the pathogenic AAGGG motif, suggesting the individuals had biallelic pathogenic RFC1 RE (Supplemental Fig. S4B,C).
Overall, manual review of LR-AS was an important step in the correct interpretation and diagnoses of very large RE. Our results also demonstrate the need for orthogonal methodologies for accurate interpretation of RFC1 alleles, which can demonstrate significant size and motif heterogeneity. LR-AS identified 29 pathogenic REs and achieved a genetic diagnosis for 26% of the cohort. This included heterozygous RE in ATXN8/ATXN8OS (2×), NOP56 (1×), and FGF14 (20×), and biallelic RE in FXN (1×) and RFC1 (5×). This is four less than the 33 pathogenic REs identified using SR-GS, all of which were individuals with SCA27B. In one of these four cases, LR-AS determined an allele size of 249 repeats, compared to 268 repeats by PCR. This falls just under the current pathogenic threshold and therefore was classified as a VUS. For the other three, the read depth was low (<7), and only a single allele (<250 repeats) was called by Straglr (Table 2).
Discussion
There are over 100 clinically recognized disorders encompassed by CA making genetic diagnosis challenging. CA can present with nonspecific and overlapping clinical features, providing minimal guidance for prioritizing genetic candidates and optimal diagnostic methodologies. RE is the predominant genetic cause of CA but until recently they were difficult to diagnose with existing molecular tools. Therefore, CA testing has been fragmented, inefficient, and often restricted to a single or a small number of RE loci. Patients often endure a long diagnostic odyssey with sequential testing, frequently to no avail (Németh et al. 2013; Daker-White et al. 2015; Rexach et al. 2019; Ngo et al. 2020). This study compares two promising technologies for comprehensive genetic diagnosis of CA, focusing on identifying pathogenic RE.
The cohort characteristics are broadly consistent with expectations for adult-onset cerebellar disorders, with similar numbers of males and females. Symptom onset (56 ± 14 years) considerably preceded mean age at testing (68 ± 13 years), reflecting a slowly progressive disease course. Diagnoses included SCA8 (2×), SCA27B (24×), SCA36 (1×), CANVAS (5×), and FRDA (1×). Our data confirm RE as the most common cause of CA in Australia, even in a cohort depleted of five commonly tested REs (SCA1, 2, 3, 6, and 7). Notable was the high frequency of SCA27B (22%). This is consistent with observations in European cohorts with unsolved ataxia, where the reported frequency ranges from 15% to 30% (Iruzubieta et al. 2023; Pellerin et al. 2023; Rafehi et al. 2023). However, there does seem to be variability associated with genetic ancestry, with a frequency of ∼1% reported in Japanese cohorts (Ando et al. 2024; Mizushima et al. 2024). It is possible that biases in cohort collection may have influenced disorder frequency compared to true Australian population prevalence. Individuals were predominantly recruited from a single center and therefore may not be broadly representative. In addition, cohort recruitment was mediated by the clinician referring patients for diagnostic testing, introducing potential bias depending on clinician expertise and enthusiasm for research.
SR-GS achieved 40 diagnoses (40/110, 36% yield) including 33 pathogenic RE and seven other non-RE causes. One advantage of SR-GS is its maturity and well-established pipelines for different types of genetic variation. It is most efficient in identifying small variants (SNV and indel) with LP/P variants accounting for 6% of the cohort. In addition, nine clinically suspicious variants, scoring 4 or 5 ACMG classification points and with a potential phenotype match were identified as VUS, requiring future clinical follow-up and functional studies to confirm pathogenicity. Overall, clinically relevant small variants were found in 15% of cases, consistent with the literature on their contribution to CA (Rudaks et al. 2024). Notably, all of these variants affected protein coding or nearby flanking sequences and therefore are detectable by standard exome sequencing.
