
Vertebrates show coordinated elevated expression of mitochondrial and nuclear genes after birth
Abstract
Interactions between mitochondrial and nuclear factors are essential to life. Nevertheless, the importance of coordinated regulation of mitochondrial–nuclear gene expression (CMNGE) to changing physiological conditions is poorly understood and is limited to certain tissues and organisms. We hypothesized that CMNGE is important for development across vertebrates and, hence, should be conserved. As a first step, we analyzed more than 1400 RNA-seq experiments performed during prenatal development, in neonates, and in adults across vertebrate evolution. We find conserved sharp elevation of CMNGE after birth, including oxidative phosphorylation (OXPHOS) and mitochondrial ribosome genes, in the heart, hindbrain, forebrain, and kidney across mammals, as well as in Gallus gallus and in the lizard Anolis carolinensis. This is accompanied by elevated expression of TCA cycle enzymes and reduction in hypoxia response genes, suggesting a conserved cross-tissue metabolic switch after birth/hatching. Analysis of about 70 known regulators of mitochondrial gene expression reveals consistently elevated expression of PPARGC1A (also known as Pgc-1alpha) and CEBPB after birth/hatching across organisms and tissues, thus highlighting them as candidate regulators of CMNGE upon transition to the neonate. Analyses of Danio rerio, Xenopus tropicalis, Caenorhabditis elegans, and Drosophila melanogaster reveal elevated CMNGE prior to hatching in X. tropicalis and in D. melanogaster, which is associated with the emergence of muscle activity. Lack of such an ancient pattern in mammals and in chickens suggests that it was lost during radiation of terrestrial vertebrates. Taken together, our results suggest that regulated CMNGE after birth reflects an essential metabolic switch that is under strong selective constraints.
Mitochondria are pivotal to cellular metabolism across all eukaryotes. In animal cells, unlike most cellular pathways, the mitochondrial genetic system is the only system with factors encoded by two genomes, in two cellular compartments: the nucleus and the mitochondria. It has been shown that genes encoded by the two genomes that participate in the same pathway, such as the energy-producing oxidative phosphorylation system (OXPHOS), have coevolved and that interference with such coevolution affects fitness (Gershoni et al. 2009; Meiklejohn et al. 2013; Bar-Yaacov et al. 2015; Barreto et al. 2018; Hill et al. 2019; Moran et al. 2024). We and others previously found that coordination of gene expression between the mitochondrial (mtDNA) and nuclear genomes, especially in the OXPHOS system, is common across most healthy human tissues (Barshad et al. 2018), a variety of cancer samples (Reznik et al. 2017), and in certain cell types (Medini et al. 2021a), yet is compromised in several disease conditions, including COVID19, Alzheimer's disease, and several cancer types (Medini et al. 2021b; Papier et al. 2022). It is thus reasonable to assume that coordinated mitonuclear gene expression (CMNGE) is important, will respond to changing environments, and will change upon alteration of physiological conditions, such as the changes that occur during prenatal development and in the transition to the neonate (Sharma et al. 2014).
Indeed, mitochondrial activity, morphology, number, and regulation of replication and transcription are critical for cell differentiation and development (Folmes et al. 2011; Hom et al. 2011; Wellen and Thompson 2012; Medini et al. 2020). Specifically, knockout of mitochondrial regulatory proteins such as mtDNA polymerase gamma (Polg) (Hance et al. 2005) and mtDNA transcription factor A (Tfam) (Larsson et al. 1998) resulted in developmental arrest between embryonic days 7.5 and 8.5 (upon Polg knockout) and lethality on day 10.5 of mouse embryos (upon Tfam knockout). In addition, knockout of mitochondrial RNA polymerase (rpom-1) resulted in decreased brood size and impaired ova development in the worm Caenorhabditis elegans (Charmpilas and Tavernarakis 2020). Second, knockout of Nrf1, a key regulator of nuclear DNA-encoded OXPHOS gene expression, led to mouse embryonic lethality (Chan et al. 1998). Third, knockout of another regulator of OXPHOS genes expression, Nfe2l2 (also known as Nrf2), led to a reduction in the levels of NADPH and the NADPH/NADP+ ratio in embryonic mouse fibroblasts (Singh et al. 2013), and ATP levels were significantly reduced upon NFE2L2 (also known as NRF2) knockdown in human cancer cell lines (Kim et al. 2011). Both NRF1 and NRF2 are involved in mitochondrial biogenesis via the regulation of peroxisome proliferator–activated receptor-gamma coactivator-1alpha (PPARGC1A), an OXPHOS master regulator (Gureev et al. 2019; Deng et al. 2020). Accordingly, targeted deactivation of Ppargc1a (also known as Pgc-1alpha) as well as a transcription factor from the same family, Ppargc1b, also known as Pgc-1beta (PGC-1αβ−/−) in the mouse heart led to reduced growth, late fetal arrest in mitochondrial biogenesis (at E16.5 and E17.5), and 70% lethality of mice within 24 h after birth (Lai et al. 2008). Fourth, dysfunctional mitochondria in the embryonic mouse and human patients’ hearts led to severe cardiomyopathy and either embryonic or neonatal lethality (Zhao et al. 2019). Taken together, transcriptional regulation of mitochondrial genes in both the nucleus and the mtDNA is pivotal for the developing embryo and for the transition to the neonate.
