
The role of transposon activity in shaping cis-regulatory element evolution after whole-genome duplication
- Øystein Monsen1,6,
- Lars Grønvold1,6,
- Alex Datsomor1,
- Thomas Harvey1,
- James Kijas2,
- Alexander Suh3,4,7,
- Torgeir R. Hvidsten5 and
- Simen Rød Sandve1
- 1Department of Animal and Aquacultural Sciences, Faculty of Biosciences, Norwegian University of Life Sciences, 1432 Ås, Norway;
- 2Aquaculture Programme, Commonwealth Scientific and Industrial Research Organisation, St. Lucia, Queensland 4067, Australia;
- 3School of Biological Sciences–Organisms and the Environment, University of East Anglia, NR4 7TU Norwich, United Kingdom;
- 4Department of Organismal Biology–Systematic Biology (EBC), Uppsala University, SE-752 36 Uppsala, Sweden;
- 5Faculty of Chemistry, Biotechnology and Food Science, Norwegian University of Life Sciences, 1432 Ås, Norway
-
↵6 These authors contributed equally to this work.
Abstract
Whole-genome duplications (WGDs) and transposable element (TE) activity can act synergistically in genome evolution. WGDs can increase TE activity directly through cellular stress or indirectly by relaxing selection against TE insertions in functionally redundant, duplicated regions. Because TEs can function as, or evolve into, TE-derived cis-regulatory elements (TE-CREs), bursts of TE activity following WGD are therefore likely to impact evolution of gene regulation. Yet, the role of TEs in genome regulatory evolution after WGDs is not well understood. Here we used Atlantic salmon as a model system to explore how TE activity after the salmonid WGD ∼100 MYA shaped CRE evolution. We identified 55,080 putative TE-CREs using chromatin accessibility data from the liver and brain. Retroelements were both the dominant source of TE-CREs and had higher regulatory activity in MPRA experiments compared with DNA elements. A minority of TE subfamilies (16%) accounted for 46% of TE-CREs, but these “CRE superspreaders” were mostly active prior to the WGD. Analysis of individual TE insertions, however, revealed enrichment of TE-CREs originating from WGD-associated TE activity, particularly for the DTT (Tc1-Mariner) DNA elements. Furthermore, coexpression analyses supported the presence of TE-driven gene regulatory network evolution, including DTT elements active at the time of WGD. In conclusion, our study supports a scenario in which TE activity has been important in genome regulatory evolution, either through relaxed selective constraints or through strong selection to recalibrate optimal gene expression phenotypes, during a transient period following genome doubling.
The two most influential mutational mechanisms that have shaped eukaryotic genome evolution are whole-genome duplications (WGDs) and transposable element (TE) activity. Both WGDs and TEs drive genome size evolution. However, as mobile genetic elements with capacity to replicate (Feschotte and Pritham 2007), TEs also impact genome evolution in numerous other ways, by generating novel genes (Elisaphenko et al. 2008; Qin et al. 2015; Cosby et al. 2019; Diehl et al. 2020), modulating chromatin looping (Diehl et al. 2020), rearranging genome structure (Bourque et al. 2018), and supplying “raw material” for gene regulatory evolution in the form of cis-regulatory elements (CREs) (Bourque et al. 2008; Feschotte 2008; Sundaram et al. 2014; Chuong et al. 2017; Diehl et al. 2020; Sundaram and Wysocka 2020; Cosby et al. 2021).
Studies of mammalian genomes have provided deep insights into the role of TEs in CRE evolution and the potency of TE-derived CREs (TE-CREs) to regulate gene expression (for review, see Fueyo et al. 2022). For example, as much as 40% of the mouse and human transcription factor (TF) binding sites have been shown to be within TEs (Sundaram et al. 2014), and as many as 19% of pluripotency factor TFs are located within TEs (Kunarso et al. 2010; Sundaram et al. 2017). Curiously, in mammals, TEs associated with gene regulation during development have been shown to be younger than those associated with regulation in adult somatic tissues (for review, see Fueyo et al. 2022), suggesting different evolutionary pressures on TEs with distinct regulatory roles.
Genome evolution through TE activity is also likely influenced by WGDs. Because WGDs result in cellular stress, TEs can escape host-silencing mechanisms following WGDs. This is supported by both experimental (Kashkush et al. 2002, 2003; Kraitshtein et al. 2010) and comparative genomic (Lien et al. 2016; Marburger et al. 2018) studies. Additionally, WGDs result in increased functional redundancy. This will reduce the average negative fitness effects of novel TE insertions and thereby allow for fixation of TE insertions following WGD (Baduel et al. 2019), including insertions that influence gene regulation. In line with this, Gillard et al. (2021) recently reported that TE insertions in promoters were associated with regulatory divergence of gene duplicates following WGD in salmonid fish. However, systematic investigations into the role of TEs in CRE evolution and genome regulatory remodeling after WGDs are still lacking.
Here we address this knowledge gap regarding the role of WGD in TE-associated genome regulatory evolution using salmonids as a model system. Salmonids underwent a WGD 80–100 Mya (Lien et al. 2016), which coincided with the onset of a burst of TE activity, particularly featuring elements belonging to the DTT/Tc1-mariner superfamily. This observation has led to the hypothesis that increased TE activity in the immediate aftermath of the WGD was a major driver of genome regulatory evolution. To explore this idea, we leverage ATAC-seq data from two tissues (brain and liver) to identify putative CREs that have evolved from TE-derived sequences. We then combine these TE-CRE annotations with analyses of the temporal dynamics of TE activity, analyses of gene coexpression, and massive parallel reporter assays. Our results support a link between WGD and TE-CRE evolution and support the idea of synergistic interactions between WGDs and TE activity to drive rewiring of genome regulation.
Results
The TE-CRE landscape of Atlantic salmon
To investigate the contributions of different TEs to CRE evolution, we first characterized the TE landscape of the salmon genome using an updated version of the existing TE annotation from Lien et al. (2016). The total TE annotation covered 51.92% of the genome. Consistent with previous findings (Goodier and Davidson 1994; Lien et al. 2016), the dominating TE group was DNA transposons from the Tc1-Mariner superfamily with more than 655,000 copies, covering 327 million base pairs, just shy of 10% of the genome (Fig. 1A–C). In general, the genomic context of TE insertions was quite similar to the genomic baseline (Fig. 1D), but with slightly more TEs in intronic regions and slightly fewer TEs in exons and intergenic regions. Of the well-represented TE superfamilies (>10 k insertions) only the Nimb retrotransposon superfamily was an exception to this pattern, for which 18% of the copies were found in promoter regions (Fig. 1D).

Overview of the genomic TE landscape. (A) Superfamily level overview of TE annotations in the Atlantic salmon genome. Number of TE subfamilies per superfamily in square brackets. (B) TE insertions per superfamily. (C) Annotated base pairs at the TE superfamily level. (D) TE annotations (base pair proportions) overlapping different genomic contexts. Genomic baseline is the proportion of the entire genomic sequence that is assigned to the four genomic contexts.