Bioinformatic tools capable of identifying pathogenic RE in SR-GS data are a relatively recent development and have yet to be widely embraced in diagnostic pipelines (Bahlo et al. 2018; Leitão et al. 2024). This study demonstrates SR-GS's utility in identifying pathogenic RE-causing CA. Analysis of 17 RE loci associated with CA yielded 33 genetic diagnoses, including both dominant and recessive disorders. Tools like EH5 estimate STR sizes and can use reported pathogenic thresholds for RE, such as ATXN8OS, which are smaller than the read length. However, larger pathogenic RE requires the empirical establishment of thresholds that maximize the specificity and sensitivity of RE identification. This is straightforward for disorders such as CANVAS, where a nonreference motif causes disease. In our experience detection of 10 or more nonreference pathogenic motifs provides strong suspicion of a pathogenic RE. Nonreference motif conditions like SCA31 and SCA37 present challenges as pathogenic motifs may be embedded within expanded nonpathogenic motifs (Rosenbohm et al. 2022). Thousands of alleles in gnomAD have nonpathogenic reference motif (AAAAT) expansions >30 repeats for SCA31 and SCA37 (out of 18,511 individuals). SR-GS methods are mostly unable to detect cases where a pathogenic RE is embedded deep within nonpathogenic motifs due to short DNA fragment libraries and reliance on sequencing reads aligning to nonrepetitive regions adjacent to a putative RE.
Our findings show that SCA27B, caused by pathogenic expansion of the reference GAA motif, is potentially the most common genetic cause of adult-onset ataxia in Australia. Although no diagnostic test is currently available in Australia for this condition, our results demonstrate SR-GS's effectiveness as a screening tool. Despite considerable population variability in nonpathogenic GAA allele size, applying an empirical threshold ≥90 repeats using EH5 provides optimal sensitivity (100%) but a false positive rate (15%). These outcomes are consistent with an independent retrospective cohort study, which showed good predictive value (70%) and excellent sensitivity (100%) using STRling for allele size estimates (Mohren et al. 2024). Collectively, the two studies suggest that while SR-GS has a high sensitivity for FGF14 RE, the false positive rate is likely to limit the utility of the method as a diagnostic test specifically for SCA27B. In the research context, there is a need to follow-up a potential positive result with targeted orthogonal analysis to accurately determine RE size in cases identified by SR-GS, especially given SCA27B's recent identification and uncertainty regarding pathogenic thresholds. While the original studies suggest a pathogenic threshold ≥250 repeats (Pellerin et al. 2023; Rafehi et al. 2023), subsequent studies suggest higher (≥300) (Méreaux et al. 2024) or lower (≥180) (Mohren et al. 2024) thresholds. We identified nine individuals (8% of the cohort) with FGF14 GAA alleles in the 180–249 range and reported them as VUS. No alternative pathogenic variants were found in other loci associated with CA in these cases, providing additional support that this range may be pathogenic.
The second most common cause of CA identified in our cohort was biallelic RE in RFC1 (5/110, 5% yield). Our results demonstrate SR-GS's ability to prioritize cases with potential pathogenic RE at this locus, but extensive orthogonal testing, expert variant interpretation, and clinical input were essential for accurate RFC1-related disorder diagnosis. Awareness of diverse pathogenic motifs and thresholds for this disorder is growing, presenting challenges for effective molecular diagnosis utilizing SR-GS (Miyatake et al. 2022b; Scriba et al. 2023). While additional bioinformatic tools can address some issues (Sullivan et al. 2024), SR-GS will likely function best as a prescreening method for RFC1 diagnostics. A long-read diagnostic tool capable of haplotype-resolved read alignments and full motif composition interrogation will be needed for effective genetic analysis and interpretation of this locus.
LR-AS is a nanopore-based LRS application that enables in silico target selection in real time and has considerable diagnostic potential. Advantages include prioritizing of likely disease-causing loci/variants, reducing curation time, and improving resource utilization by enabling higher sequence multiplexing. In RE disorders, including CA, sequencing depth is critical as knowledge of RE size and composition affects disease prognosis and management (Hannan 2018; GeM-HD 2019). This study achieved a mean approximately eightfold target enrichment and on-target mean sequencing depth of 22 (95% CI 12.7, 31.0) (Fig. 3). This was insufficient for local assembly of diploid alleles and motif composition interrogation for a majority (75%) of the 2420 RE (22 × 110) targeted. Our results contrast with reports of successful haplotype-resolved allele interrogation with similar metrics (∼5× enrichment, ∼8–40× target coverage) (Miller et al. 2021; Stevanovski et al. 2022). Reliable and reproducible data are essential for the accreditation of a diagnostic test, and it is possible these divergent results are due to the choice of bioinformatic tools implemented by the different studies. A current limitation of LR-AS and indeed the ONT platform more broadly, is the lack of widely accepted best-practice pipelines for bioinformatic analysis of data. The Epi2Me Labs wf-human-variation pipeline produced by ONT's Customer Workflows group is the closest analog to the GATK best practices broadly used in processing short-read sequencing data. We designed our analysis after the Epi2Me pipeline, using the most recent, highest-accuracy basecalling model available, and selecting minimap2 (Li 2018) as recommended by recent benchmarks of alignment tools (LoTempio et al. 2023; Helal et al. 2024). Similarly, we selected Straglr (Chiu et al. 2021) as our primary analysis algorithm for its compatibility with minimap2-aligned data, as it is the currently recommended STR caller in Epi2me's human-variation pipeline.