Studies in model organisms support the occurrence of a metabolic shift after birth in certain tissues. Quantification of lactic acid in rat livers revealed that glycolysis is decreased after birth (Burch et al. 1963). Consistent with this finding, the human postnatal liver and heart collected 6 and 36 weeks after birth displayed an increase in mitochondrial content and respiratory chain activities compared with fetal samples collected after 11–17 weeks of gestation (Minai et al. 2008). Finally, an increased level of the mitochondrial electron carrier cytochrome c was observed in newborn human and rat livers compared with fetal samples (Krizova et al. 2021).
These pieces of evidence led us to hypothesize that there is a regulated increase in OXPHOS function after birth, which is conserved in evolution. Our analysis relies on the notion that genes belonging to the same pathway, including OXPHOS, tend to be coregulated (van Waveren and Moraes 2008; McShane et al. 2024). To test this hypothesis, we interrogated N = 1482 RNA-seq experiments from samples collected during embryo and fetal development as well as after birth from a variety of metazoans in several different tissues (Fig. 1).
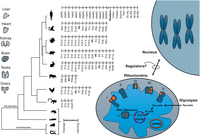
Roadmap to assessment of mitonuclear gene expression (RNA-seq) during the course of development and evolution. Tissues analyzed (indicated) were collected from six mammals (Homo sapiens, Macaca mulatta, Mus musculus, Rattus norvegicus, Oryctolagus cuniculus), as well as from Gallus gallus (chicken), during development. The developmental stages are indicated per organism, and P0 (birth/hatching) is highlighted for mammals and birds. Missing stages (owing to lack of samples) are indicated by “/.” To avoid sample size issues, gene expression in Anolis carolinensis (brain, heart, kidney, liver) was compared between a group of embryonic versus adult samples (e.g., not divided into further stages). Analysis of additional nonmammalian vertebrates, for example, Xenopus tropicalis (tropical frog) and Danio rerio (zebrafish), as well as two invertebrates, for example, Drosophila melanogaster and Caenorhabditis elegans, was limited to the prenatal stages. mtDNA and nuclear DNA–encoded subunits of OXPHOS complexes I, III, IV, and V are indicated in orange and dark blue, respectively. Additional selected metabolic pathways are indicated, including the mitochondrial ribosome (mito-ribosome), tricarboxylic acid cycle (TCA), and glycolysis. The lack of knowledge of regulators of mitonuclear gene expression is indicated.