Active CREs in tissues and cells are associated with increased chromatin accessibility (Keene et al. 1981; McGhee et al. 1981; Buenrostro et al. 2013). Thus, to study the contribution of TEs to the salmon CRE landscape, we integrated our TE annotation with annotations of accessible chromatin regions identified using ATAC-seq data from the liver and brain. Analysis of the overlap between TEs and accessible chromatin revealed a large depletion of TEs in accessible chromatin. Although TEs represent ∼52% of the genome sequence, TE insertions only represent <20% of the regions of accessible chromatin (Fig. 2A), with the liver having a higher proportion of annotated TEs in accessible chromatin than the brain.
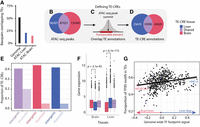
TE-CRE landscape. (A) The proportion of base pairs overlapping TEs, either from out of all genome-wide base pairs or from those within an ATAC-seq peak. (B–D) Pipeline to define putative TE-CREs. (B) Venn diagram of tissue-specific and shared ATAC-peaks from the liver and brain. (C) Cartoon showing how TE-CREs are defined as ATAC-seq peak summits when overlapping with a TE. (D) Venn diagram of tissue-specific and shared TE-CREs from liver and brain. (E) Proportion of shared and tissue-specific TE-CREs in promoter versus intergenic regions. (F) Gene expression levels of the nearest genes to tissue-specific and shared TE-CREs in the brain and liver. P-values from a Wilcoxon test are indicated above tissues. (G) Correlation between TF tissue specificity and the proportion of genome wide TF motif matches located in TEs. Each point represents a TF motif. Tissue specificity is based on differential TF binding score from TOBIAS (Bentsen et al. 2020), which essentially summarizes the relative ATAC-seq footprint signal across all potential binding sites.
To define a set of TEs that contribute to putative CREs, we narrowed in on those TE annotations overlapping chromatin accessibility peaks (Fig. 2B–D). These were defined as putative TE-CREs. Because TEs by definition are multiple copies of the same sequence, multimapping of ATAC-seq reads and bias in the peak calling is a potential issue. We therefore assessed the level of multimapped ATAC-seq reads in TEs versus other genomic regions for one of the brain samples. This analysis showed that genome-wide multimapping levels were 14%, whereas reads mapping to TEs from larger subfamilies (more than 500 insertions) had an average multimapping level of 12%. As the majority of TEs in the Atlantic salmon genome are quite old (see analysis in Fig. 4), a low level of multimapped reads is, in fact, expected. Hence, we conclude that problems with assigning ATAC-seq reads to individual TE insertions do not impact our TE-CRE results to a large degree.
Although the majority (55%) of TE annotations (excluding “unknown” repeats without classification) were DNA elements (1,335,051 insertions), TE-CREs from DNA elements were a minority (27%). Both the proportion of base pairs in accessible chromatin (Fig. 2A) and the number of putative TE-CREs (Fig. 2D) were higher in the liver compared with the brain. Of a total of 55,080 TE-CREs, 20% were shared between tissues, 37% were brain specific, and 43% were liver specific (Fig. 2D). Tissue-shared TE-CREs were overrepresented about fourfold in promoters compared with the tissue-specific TE-CREs (Fig. 2E). We also found that tissue-specific TE-CREs were associated with tissue bias in gene expression (Fig. 2F), supporting a regulatory effect of TE-CREs.
One reason for TE-CREs tending to be specific to the liver rather than the brain (Fig. 2D) could be owing to differences in purifying selection pressure on regulatory networks important for brain function compared with liver function. One expectation from this hypothesis would be that TF binding motifs with a strong brain bias in TF binding would be depleted in TE sequences. To test this, we first inferred tissue bias in TF binding using genome-wide TF binding motif occupancy signals through TF footprinting. We then correlated these signals with the proportion of TF binding motifs found in TE sequences. In line with our expectations, motifs for brain-biased TFs were less frequently found in TEs (regression line in Fig. 2G). The most highly liver-biased TFs, such as HNF1A, were exceptions to this general trend, although these liver-biased TFs were fewer and much less depleted in TEs compared with the most highly brain-biased TFs such as NEUROD1 (Fig. 2G). Taken together, our results are in line with a model in which TEs with smaller chances of having a negative fitness consequence for the host are evolutionarily more successful.
A minority of TEs have CRE superspreader abilities
Next we wanted to understand the contribution of specific TEs to the TE-CRE landscape. Overall, there was a positive linear relationship between the genomic copy number and the number of TE-CRE for TE superfamilies (Fig. 3A). However, some superfamilies (Fig. 3A, see data points outside 95% CI), contributed significantly less (RIC and DTT) or more (RIN, RLG, DTA, RIJ) to the TE-CRE landscape than expected based on the genomic copy numbers (Fig. 3A). Particularly striking was the DTT superfamily elements, which are dominating in terms of numbers of insertions (∼27% of all TE copies with an assigned taxonomy) but only represented ∼4% of the TE-CREs.
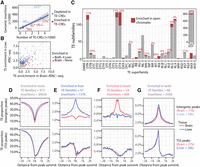
TEs enriched in open chromatin. (A) The number of insertions per superfamily plotted against the number of CREs in each superfamily. The shaded area is a 95% confidence-level interval. Superfamilies falling outside the 95% confidence interval are annotated with the three-letter superfamily code. (B) TE families (more than 500 genomic insertions) plotted according to fold enrichment within ATAC-seq peaks in the brain and liver. TE subfamilies are assigned into categories based on enrichment in liver, brain, or both. (C) Proportion of TE subfamilies enriched in open chromatin per superfamily. A manual curation step of the TE subfamilies enriched in open chromatin resulted in a slightly different superfamily list than the initial machine-predicted annotations presented in Figure 1. Note also that only TE subfamily sequences with more than 500 insertions have been included. The percentage of enriched TE subfamilies per superfamily are indicated above bars. (D–G) Proportion of base pair overlapping TEs from each enrichment category around peak summits in intergenic or promoter regions (summit within 500 bases of a TSS). Peaks in promoter regions are oriented according to the corresponding TSS with gene bodies to the right in figures.
To characterize the TE-CRE landscape in more detail, we identified individual TE subfamilies enriched in open chromatin (see Methods) (Fig. 3B). TE subfamilies significantly enriched in open chromatin are hereafter referred to as CRE superspreaders. After removing TE subfamilies with few genomic insertions (500 or fewer), we found that only 178 (16%) out of 1119 TE subfamilies were defined as CRE superspreaders (Fig. 3B). Forty-nine percent of these superspreaders were enriched in open chromatin in both tissues (88), whereas 39% (69) and 12% (21) were tissue specific and enriched in accessible chromatin only in the liver or brain, respectively. The proportion of taxonomically unclassified TEs (three-letter code “XXX”) was >50% among the identified CRE superspreaders (101 out of 178 families). For these TE subfamilies, we therefore performed manual curation, resulting in four TE subfamilies being discarded from further analyses, and only 34 families remained taxonomically unclassified (Supplemental Table S1).
We find that CRE superspreaders were taxonomically diverse, belonging to 18 different TE superfamilies, but that very few TE subfamilies in the DTT superfamily evolved into CRE superspreaders (Fig. 3C). Note that superfamilies consisting only of CRE-superspreader TEs are a technical artifact stemming from the manual curation of the taxonomically unknown (three-letter code XXX) superspreaders.