Despite read depth limitations associated with some targets, LR-AS performed exceptionally well in this trial, achieving 29 diagnoses (29/110, 26% yield). Greater manual review and interpretation of LR-AS data were required to support a diagnosis at specific loci compared to SR-GS. For example, Straglr analysis did not identify potential RE in NOP56, despite 4/8 reads indicating a pathogenic RE was likely present (Supplemental Fig. S4). Similarly, while Straglr identified six individuals with potential biallelic pathogenic RE in RFC1, extensive manual curation and orthogonal testing showed that only three had true biallelic RE. Moreover, Straglr was unable to generate an allele size estimate for two individuals that SR-GS and orthogonal analysis identified as biallelic. Pathogenic RE was identifiable in aligned reads, but low read depth and few spanning reads compromised allele identification and sizing. However, LR-AS data provided superior diagnostic outcomes at other loci, including FGF14.
This study was not designed to efficiently benchmark STR callers and indeed different tools have strengths and weaknesses dependent on the program design. However, we did reanalyze the LR-AS data using an alternate algorithm (vamos), which can interrogate both motif size and composition (Ren et al. 2023). Vamos and Straglr results were broadly concordant (Supplemental Fig. S5) and as a general comment, neither tool could be described as superior to the other. Missing calls were observed with both and these were predominantly due to low sequencing depth; manual review and interpretation were similarly required for vamos. Comparison of Straglr and vamos calls for the 22 loci where there is a biallelic or heterozygous pathogenic RE revealed some discordant results (Supplemental Table S7), which likely reflect the underlying differences in how the algorithms function. Straglr uses a statistical model of length alone that fits Gaussian mixtures to repeat length estimates to produce allele sizes. Straglr may miscall a heterozygote as a homozygote if depth is low. Vamos performs assembly on phased reads and then assesses repeat composition and length from a catalog derived from reference assemblies. Therefore, vamos may perform better where two alleles have repeat sizes that are close to one another, as these are difficult to distinguish from homozygous alleles in the absence of phased reads. We believe vamos is more sophisticated than Straglr when used on aligned reads that have been phased, but is much more sensitive to the specification of the catalog parameters. For example, vamos’ calls on the nonexpanded HTT locus using the “efficient motif catalog” provided by the authors were 3× longer than expected; the use of a CAG-only catalog resulted in HTT calls in the normal range. Similarly, vamos did not produce calls for FGF14 until we used a simplified motif catalog testing GAA only at this locus (Supplemental Table S8). In addition, standard implementation of vamos requires phased reads (e.g., from WhatsHap), and the variation in sequencing depth at RE loci can result in vamos performing assembly with as few as two reads. Our experience suggests that discordancy between algorithms is minimized by greater read depth and the best practice may be to use a pipeline with multiple algorithms. For example, Straglr can be used to identify expanded repeats, while vamos can provide redundancy for expansion detection and potentially provide information about motif composition.
RE identification by LR-AS and Straglr analysis was concordant with SR-GS, except for four individuals with SCA27B (Table 2). These diagnoses were missed due to low read depth at the FGF14 locus, and do not constitute a systemic issue with interrogation of this locus. Indeed, LR-AS appears to have significant advantages over SR-GS for SCA27B diagnosis. No false positive results were observed and LRS can easily span the largest FGF14 alleles reported to date (>900 repeats) (Mohren et al. 2024) using current diagnostic workflows for gDNA extraction. High correlation in repeat length estimates with LR-PCR (Fig. 5) (R2 = 0.998) suggests orthogonal validation will not be required when suitable read depth is achieved consistently.