Results
Postnatal mtDNA gene expression is generally elevated across evolution
To assess the dynamics of mitochondrial transcript regulation during metazoan development, we have analyzed publicly available bulk RNA-seq data from six mammals—Homo sapiens, human; Macaca mulatta, rhesus monkey; Mus musculus, mouse; Rattus norvegicus, rat; and Oryctolagus cuniculus, rabbit—and from Gallus gallus (chicken). The data were generated from seven tissues collected during prenatal and postnatal development (Data set I; Methods) (Table 1; Cardoso-Moreira et al. 2019; Chen et al. 2022). This analysis was augmented by analysis of RNA-seq from samples collected from Sus scroffa (pig) liver during prenatal stages and from the neonate (Data set II). To identify the specific time frame in which potential transition in gene expression occurred, the data were analyzed in sliding windows of available developmental stages, while testing a variable number of consecutive stages (e.g., windows containing either two, four, or six of the tested developmental stages; see Methods). Window sizes w = 4 and w = 6 showed similar trends (Supplemental Figs. S1–S6); however, given the limited number of time points in chicken samples (mostly n = 9), we reasoned that using w = 6 would reduce resolution and potentially mask key shifts in gene expression. Window w = 2, on the other hand, increased the noise. Therefore, for consistency, w = 4 was chosen in the analyses across species, offering sufficient resolution in all tested organisms. For each time point, we computed the average fold-change (FC) of mtDNA genes’ expression and identified the time points with the highest mean FC. Our findings revealed significantly elevated mtDNA gene expression, which was observed in postnatal samples from five out of seven tissues across all tested species, with one exception: reduction in mtDNA gene expression in chicken liver after hatching (see below) (Fig. 2A,B; Supplemental Fig. S7). Specifically, we found elevated mtDNA genes’ expression right after or at 1–2 weeks after birth/hatching in the heart in six of six of the tested organisms, in the forebrain/cerebrum in five of six organisms (human, rat, rabbit, mouse, and rhesus monkey), in hindbrain/cerebellum samples in six of six organisms, in kidney samples in four of six organisms (human, mouse, chicken, and rhesus), in liver samples in five of seven organisms (human, rat, mouse, rabbit, and pig—the only tissue tested in the latter), and in testes only in rhesus monkey (Table 2). Further support for the elevation of mtDNA gene expression after birth came from the analysis of other additional RNA-seq data sets collected from postnatal mouse heart samples compared with embryonic samples (Data set IV, embryos collected from gestational day 13.5 vs. samples collected 6 weeks after birth) (Supplemental Fig. S8; Matkovich et al. 2014). All these results reflect a conserved elevation of mtDNA gene expression across tissues, implying a conserved regulatory mechanism.
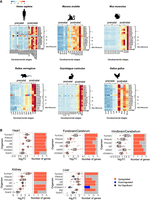
mtDNA gene expression is elevated in postnatal vertebrate samples. (A) Heatmaps representing mtDNA gene expression in heart from five mammals and in the chicken. Color bar indicates the scaled expression. Red scale indicates elevated expression; blue scale, reduced expression. The x-axis indicates prenatal and postnatal stages of sample collection; y-axis, mtDNA genes and their OXPHOS complex assignments. (B, left) Box plots indicating the log2FC of mtDNA genes. The x-axis indicates Log2FC; y-axis, the tested organisms: H. sapiens, human; M. mulatta, rhesus; M. musculus, mouse; R. norvegicus, rat; O. cuniculus, rabbit; G. gallus, chicken; Sus scroffa, pig; and A. carolinensis, lizard. Color bar: blue dots indicate downregulation (log2FC < −0.2, P-value < 0.05); red dots, upregulation (log2FC > 0.2, P-value < 0.05); and gray dots, nonsignificant. Dashed vertical red line displays the value of zero log2FC on x-axis. (Right) Bar plots representing the number of significant (indicated in red or blue) and nonsignificant (NS) mtDNA genes expression (colored in gray). Blue indicates statistically significant genes with decreased expression between the time points (P < 0.05); red, number of genes with significantly increased expression. The x-axis indicates number of genes; y-axis, tested organisms.
Resources for RNA-seq data per tissue and organism
The developmental time point presenting the highest fold-change of either upregulation or downregulation of mtDNA gene expression in samples from Data sets I and II
While considering the liver, we noticed that, unlike mammals, chicken liver displayed a reduction in mtDNA gene expression during the first week after hatching. This finding gained support from the analysis of an additional data set (Data set III), which includes chicken liver samples from embryonic day 13, compared with samples collected 5 weeks after hatching (Supplemental Fig. S9; Xu et al. 2019). We interpret the divergent mtDNA gene expression pattern in the chicken liver compared with the mammalian liver as likely associating with the accumulation of fat in the chick liver but not in mammals, thus offering a different carbon source for OXPHOS activity in chicken liver after hatching (see Discussion).
Our analysis so far focused mostly on mammals with one nonmammalian vertebrate: the chicken. To assess whether our observed elevated mtDNA gene expression after birth is shared across other vertebrates, we sought to extend our analysis to other additional nonmammalian vertebrates. Accordingly, analysis of RNA-seq experiments generated from 48 embryonic samples from the lizard Anolis carolinensis (collected from developmental stages 15–16) compared with 30 adult samples from four organs (brain, heart, kidney, liver; Data set V) (Marin et al. 2017) revealed elevated mtDNA genes expression in the adult heart, brain, and kidney. The A. carolinensis liver did not show any consistent pattern (Fig. 2B; Supplemental Fig. S10). This finding suggests that elevated mtDNA gene expression after birth is conserved across mammalian and terrestrial nonmammalian vertebrates.