Next, we explored the local TE landscape around the open chromatin peaks in various genomic contexts (intergenic and promoters) and tissues (Fig. 3D–G). We find that in promoters the proportion of TEs in open chromatin decreases toward a transcription start site (TSS) and the gene body, probably reflecting increased purifying selection pressure (less tolerance for TE insertions) inside the gene body.
Furthermore, we find that close to genes (i.e., in promoters), TE proportions were higher for tissue-shared (Fig. 3G) compared with tissue-specific (Fig. 3E,F) TE-CREs. In intergenic regions (i.e., putative enhancers), we find very strong tissue-specific TE enrichment signals not present in promoter TE-CREs (Fig. 3E,F). In sum, we find that CRE-superspreader TEs are biased toward certain taxonomic groups of TEs and that these TEs are enriched in accessible chromatin with distinct patterns and effect sizes across tissues and genomic contexts.
The temporal dynamics of TE-CRE evolution
The main hypothesis we set out to test in this study was whether the increase in TE activity associated with salmonid WGD was instrumental in driving TE-CRE evolution. To explore this hypothesis, we first calculated sequence divergence between individual TE insertions and their TE-subfamily consensus sequence in the form of CpG-normalized Kimura distances. Sequence divergence estimates can be used as a proxy for time and then be compared with the Kimura distance expectation for TEs being active before or after the salmonid WGD event (for details, see Methods). One challenge with sequence divergence–based comparisons is, however, the intrinsic connection between sequence divergence and purifying selection pressure that can bias our results. Here we used the entire TE insertion (not only the part that is in open chromatin) to estimate divergence to TE-subfamily consensus; hence, we expect this bias to be negligible. Nevertheless, we first analyzed the sequence divergence distributions of different classes of TE-CREs as well as TE sequences not in accessible chromatin (Fig. 4A). As expected, we do not find that TE-CREs are more similar to their TE-subfamily consensus than to other TE insertions, supporting that putative purifying selection on TE-CRE function does not transfer to our sequence divergence–based age-proxy. If anything, the TE sequences giving rise to tissue-shared CREs are older than TEs not giving rise to TE-CREs (see “both” in Fig. 4A).
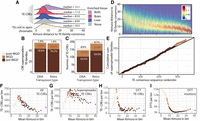
Temporal dynamics of TE-CRE insertion activity by TE taxonomy. (A) Distribution of sequence divergence of TE-CREs from their TE-subfamily consensus sequence. Colors represent if TE-CREs are from TE subfamilies with superspreader ability (liver, brain, or both) or not (gray). (B) Number of TE subfamilies with superspreader ability subdivided into DNA elements and retroelements. Colors represent the TE-subfamily age proxy calculated as mean divergence between genomic insertions and their consensus TE sequence. (Post-WGD) <7 Kimura distance, (WGD) 7–10 Kimura distance, (pre-WGD) >10 Kimura distance. (C) Number of TE-CREs from TEs with a taxonomic classification subdivided into DNA elements and retroelements. Colors represent the TE-subfamily age proxy. (D) Heatmap of the divergence distributions of all insertions per TE subfamily (with more than 500 insertions) to their consensus sequence. TE families are ordered based on mean divergence from consensus. (E) Cumulative distribution of CRE-superspreader TE families ordered by mean Kimura distance between genomic copies and TE-subfamily consensus sequence. Colors represent age proxy as defined by mean divergence to TE-subfamily consensus sequence (F–I) The number of TE-CREs (F–H) and TE insertions (I) per “age”-bin of Kimura distances for all TE-CREs, TE-CREs from superspreader families, and TE-CREs from the DTT superfamily.
Next, we stratified the TE-CREs on their transposition age-proxy relative to the WGD and their taxonomic order (Fig. 4B,C). Seventeen percent (189) of TE subfamilies had a mean Kimura distance between insertions and the TE-subfamily consensus reflecting activity in the approximate range around the time of the WGD (Kimura distance of 7–10). Among the TE subfamilies with CRE-superspreader ability, there was about the same proportion of TE subfamilies active around the time of WGD classified as DNA elements (19.6%) compared to retroelements (20.0%) (Fig. 4B). Slightly higher proportion of TE-CRE insertions from DNA elements were assigned to the time around WGD based on the mean Kimura distance to TE-subfamily consensus (26.8% vs. 22.8%) (Fig. 4C); however, the retroelement-derived TE-CREs dominated over DNA-element TE-CREs in absolute numbers (Fig. 4C).
Even if most TE-CREs are older than the salmonid WGD, it is still plausible that the WGD triggered a shift in the evolutionary rates of TE-CREs. To explore this in more detail, we first analyzed the temporal dynamics of all TE subfamilies (more than 500 genomic copies) (Fig. 4D) by plotting the cumulative sum of TE-CRE superspreader families against all TE subfamilies ordered by mean divergence to consensus (Fig. 4E). If WGDs were associated with a general burst of CRE-superspreader activity, we expect to see a steeper slope in the cumulative sum distribution around mean Kimura distances of 7–10 to the TE-subfamily consensus. Although we find a slight change in CRE-superspreader accumulation rates around this divergence range (Fig. 4E), most of the data points lie on or close to the diagonal (null model), and the age distribution of CRE-superspreader TE subfamilies was not significantly different from other TE subfamilies (two-sided Kolmogorov–Smirnov test, P-value = 0.5). Nor did we find a significant increase in the proportion of TE-CRE superspreader families among TE subfamilies with predicted activity around the WGD (mean Kimura distance 7–10, Fisher's exact test, P-value = 0.45). Taken together, these results do not support a model in which the WGD caused a large shift in TE-CRE superspreader subfamily activity.
Our subfamily-level analyses showed that CRE-superspreader activity was evenly distributed in time and was mostly pre-WGD (Fig. 4B,E). Yet, the distributions of Kimura distances for TE-CRE insertions (except those from families enriched in both tissues) suggests that TE-CREs are biased toward insertions happening around the time of, or more recent than the WGD (median Kimura distance <10) (Fig. 4A). One explanation for this seeming contradiction could have been that younger TE subfamilies have more insertions that evolve into TE-CREs, but this is not the case (Fig. 4C). Another explanation could be that using the mean of each TE subfamily obscures within subfamily heterogeneity in Kimura distances, which can arise from multiple activity bursts or prolonged activity across time. As an alternative approach to associate WGD with TE-CRE evolution, we therefore used the Kimura divergence from individual TE insertions. We divided TE insertions into 100 equally large “age” bins based on Kimura distance and plotted the distribution of TE-CREs per bin. For all TE-CREs, we found a significant enrichment (Fisher's exact test, P = 3.4 × 10−100) of TE-CREs with a Kimura distance to consensus reflecting activity around the time of the WGD (Fig. 4F). Using only TE-CREs from superspreader TE subfamilies, a much weaker association with the WGD was found (Fig. 4G), with more TE-CREs coming from TE activity prior to the WGD (in line with Fig. 4B). Tallying TE-CREs per superfamily into our three defined temporal activity periods (Supplemental Fig. 2), we find that the DTT superfamily has the largest proportion of TE-CREs arising from insertions at the time of the WGD. In fact, the Kimura distances of DTT-derived TE-CREs (Fig. 4H) reflect an extremely strong clustering of TE-CREs with insertion ages close to the WGD. This pattern could be caused by a simple correlation between the number of DTT insertions and number of TE-CREs, but this is not the case as the total number of DTT insertions continues to increase as Kimura distances decrease toward zero, whereas TE-CRE numbers drop for DTT insertions after the WGD (Fig. 4I). In conclusion, we find that the WGD is strongly associated with increased rates of TE-CRE evolution, particularly from DTT elements, but that this association is not driven by higher transposition activities of CRE-superspreader TE subfamilies.