Read depth is a major technical impediment to the diagnostic implementation of LR-AS. While there is a lack of consensus in the field, a read depth of ∼33× supports phased de novo genome assembly (Porubsky et al. 2021). For diagnostic purposes 33× is likely the lower bound; ideally, all would be spanning reads to support accreditation benchmarks for consistent and accurate characterization of RE (and small variants). Reliably achieving these targets may be challenging given the competing need to minimize the target panel for maximum enrichment versus increasing panel size to reduce nanopore destruction due to polarity reversal associated with off-target rejection. Additional enrichment strategies, including CRISPR–Cas9, have been proposed with LR-AS, or indeed as an alternative enrichment protocol (Mizuguchi et al. 2021; Lopatriello et al. 2023; Leitão et al. 2024). This developing technology has now been deployed to characterize RE in both the research and diagnostic settings, for example, the PureTarget RE panel developed by PacBio. However, in the context of diagnostic application, CRISPR–Cas9 approaches are bespoke, require considerable optimization, and are limited in the number of targets that can be successfully multiplexed. A second limitation of LR-AS and indeed PCR-free ONT sequencing more broadly for diagnostic implementation is the preparation of the genomic DNA and sequencing library. Up to 10 times the amount of gDNA is needed compared to SR-GS and depending on the platform and experimental design shearing of the DNA may be required for optimal results. This is most reproducible using physical techniques such as Adaptive Focused Acoustics (Covaris) or hydropore shearing (Megaruptor), however, these techniques require additional experimental protocols, leading to potential increases in both the cost and time to return of results.
One major point of difference between SR-GS and LR-AS is the diversity of mutation types that can be interrogated. We used existing diagnostic grade pipelines for the identification of small variants, CNV, and alterations in mtDNA in the SR-GS data, showing pathogenic small variants as the second most common CA mutation class. We did not formally interrogate the LR-AS data for small variants given the read depth achieved and ONT sequence data error rates (Ni et al. 2023). However, visual inspection of aligned reads spanning pathogenic small variants identified by SR-GS revealed seven of eight variants were identifiable in the ONT reads, suggesting that LR-AS data analysis for RE and small variants could improve diagnostic yield (Supplemental Fig. S6). Notably, the HEXA variants in P018 were observed to be in trans. These variants are separated by 22 kb of genomic sequence, making phasing with singleton SR-GS challenging. Indeed, phasing of variants in late-onset conditions such as CA can often be difficult as parental samples cannot be obtained for orthogonal testing. While the accuracy of ONT base calling has been improving over time (>99%) it remains more error-prone than the alternative long-read technology PacBio (>99.9%) (Espinosa et al. 2024) and this may be a consideration for RE testing. Very recently a Tandem Repeat Genotyping Tool (TRGT) and an accompanying Tandem Repeat database have been developed specifically for RE characterization using PacBio HiFi data and this is likely to have utility for diagnostic testing of RE (Dolzhenko et al. 2024). A second major point of difference between SR-GS and long-read technologies more broadly is the ability of the latter to detect base modifications in single molecules of genomic DNA. Methylation of cytosine molecules, most commonly 5-mCpG or 5-hydroxymethylcytosine (5hmC), plays a key role in complex regulatory mechanisms controlling gene expression, ultimately impacting development and disease (Greenberg and Bourc'his 2019). Clinical epigenomics is also playing an increasingly important role in disease diagnosis and prognostication, driving the development of an array of tools to accurately assess genome-wide methylation in long-read sequence data (Sigurpalsdottir et al. 2024).
In conclusion, diagnosing CA remains challenging due to its complexity and variability. This study compared SR-GS and LR-AS for their efficacy in identifying pathogenic REs, the predominant genetic cause of CA. SR-GS demonstrated a higher diagnostic yield (36%) and benefits from its mature, established clinical use, capable of identifying various genetic variants. It also supports future reanalysis as new RE and CA-associated genes are identified. However, its reliance on orthogonal validation for certain RE and limitations in mapping large, complex RE limit its use primarily as a screening tool. Conversely, LR-AS showed promise with a 26% diagnostic yield and potential for more precise haplotype resolution and targeted analysis of clinically relevant regions. Despite requiring extensive manual data interpretation and facing challenges with read depth and error rates, LR-AS has the potential to develop into a standalone frontline diagnostic test for RE. Both technologies represent significant advancements in CA diagnostics that require continued investment in technological development and computational resources. Cost will also significantly impact clinical implementation. SR-GS capacity is established in many diagnostic facilities but reagent costs per test are unlikely to decrease significantly. LR-AS will require investment to establish equipment and computational infrastructure but reagent costs per test will likely decrease due to sample multiplexing and targeted array analysis. Ultimately, a substantial proportion of the cost for both technologies lies in variant curation and interpretation. The future integration of enhanced variant interpretation algorithms, including artificial general intelligence capability, into diagnostic pipelines will benefit both platforms.
Methods
Cohort recruitment and clinical phenotype
The Royal Children's Hospital Human Research Ethics Committee (HREC #28097) and the Walter and Eliza Hall Institute of Medical Research (HREC 18/06) approved the study. Multiple health practitioners in Australia referred individuals with a clinical diagnosis of CA for standard diagnostic testing of five RE loci (SCA1/2/3/6/7), mostly to a single center (VCGS). Informed consent for research genetic testing was obtained from 110 participants undergoing clinical testing and phenotype details were derived from the pathology test request form or the referring clinician. Blood-derived genomic DNA was isolated and quality control was performed using standard techniques as described in Supplemental Methods.
Short-read genome sequencing and analysis
Genome sequencing was performed by the VCGS in Melbourne, Australia with the TruSeq PCR-free DNA HT Library Preparation Kit and sequenced on the Illumina NovaSeq 6000 platform at targeted mean coverage of 30×. For the detection of pathogenic non-RE variants, data were bioinformatically processed by VCGS using commercially available pipelines. Alignment to the reference genome (GRCh38) and calling of nuclear/germline DNA variants was performed using the Dragen v.3.3.7 (Illumina) workflow. Alignment to the revised Cambridge Reference Sequence (rCRS) mitochondrial genome (NC_012920.1) and calling of mtDNA variants was performed using an in-house analysis pipeline based on the Broad Institute best practice workflow (Mitoreport V1.2.1, see Data access). We used a research instance of seqr (Pais et al. 2022) implemented by Murdoch Children's Research Institute, Melbourne, Australia, to annotate and filter SNV and indel variants. Variants were prioritized for curation using the PanelApp Australia Ataxia_Superpanel gene list (Version 3.13). Candidate variants were reviewed at a multidisciplinary team (MDT) meeting comprised of clinicians, genomic laboratory staff, and bioinformaticians and classified based on ACMG guidelines (Richards et al. 2015). CNV was screened for and interpreted using the CNV detection tool CXGo (Sadedin et al. 2018). mtDNA variant interpretation was performed by manual filtering, with a variant regarded as homoplasmic or apparently homoplasmic when it was present in at least 97%, respectively, of sequence reads aligned to the genomic position. For RE detection, alignment and variant calling were performed according to the GATK best practice pipeline, and analysis of 67 RE loci was performed utilizing exSTRa (v.1.1.0) (Tankard et al. 2018) and ExpansionHunter (v.5.0.0) (Dolzhenko et al. 2019) as previously described (Rafehi et al. 2023).
Targeted long-read sequencing and analysis
LRS was performed on ONT MinION Mk1B sequencer using standard methods as described in Supplemental Methods. Libraries were loaded onto FLO-MIN106D flow cells (ONT) and run for 16–20 h before performing a wash and reload using the Flow Cell Wash Kit (EXP-WSH004, ONT) and run for an additional 24–36 h. Targeted sequencing was performed using the Readfish software package (Payne et al. 2021). A panel of 334 regions of genome sequence was targeted, including 67 genes with a pathogenic RE associated with neurological/neuromuscular conditions, genes identified in the Ataxia_Superpanel (PanelApp Australia Version 3.13), and other candidate genes potentially associated with CA. A full list of target genes and co-ordinates (including 100 kb buffer) is provided in Supplemental Table S5. Libraries were processed using the wf-human-variation pipeline (ONT, v1.8.3), with super-accuracy basecalling model dna_r9.4.1_e8_sup from Dorado v0.3.1 on NVIDIA A100 and A30 GPUs. Minimum depth for the pipeline was set at 2× coverage. Reads were aligned to GRCh38 (GRCh38_full_analysis_set_plus_decoy_hla) using minimap2 (v2.24-r1122). STRs were typed against a custom locus catalog containing 154 locus-motif pairs at 67 unique loci using Straglr (v1.4.1) or against a subset of loci from vamos efficient motif set (v2.1) using vamos (v2.1.3) on the haplotagged CRAM files produced by the wf-human-variation pipeline. Loci were manually reviewed with IGV (v.2.16.2).