Coexpression analysis support TE-CRE driven regulatory network evolution
If TEs are spreading CREs with sequences that either have a potent TF binding motif or are prone to mutate into a TF motif, we expect different genes with similar TE-CREs (TEs insertions belonging to the same consensus sequence) to be more similarly regulated than random gene pairs. To identify such putative cases of TE-CRE driven evolution of gene regulation, we assigned each TE-CRE to the closest gene and tested if genes with similar TE-CREs were more coexpressed than expected by chance.
We first used RNA-seq data from the liver of 112 individuals spanning different ages, different sexes, and different diets in fresh water (Gillard et al. 2018). In the context of this liver coexpression network, significant coexpression (low P-values) indicates that TE-CREs from one particular TE subfamily are candidates for modulating the gene regulation in the liver depending on developmental and physiological states. Using only TE-CREs from liver, 41 TE subfamilies (41 of 1387 = 3%) were associated with genes that were significantly coexpressed (false-discovery rate [FDR]-corrected P-value < 0.05) (Fig. 5A). Of these significant TE subfamilies, 20 (49%) were CRE superspreaders. The significant TE subfamilies came from 12 superfamilies and TEs of unknown origin (XXX), accounting for 29% (Fig. 5B). The cumulative distribution of TE-CREs associated with gene coexpression did not suggest a temporal co-occurrence of WGD and the TE-CREs with putative gene regulatory effects (Fig. 5C).
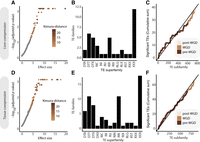
TE-CREs driving coexpression. (A–C) Results from liver coexpression. (D–F) Results from tissue atlas coexpression. (A,D) Significance (FDR-adjusted P-values) plotted against effect size (standard deviations) for each TE subfamily, indicating the strength of coexpression of their associated genes in the liver (A) and tissue atlas (D) coexpression networks, respectively. Points with FDR-adjusted P-value < 0.05 are colored by Kimura distance to TE-subfamily consensus. (B,E) Distribution of significant TE subfamilies grouped by superfamilies in liver (B) and tissue atlas (E) data sets. (C,F) Cumulative distribution of TE subfamilies with significant effect on gene coexpression in liver (C) and tissue atlas (F) data sets. Temporal classification was based on the mean divergence of all TE insertions to their TE subfamily consensus sequence, for which post-WGD was defined as Kimura distance < 7, WGD as 7–10, and pre-WGD as >10.
TE-CREs are also known to induce tissue-specific regulatory effects (Karttunen et al. 2023). We therefore conducted the same analyses using RNA-seq data from 13 different tissues (Lien et al. 2016). Using TE-CREs from both the liver and brain, 71 TE subfamilies (71 of 1465 = 4.8%) were associated with significant coexpression (Fig. 5D), of which 29 (41%) were superspreaders. The significant TE subfamilies came from 13 TE superfamilies (Fig. 5E). Each significant TE subfamily was associated with a tissue TE-CRE profile (fraction of TE-CREs found in liver, brain, or both), and these profiles generally agreed with the tissue expression profiles of the associated genes (RNA-seq expression values across 13 tissues), thus corroborating that our approach indeed identified regulatory-active TE-CREs. Similar to the liver coexpression analyses, the cumulative distribution of TE-CREs impacting tissue regulation did not suggest any link between the WGD and the TE-CRE evolution shaping gene coexpression (Fig. 5F). Taken together, we find evidence for a small proportion (3%–5%) of the TE subfamilies spreading CREs that regulate nearby genes.
Functional validation of TE-CREs using massively parallel reporter assay
To be able to directly assess regulatory potential of TE-CREs in Atlantic salmon, we carried out massive parallel reporter assays (MPRAs), specifically an ATAC-STARR-seq experiment, in salmon primary liver cells (Fig. 6A). This method assesses the ability of random DNA fragments from accessible chromatin to modulate transcription levels (Wang et al. 2018). In total, 4,267,201 million unique DNA fragments from open chromatin in liver were assayed. Thirty-four percent of these fragments (1,456,914) could be assigned to a specific TE insertion site (>50% overlap with a TE annotation) (Fig. 6B). Of the TE-derived sequence fragments assayed, 1.2% had transcriptional regulatory activity, a slightly lower proportion than non-TE fragments (1.6%), and TE-derived regulatory active fragments were more likely to induce transcription compared with non-TE sequences (see “Up” in Fig. 6C).
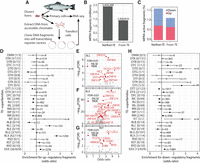
Massive parallel reporter assay screening of regulatory activity. (A) Schematic overview of the ATAC-STARR-seq MPRA experiment. (B) Barplot of the origin of sequence fragments included in the analyses. (C) Regulatory activity (inducer or repressor) of MPRA sequence fragments from TE and non-TE sequences. (D) Fisher's exact test results for enrichment of transcriptional-inducing MPRA fragments within a TE superfamily compared with all other TEs. Unknown taxonomy and DNA/retrotransposons of unknown origin (DTX/RLX) are considered separate groups. A similar test is also done on the subfamily level, and the number of significant TE subfamilies and total number of subfamilies tested are given in square brackets next to the superfamily codes. Number of regulatory active fragments are given for each category (n). (E–G) TF motif enrichment in transcriptionally inducing MPRA fragments from TE superfamilies enriched in regulatory active fragments. TF names are from the JASPAR database, and the nomenclature reflects whether it came from human or mouse. (H) Fisher's exact test results for enrichment of transcriptional-repressing MPRA fragments within a TE superfamily compared with all other TEs. A similar test is also done on the subfamily level, and the ratio of number of significant TE subfamilies to total number of subfamilies tested is given in square brackets next to the superfamily codes. Unknown taxonomy and DNA/retrotransposons of unknown origin (DTX/RLX) are considered separate groups. Number of regulatory active fragments are given for each category (n).