Molecular genetic studies
FGF14 LR-PCR and RP-PCR were performed as previously described (Rafehi et al. 2023), but using LongAmp HotStart Taq (New England Biolabs). RFC1 flanking PCR was performed as previously described (Rafehi et al. 2019). RFC1 RP-PCR and LR-PCR were performed using Q5 High-Fidelity PCR Master Mix (New England Biolabs) and previously published primers (Cortese et al. 2019) ATXN8/ATX8OS sizing PCR and RP-PCR were performed as previously described (Zhou et al. 2019). NOP56 RP-PCR was performed as previously described (García-Murias et al. 2012). For all PCRs, fragment analysis of FAM-labeled PCR products was performed on the capillary array (ABI3730xl DNA Analyzer, Applied Biosystems) with a LIZ1200 size standard, and visualized using PeakScanner 2 (Applied Biosystems).
Statistical analyses
Pearson's correlation performed in R (version 4.0.5) (R Core Team 2024) was used to determine concordance for FGF14 size estimates between SR-GS, LR-AS, and PCR. RE consisting of non-GAA motifs and low-coverage LR-AS calls were excluded from the calculation. Sensitivity and specificity for FGF14 for SR-GS and LR-AS were calculated only for individuals with pure GAA motifs. For LR-GS, samples with read depth ≤7 were excluded from the calculations.
Data access
All raw sequence data generated in this study have been submitted to the European Genome-phenome Archive (EGA) (https://ega-archive.org/) under accession numbers EGAS50000000573 (study ID) and EGAD50000000815 (data set ID). Source code for mitoreport is publicly available at GitHub (https://github.com/bioinfomethods/mitoreport) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the Australian Government National Health and Medical Research Council grants (GNT2001513 and MRFF2007677) to P.J.L., M.B.D., H.R., M.B., and D.S. H.R. was supported by a NHMRC Emerging Leadership 1 grant (1194364) and M.B. was supported by an NHMRC Investigator grant (1195236). Additional funding was provided by the Independent Research Institute Infrastructure Support Scheme, the Victorian State Government Operational Infrastructure Program, the Serp Hills Foundation, and the JTM Foundation. K.R.K. reports financial support was provided by the Ainsworth 4 Foundation unrelated to the current study.
Author contributions: H.R., L.G.F., M.B., and P.J.L.: conceptualization and funding acquisition; H.R., L.G.F., J.R., P.S., K.C.D., L.S., G.G., G.C.T., T.A.F., A.E., S.B., E.B., L.C., J.D., H.G., A.H., G.M.H., A.H., K.K., K.R.K., S.K., M.L-A., A.M., M.Pa., M.Po., K.P., K.R., S.R., A.N., M.S., R.S., C.S., M.W., J.W., T.W., K.W., C.W., K.H.C.W., D.J.S., M.B.D., M.B., and P.J.L.: investigation; M.B. and P.J.L.: supervision; P.J.L.: writing—original draft; H.R., L.G.F., J.R., P.S., K.C.D., L.S., G.G., G.C.T., T.A.F., A.E., S.B., E.B., L.C., J.D., H.G., A.H., G.M.H., A.H., K.K., K.R.K., S.K., M.L-A., A.M., M.Pa., M.Po., K.P., K.R., S.R., A.N., M.S., R.S., C.S., M.W., J.W., T.W., K.W., C.W., K.H.C.W., D.J.S., M.B.D., M.B., and P.J.L.: writing—review and editing.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279634.124.
- Received June 11, 2024.
- Accepted November 21, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