To test if CRE sequences from particular TE insertions were more likely to increase gene expression (i.e., act as enhancers), we compared the ratio of regulatorily active versus inactive fragments. This analysis was done at the TE-superfamily level (including groups with partially assigned and unknown taxonomy) and at the level of each TE subfamily (Fig. 6D). Three retrotransposon superfamilies were significantly enriched for regulatorily active fragments (Fisher's exact test, FDR-corrected P-value < 0.05). Two of these were LTRs (RLC and RLX) which had a more than twofold higher ratio of fragments acting as enhancers, whereas another SINE superfamily (RST) was significantly enriched but with a much lower effect size estimate (Fig. 6D). The transcriptionally inducing fragments from these three superfamilies were enriched for a total of 38 unique TF binding motifs (RLC = 12, RST = 8, and RLX = 19) (Fig. 6E–G). Many of these top-enriched motifs are known to be bound by liver active TFs (i.e., SREBF2, KLF15, FOXA2, THRB) (Tao et al. 2013; Lau et al. 2018; Chaves et al. 2021; Yerra and Drosatos 2023), including the TFCP2L1 binding motif (Wei et al. 2019), which was enriched in all three superfamilies (Fig. 6E–G). We also tested for enrichment of transcriptionally repressing activity and found 6 superfamilies (in addition to the XXX and DTX groups) that were enriched for transcription repressing fragments (Fig. 6H). Fragments from the WGD-associated DTTs were significantly less likely to be regulatorily active (both to induce and to repress transcription) compared with other TE-derived sequences in the MPRA experiment (i.e., odds ratio < 1 in Fig. 6D,H). This is consistent with our findings that DTTs are depleted in TE-CREs compared with random expectations (Fig. 3A).
Enrichment tests of regulatory active fragments at the level of each individual subfamily (Supplemental Table S2) highlighted some subfamilies with particularly potent transcriptionally inducing and repressing activities (see square brackets in Fig. 6D,H). For most subfamilies, the number of fragments assayed in the MPRA experiment were low; hence, the power to detect enrichment at the subfamily level was generally also low. However, in the RLX superfamily group as many as 36% (42/114) of subfamilies were enriched for transcriptionally inducing fragments.
Discussion
The Atlantic salmon TE-CRE landscape
Most in-depth characterizations of TE-associated CREs have so far been carried out in mammalian cells and tissues. Our investigations into the Atlantic salmon genome revealed similarities with mammals, but also highlighted some unique features of the salmonid TE-CRE landscape. About ∼15%–20% of CREs were derived from TE-sequences (Fig. 2A,C), which is in the lower bound of what has been found in mammals using similar methods to identify TE-CREs (Bourque et al. 2008; Kunarso et al. 2010; Sundaram et al. 2014). Consistent with studies of mammalian genomes (Simonti et al. 2017; Nishihara 2019), the majority of putative TE-CREs in Atlantic salmon were associated with enhancer function rather than promoters (Fig. 2E).
Mammalian TE repertoire (Feschotte and Pritham 2007) and TE-CRE landscapes (Nishihara 2019; Pehrsson et al. 2019; Roller et al. 2021) are dominated by retroelements. In most fish (Shao et al. 2019), including Atlantic salmon (Fig. 1, DNA transposons = 55% of the TEs), DNA transposons are the dominating TEs. However, similar to mammals the majority of Atlantic salmon TE-CREs (73%/45,419) were derived from retroelements (Fig. 4C). Our MPRA data (Fig. 6) also pointed to retroelements being more likely to induce transcription compared to DNA transposons (Fig. 6D) and that transcription-inducing fragments from these TEs were enriched for TF binding motifs known to be bound by liver-active TFs (Tao et al. 2013; Lau et al. 2018; Chaves et al. 2021; Yerra and Drosatos 2023). Only one TF binding motif, the tfcp2l1, was enriched across all three superfamilies enriched for transcription-inducing fragments (Fig. 6E–G). TFCP2L1 has previously been found to bind LTRs in human stem cells (Wang et al. 2014) and is proposed to be a top regulator of human hepatocyte differentiation (Wei et al. 2019), hence it is also likely a key player in shaping evolution of retroelement-associated TE-CRE landscapes in Atlantic salmon.
Although retroelements dominate the salmon TE-CRE landscape, the role of DNA transposons such as DTA and DTT elements in TE-CRE evolution cannot be neglected owing to their high genomic copy numbers (Figs. 1, 3A). Indeed, the TE superfamily contributing the highest number of TE-CREs was the DTA (hATs) superfamily of DNA elements (Fig. 3A). DTAs have also been found important for TE-CRE evolution in several other species. Enrichment of DTA element-insertions in accessible chromatin has also been found in maize (Noshay et al. 2021), and DTA elements make up a significant proportion (15%) of the TE-derived CTCF sites associated with TAD loop anchoring in certain human cell types (Choudhary et al. 2023). In this study we also find families of DTA (as well as DTT TEs) driving rewiring of tissue gene regulatory networks (Fig. 5B,E). Furthermore, even though DTA sequences were not significantly more likely to drive transcription compared to any other TE superfamily (FDR-corrected P-value = 0.18) (Fig. 6D), DTA sequences were more likely to induce transcription (0.72% of fragments were up-regulatory active) compared to sequence fragments derived from DNA transposons in general (0.51% up-regulatory active). Hence, DNA transposons have been a considerable source of novel CREs sequences and likely played an important role in the evolution of genome regulation in Atlantic salmon and other salmonids.
Selection on TE-CRE repertoire
Studies examining how evolutionary forces mold the genomic TE-landscape underscore the significant role of purifying selection in limiting TE accumulation within protein-coding gene sequences (Bartolomé et al. 2002; Rizzon et al. 2003), but also in noncoding regions (Hollister and Gaut 2009; Bergthorsson et al. 2020; Langmüller et al. 2023). These selection signatures on TE insertions in noncoding regions indicate selective forces on TE-CRE evolution, which is also evident from several analyses in our study.
We find clear underrepresentation of TE sequences in accessible chromatin (Fig. 2A), and in particular near the peaks in accessible chromatin in promoters and intergenic regions (Fig. 3D), consistent with purifying selection against TE accumulation in regulatory active regions (Bergthorsson et al. 2020; Langmüller et al. 2023).
In mammals, TEs-CRE are typically from older TE insertions (Simonti et al. 2017; Pehrsson et al. 2019) suggesting that selection pressure on TEs depend on TE insertion age, which is likely related to deterioration of transposition ability as TEs age and accumulate mutations. In Atlantic salmon however, we do not find a general trend of older TE-sequences giving rise to TE-CREs (Fig. 4A). This could be linked to a general relaxation of purifying selection pressure after WGD (Ronfort 1999; Baduel et al. 2019), see section below for in depth discussion. However, we do find that tissue-shared TE-CREs clearly have an older origin compared to tissue-specific TE-CREs (Fig. 4A). One way to interpret this age bias is that tissue-specific TE-CREs have on average more neutral fitness effects. Conversely, older and tissue-shared TE-CREs are more likely to be advantageous, fixed by selection, and maintained for longer under purifying selection. Under this model we expect higher TE-CRE turnaround rates (loss and gain) for tissue-specific compared to tissue-shared TE-CREs, which has been described in mammals (Roller et al. 2021). Higher evolutionary turnaround rates of tissue-specific TE-CREs is also expected if tissue- or cell type–specific CREs are “easier” to evolve than tissue-shared CREs, which has recently been suggested to be the case (Luthra et al. 2024).
Because gene regulation is under tissue-specific selection pressure (Brawand et al. 2011; Berthelot et al. 2018), we expect CRE evolution to be under different selection pressures in different tissues. From mammalian studies, we know that purifying selection on gene regulation is stronger in the brain than liver (Wang et al. 2020); hence, we expect TE-CRE evolution to reflect this asymmetry in selection pressure. Consistent with this expectation, we find clear tissue differences in TE-CRE numbers (Fig. 2D), and TE sequences were consistently depleted in highly brain-biased TF binding motif (Fig. 2G). One interpretation of these results may be that the evolutionary arms race between genomic “parasites” and the host results in selection pressure to “avoid” having sequences that function as, or can evolve into, CREs that impact gene regulatory networks related to critical brain-functions. Because the liver is a key organ for nutrient conversion and detoxification, it is also possible that higher rates of liver-biased TE-CRE evolution (compared to the brain) reflect adaptive evolution of liver function as a response to continuous changes in the environment through macroevolutionary timescales.
TE-CRE evolution in aftermath of the WGD
A premise of this study is our ability to estimate the relative age of TE activity compared with the WGD using a “distance to consensus” approach (for details, see Methods). One potential weakness with this method is that the host genomes’ silencing of TE activity through methylation (Zhou et al. 2020a,b) also impacts mutation rates (Fryxell and Moon 2005; Zhou et al. 2020a,b), which could bias age estimates. To minimize this bias, we applied a CpG-content normalization. It is however plausible that some TEs are able to escape silencing and accumulate less methylation-driven mutations, independent of their CpG content. In such cases, these TEs will appear younger than they actually are. Although we lack empirical data to test this, some results indirectly address this question. The TEs that have been most effective at evading silencing during salmonid evolution are likely the CRE superspreaders, yet these TE-CREs do not have a “younger” age profile compared with TE-CREs from nonsuperspreaders (Fig. 4A). Additionally, our results show that TE insertions from historically highly active DTT elements spiked around the time of WGD (Fig. 4H), which aligns with previous findings (Lien et al. 2016) and support the robustness of our aging approach.
The WGD in the ancestor of salmonids resulted in large-scale gene regulatory rewiring (Lien et al. 2016; Varadharajan et al. 2018). These novel gene regulatory phenotypes have been partly linked to divergent TE insertions in promoters of gene duplicates (Gillard et al. 2021; Sahlström et al. 2023), but the link between WGD and TE-CRE evolution has remained elusive. One hypothesis is that WGDs induce a genomic shock that results in bursts of TE activity (the “genomic shock” model) (McClintock 1984), and that these novel TE insertions allow for rapid TE-CRE evolution and rewiring of gene regulatory networks in the initial aftermath of a WGD. Another hypothesis is that relaxed purifying selection in polyploids allows for higher rates of TE accumulation (Baduel et al. 2019), which in turn will lead to higher rates of neutral and nearly-neutral TE-CRE evolution. In this scenario, however, there is no expectation of a temporal link between bursts of TE activity and bursts of TE-CRE evolution.
Our results do lend support to the “genomic shock” model for TE-CRE evolution following WGD, as we find an increase in the rate of TE-CRE evolution from insertions happening around the time of WGD (Fig. 4F). It is, however, important to note that the WGD-associated TE-CRE evolution is not driven by specific TE subfamilies that were particularly effective at evolving into TE-CREs (Fig. 4B,C,G). DTTs, which exploded in numbers around the WGD (Lien et al. 2016), were poor at evolving into TE-CREs (Fig. 3A) and were significantly less likely to impact transcription compared with other TEs (Fig. 6D,H), in line with other studies showing that the DTT superfamily does not contain many TF binding sites (Simonti et al. 2017; Zeng et al. 2018). Despite this, the WGD-associated rate shift in the evolution of TE-CREs (Fig. 4H,I) was particularly strong for DTTs, and DTTs were associated with rewiring of gene regulatory networks after the WGD (Fig. 5). These results could be explained by a transient period of extreme relaxation of selective constraints, or a confined period of strong selection to recalibrate optimal gene expression phenotypes in the aftermath of the genome doubling. However, to further quantify the importance of selection on TE-CRE evolution, a larger comparative approach (Andrews et al. 2023) is warranted.
Methods
TE annotation
The TE library (ssal_repeats_v5.1) used to annotate TEs in this study is described in detail by Minkley (2018). To generate a TE annotation of the salmon genome (ICSASG v2 assembly), we used RepeatMasker version 4.1.2-p1 (Smit et al. 2015) under default settings with the ssal_repeats_v5.1 library. RepeatMasker takes a library of TE consensus sequences and detects whole and fragmented parts of these consensuses across the genome using a BLAST-like algorithm. The output file contains the genomic coordinates of the annotation, as well as various quality measures such as completeness and divergence from consensus. The latter measure was used to estimate relative ages of TE activity. TE superfamilies were assigned a three-letter tag based on the classifications from Figure 1 in the work of Wicker et al. (2007). Where there was no obvious categorization, a literature review was conducted to determine the taxonomic status of a superfamily, and a new tag name introduced based on available letters (so, e.g., Nimb is here called RIN as a superfamily of LINE elements).
Manual curation of specific TE families was done following an adapted version of the process of Goubert et al. (2022), under inspiration from Suh et al. (2018): Using BLASTN (Altschul et al. 1990), we aligned each TE-consensus to the genome, extracted the 20 best matches, and extended them by 2000 bp upstream and downstream. We checked the extended matches against the Repbase (Bao et al. 2015) database using BLASTN and xBLAST with standard settings, before we aligned them using MAFFT's “einsi” variant (Katoh and Standley 2013). Then, we inspected these alignments for structural features in BioEdit (Hall 1999) for sequence conservation in JalView (Waterhouse et al. 2009). In addition, we ran the TE-Aid package (https://github.com/clemgoub/TE-Aid) on each consensus to help guide curation efforts and check each consensus according to its annotation profile and self-alignment. This helped screen for technical noise such as microsatellite sequences near sites of local annotation enrichment. If the annotating consensus was deemed to be incomplete (i.e., if parts of the extended sequence aligned well outside of the consensus), we used the Advanced Consensus Generator (https://www.hiv.lanl.gov/content/sequence/CONSENSUS/AdvCon.html) to generate a new consensus from the most complete of the extracted alignments for classification.
We produced plots using base R's (R Core Team 2021) plot function, as well as the packages ggplot2 (Wickham 2016) and cowplot. Both the tidyverse (Wickham et al. 2019) and data.table packages were used for analysis, summary statistics, and data wrangling.
Defining genomic context
Based on the NCBI gene annotation (RefSeq ID: GCF_000233375.1), each part of the genome was assigned as promoter, exon, intron, or intergenic. For Figure 1D, the promoter was defined as 1000 bp upstream of to 200 bp downstream from each TSS. Gene annotations can overlap, for example, because of multiple transcript isoforms, so overlapping annotations were merged by prioritizing promoter > exon > intron > intergenic. For TE-CREs (Figs. 2E, 3D–G), each peak was classified as promoter if the summit is <500 bp upstream of or downstream from the start of gene (i.e., first TSS per gene) or intergenic if summit is >500 bp from any gene (exon and intron TE-CREs are not specifically mentioned).
ATAC-seq peak calling
To annotate regions of accessible chromatin, we used ATAC-seq data from four brains and livers from Atlantic salmon (European Nucleotide Archive [ENA; https://www.ebi.ac.uk/ena/browser/home] accession number PRJEB38052). The ATAC-seq reads were mapped to the salmon genome assembly (ICSASG v2, RefSeq ID: GCF_000233375.1) using BWA-MEM (Li and Durbin 2009). Genrich v.06 (https://github.com/jsh58/Genrich) was then used to call open chromatin regions (also referred to as “peaks”) with default parameters, apart from “-m 20 –j” (minimum mapping quality 20; ATAC-seq mode). Genrich uses all four replicates to generate peaks, resulting in one set of peaks for each tissue. The summit of each peak is identified as the midpoint of the peak interval with highest significance.
Estimating proportion of multimapped reads
When a read maps equally well to several loci, BWA-MEM will assign it randomly to one of the loci and give it a mapping quality of zero (MAPQ = 0) in the BAM file. Using the MAPQ field in the BAM file, we extracted the multimapped reads to a separate BAM file and used subread featureCounts (Liao et al. 2014) to get the number of reads that overlap TEs. The counts were summed per TE subfamily, and the proportion of multimapped reads was calculated by dividing the multimapped with the total. This was performed on only one of the brain ATAC-seq samples.
TE-CRE definition
To define TE-CREs, we combined the ATAC-seq peak set with our TE annotations and classified an ATAC-seq peak as a TE-CRE if the peak summit is inside a TE annotation. TE-CREs were defined as shared between tissues if (1) the brain ATAC-seq peak summit was within the liver ATAC-seq peak interval and (2) both the liver and brain peak summits are inside the same TE annotation.
TF motif scanning
To identify potential TF binding sites in the genome, we scanned the entire genome for motif matches using FIMO (Grant et al. 2011). The TF motifs were downloaded from the 2022 JASPAR CORE vertebrate nonredundant motif database (Castro-Mondragon et al. 2022).
Differential TF binding score between liver and brain
To identify TFs that are specific to the liver or brain, we used the differential binding score that was generated by the TOBIAS software (Bentsen et al. 2020) in an earlier study (Gillard et al. 2021) the same ATAC-seq data. In short, the differential binding score quantifies the change in TF binding activity between the two tissues by comparing footprints in accessible chromatin regions. TOBIAS first identifies TF footprints based on ATAC-seq signal, representing areas protected by bound TFs. It then performs motif matching against the underlying genomic sequence to associate footprints with specific TFs. The differential binding score is then summarized across individual binding sites to provide a global measure of TF activity. In our case, positive scores indicate higher TF binding in liver, whereas negative scores indicate higher binding in brain.
Identification of TE families enriched in open chromatin
To identify TE families that had contributed more to TE-CREs than expected by chance, we counted the observed number of ATAC-seq peak summits that are inside an annotated TE for each subfamily and compared that to the expected count if the summits were randomly distributed along the genome. However, because some TE families tend to have certain preferences to where they are inserted in the genome (Chuong et al. 2017), we take into account the genomic context when calculating the expected counts following the methodology described by Bogdan et al. (2020). Specifically, the genome is divided into different regions based on distance from the annotated genes. The regions are (in order of priority): 5UTR, exon, TSS (<1 kb upstream), promoter (1–5 kb upstream), intron, proximal (5–10 kb upstream or <10 kb downstream), distal (10–100 kb upstream or downstream), and desert (>100 kb upstream or downstream). Within each genomic context, the expected count was determined by multiplying the number of summits with the proportion of bases covered by the TE subfamily. Note that because the summits are single base pair points, this straightforward calculation is equivalent to shuffling the summits like they did in Bogdan et al. (2020). After determining the context-specific expected count, the total expected count was calculated as the sum of those. We then used a one-sided binomial test to determine if the observed overlap was significantly greater than expected. Note that the sum of binomial distributions is not exactly a binomial distribution but with large enough numbers, as in our case, the difference is insignificant. The test was performed for each TE subfamily with more than 500 insertions, and P-values were corrected for multiple testing using the Benjamini–Hochberg method. This procedure was repeated for both the liver and brain.
Estimating the temporal activity of TEs
Estimating an “age” of TE activity is notoriously difficult. For some TE-types, it is possible to use a molecular clock-based age estimate (with an assumed mutation rate) (Marchani et al. 2009), but these advanced methods are not applicable to all types of TEs. Hence, in this work we took a different, practically feasible, but more naive approach, in which we compared the similarity between TE insertions to the subfamily consensus sequence. TE insertions will accumulate individual substitutions and diverge in sequence similarity over time. Hence, the sequence divergence between insertions and their TE-subfamily consensus sequence can be used as a proxy for the time since transposition activity.
Sequence divergence estimates were extracted from RepeatMasker software output (Smit et al. 2015). Note that we did not use the default divergence measure reported by RepeatMasker, but rather Kimura distances normalized for CpG content. CpG normalization was chosen because TEs vary in their CpG content, and because CpG sites have higher mutation rates (Coulondre et al. 1978; Fryxell and Moon 2005), CpG content could thus bias estimates of transposition activity “age.” Specifically, two transition mutations at a CpG site are counted as a single transition, one transition is counted as 1/10 of a standard transition, and transversions are counted as usual. These CpG-adjusted Kimura values can be found in the “.align” files output by RepeatMasker when running with the “-a” option.
Gene conversion could also impact sequence divergence estimates; however, it is unlikely to have a major impact on our estimates of TE activity results. Although the conversion process will influence the pairwise divergence between two insertions, each insertion's distance to the TE-subfamily consensus should not be affected to a large degree.
Because our TE activity “age” estimates in the form of Kimura distances cannot easily be converted to absolute time, we used an empirically driven approach to define a Kimura distance interval that represented the approximate time of WGD. All TE insertions happening prior to WGD would become represented on two duplicated chromosomes after the WGD. We therefore reasoned that TE subfamilies with a high proportion of their TE-copies in genomic alignments between duplicated regions of the salmon genome would likely represent TE activity from before the WGD event. Conversely, transposition events occurring after the WGD would likely not end up in the paralogous genomic region in the genome and, thus, not be included in genomic alignments between duplicated chromosomes. Finally, we plotted the mean Kimura distance between the TE-subfamily consensus sequences and all genomic insertions for that subfamily against the proportion of TE insertions in duplicate region alignments (Supplemental Fig. 1). This plot indicated that the proportion of TE insertions in alignments started increasing at a Kimura distance around 7–10, which we then used as a cutoff for classifying TE activity temporally associated with the salmonid WGD.
Coexpression analysis
We used two RNA-seq expression data sets to analyze the effect of TE-CREs on gene expression: (1) a liver data set comprising 112 samples spanning different diets and life stages in fresh-water (27,786 expressed genes) (Gillard et al. 2018) and (2) a tissue atlas comprising 13 different tissues (24,650 expressed genes) (Lien et al. 2016).
TE-CREs in liver, brain, or both (the ATAC-seq peak summits of the liver, and brain TE-CREs reciprocally overlapped the peak in the other tissue) were assigned to genes with the closest TSS (no distance threshold was enforced). For each TE subfamily with insertions associated with at least five expressed genes, we computed the network density of the associated genes (i.e., the mean Pearson's correlation between all gene pairs associated with that TE subfamily). FDR-corrected P-values were obtained by comparing these network densities to those of randomly selected expressed genes. We ran 100,000 simulations drawing the same number of genes, containing the same number of WGD-derived duplicates (which are often coexpressed), as found in the original data. Effect sizes were calculated as the number of standard deviations away from the mean of randomized network densities.
Massive parallel reporter assay
The transcriptional regulatory potential of TE-CREs in Atlantic salmon was assessed using ATAC-STARR-seq as previously described by Wang et al. (2018). We used the pSTARR-seq reporter plasmid with the core promoter of Atlantic salmon elongation factor 1 alpha (EF1α; NC_027326.1: 7785458–7,785,702) instead of the super core promoter 1 (SCP1) originally adapted in human cells (Arnold et al. 2013). ATAC DNA fragments were extracted from Atlantic salmon liver cell nuclei following the Omni-ATAC protocol (Corces et al. 2017). A clean-up step was performed using a Qiagen MinElute PCR purification kit, and PCR-amplification was done using NEBNext Ultra Q5 DNA polymerase master mix (New England Biolabs) with forward primer (5′-TAGAGCATGCACCGGCAAGCAGAAGACGGCATACGAGAT[N10]ATGTCTCGTGGGCTCGGAGATGT-3′, where N10 corresponds to a random 10 nucleotide i7 barcode sequence) and reverse primer (Rv:5′-GGCCGAATTCGTCGATCGTCGGCAGCGTCAGATGTG-3′). Thermo cycling conditions were 5 min at 72°C, 30 sec at 98°C, eight cycles of 10 sec at 98°C, 30 sec at 63°C, and 1 min at 72°C. PCR products were purified using a Qiagen MinElute PCR purification kit and were size-selected (∼30–280 bp) using Ampure XP beads (Beckman Coulter). Reporter plasmid libraries were made by cloning amplified ATAC fragments into AgeI-HF- and SalI-HF-linearized pSTARR-seq plasmid using InFusion HD cloning kit (Takara) and then propagated in MegaX DH10B T1R electrocompetent bacteria. Plasmids were isolated using the NucleoBond PC 2000 mega kit (Macherey-Nagel). An aliquot of plasmid library was PCR-amplified with i5 and i7 primers and sequenced on NovaSeq (150 bp paired-end) and aligned to the salmon genome to ensure sufficient complexity and proportions of cloned fragments within open chromatin region. The plasmid library was electroporated into primary salmon hepatocytes as previously described (Datsomor et al. 2023). Total RNA was isolated 24 h post-transfection using the Qiagen RNeasy midi columns. Poly(A)+ RNA from total RNA was extracted using the mRNA isolation kit (Roche). Remaining genomic DNA in isolated mRNA were digested with Turbo DNase (Thermo Fisher Scientific). Complementary DNA (cDNA) from mRNA was generated using the Superscript III reverse transcriptase (Thermo Fisher Scientific) with a gene-specific primer (5′-CAAACTCATCAATGTATCTTATCATG-3′). Sequencing-ready libraries from cDNA and the input (reporter plasmid library) were prepared as previously described by Wang et al. (2018) and Tewhey et al. (2016).
Sequenced reads were mapped to the salmon genome assembly (ICSASG v2, RefSeq ID: GCF_000233375.1) using BWA-MEM (Li and Durbin 2009). The number of read pairs mapped to each unique location was counted. Each unique location, that is, having a specific start and end, was assumed to come from a unique fragment. These counts were fed into DESeq2 (Love et al. 2014) using the DNA (input plasmid library) as control and contrasted with the RNA (cDNA) samples. Fragments with a significant RNA-to-DNA ratio were used to define fragments with significant regulatory activity. Prior to DESeq2, the fragment counts were split into bins by length.
Enrichment analysis of regulatory-active fragments in TE families
To test whether specific TE superfamilies were enriched for regulatory-active fragments in our MPRA data, we first classified each assayed fragment as “up-regulatory,” “down-regulatory,” or “no effect” based on its differential abundance in RNA (cDNA) versus DNA (plasmid input; i.e., log2FoldChange and adjusted P-value from DESeq2). We retained only fragments that overlapped a TE annotation by at least 50% of their length. For each TE superfamily, we then performed a Fisher's exact test comparing the ratio of (1) regulatory-active (up- or down-regulatory) versus nonactive fragments in that superfamily to (2) regulatory-active versus nonactive fragments in all other TE superfamilies. Odds ratios and two-sided P-values were calculated for each comparison, and P-values were FDR-adjusted for multiple testing. TE superfamilies with FDR-adjusted P-values below 0.1 and an odds ratio greater than one were considered significantly enriched for either up- or down-regulatory fragments. The same approach was repeated at the subfamily level, considering each TE subfamily separately.
TF motif enrichment in TE-derived active MPRA fragments
To investigate what TF motifs that drive the enhancer activity in TE-CREs we performed a Fisher's exact test to test the dependence between having a motif in the MPRA fragment and the fragment being regulatory active. For each TE superfamily (RLX, RLC, or RST), we considered the MPRA fragments that overlap a TE insertion (with at least 5 bp) and checked if it had any TF motif matches in the overlapping region. The Fisher's exact test was performed for each motif, testing the dependency between the fragment being classified as up-regulating and a motif match. The FDR was controlled with the Benjamini–Hochberg method in R (R Core Team 2021).
Data wrangling and visualization
Analyses of processed raw data (i.e., tabulated results data) were done in R (R Core Team 2021) using base-R functions and tidyverse (Wickham et al. 2019). Data visualizations were done in R using the libraries ggplot2 (Wickham 2016) and pheatmap (https://github.com/raivokolde/pheatmap).
Data access
Raw sequencing data from the ATAC-STARR-seq MPRA experiment generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB81135. All scripts to reproduce figures and analysis can be found as Supplemental Code. The code is also available on GitLab (https://gitlab.com/sandve-lab/TE-CRE) and Zenodo (https://doi.org/10.5281/zenodo.13907938). Data used to generate all figures and analyses are also at Zenodo (https://doi.org/10.5281/zenodo.13903583).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was funded by Norwegian University of Life Sciences (NMBU) and the Norwegian Research Council through the projects Transpose (275310), Rewired (274669), and DigiSal (248792). We thank Sigbjørn Lien for comments on earlier versions of the manuscript and the anonymous reviewers for a thorough and fair review process that significantly improved the quality of the paper.
Author contributions: S.R.S. and T.R.H. conceived the study. S.R.S. and T.R.H. acquired funding. A.D. and T.H. performed laboratory experiments related to the massive parallel reporter assays. Ø.M., L.G., S.R.S., and T.R.H. performed analyses. Ø.M., L.G., T.R.H., and S.R.S. drafted the manuscript. All authors took part in critical discussions of various aspects of laboratory work and/or analytical approaches relevant to their expertise. All authors critically reviewed the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278931.124.
- Received January 2, 2024.
- Accepted February 6, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