Expression of postnatal nuclear DNA–encoded OXPHOS gene is elevated across evolution
As mtDNA-encoded proteins interact with nuclear DNA–encoded subunits of the OXPHOS system, we sought to assess the expression pattern of the latter during fetal development and after birth. Our findings indicate that the same tissues that showed elevated mtDNA gene expression also showed clear elevation of nuclear DNA–encoded OXPHOS gene expression in Data sets I, II, and IV (Table 2; Fig. 3A,B; Supplemental Figs. S11, S12). It is worth noting that we used the sliding windows approach as in the mtDNA analyses (Supplemental Figs. S13–S18). This finding supports the hypothesis that the elevation of OXPHOS genes’ expression after birth/hatching is coordinated between the mtDNA and the nucleus (mitonuclear). To quantify the level of CMNGE in each of the tested organisms and tissues, we calculated the number of significantly altered expression of mtDNA genes and nuclear DNA-encoded OXPHOS genes and divided them by the number of genes with available expression information (in each available data set, separately). This analysis revealed that the heart displayed high coordination level in all organisms analyzed (average 0.8, SD 0.08) (Table 2; Fig. 3B; Supplemental Table S1). Moreover, the organisms that displayed change in mtDNA gene expression during the first weeks after birth/hatching also displayed CMNGE in the same days, including chicken liver that showed coordinated downregulation in OXPHOS genes (level of coordination = −0.6, e.g., downregulation; Data set I) (Table 2; Fig. 3B; Supplemental Table S1; Supplemental Figs. S11, S19). An exception was the chicken hindbrain/cerebellum, which displayed elevated mtDNA gene expression between P0 and P7 days after birth yet decreased OXPHOS nuclear genes’ expression during the same days (level of coordination = −0.19; e.g., downregulation). This observation awaits confirmation once additional RNA-seq data sets become available from chicken samples collected from the hindbrain during development. In the A. carolinensis (hereon lizard) heart, and to a lower extent in the kidney, we observed a tendency toward an elevated expression of structural nuclear DNA–encoded OXPHOS genes (Supplemental Fig. S20). The lizard brain did not show a consistently elevated OXPHOS gene expression in the adult. These results suggest that CMNGE is observed in the lizard heart and in the kidney. Finally, it is worth noting that while considering mammals and the chicken, structural subunits of the OXPHOS showed a sharper elevation in gene expression after birth than did OXPHOS assembly subunits (Fig. 3; Supplemental Figs. S21–S27). This suggests that for OXPHOS, elevated CMNGE after birth/hatching is more coordinated among the inherent structural members of the OXPHOS system.
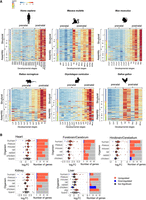
Nuclear DNA-encoded OXPHOS genes expression is elevated after birth/hatching across vertebrates. (A) Heatmaps representing expression of nuclear DNA-encoded OXPHOS gene in the heart from five mammals and in birds. Color bar: red scale indicates elevated expression; blue scale, reduced expression. Above each panel are the labels of the prenatal and postnatal samples. The x-axis indicates developmental stages; y-axis, nuclear DNA-encoded genes, divided according to OXPHOS complexes, structural and assembly genes, and Cytochrome C (CYCS). (B, left) Box plots indicating the log2FC of nuclear DNA-encoded structural subunits of the OXPHOS. The x-axis indicates Log2FC; y-axis, the tested organisms. Color bar: blue dots indicate downregulation (log2FC < −0.2, P-value < 0.05); red dots, upregulation (log2FC > 0.2, P-value < 0.05); and gray dots, nonsignificant. Dashed vertical red line displays the value of zero log2FC on the x-axis. (Right) Bar plots representing the number of significant (indicated in red or blue) and nonsignificant (NS) nuclear OXPHOS genes expression (colored in gray). Blue indicates statistically significant genes with decreased expression between the time points (P < 0.05); red, number of genes with significantly increased expression. The x-axis indicates number of genes; y-axis, organisms.
Altered mitonuclear OXPHOS gene expression after birth is part of a wider metabolic switch
Because elevation in OXPHOS function after birth could be part of a general functional mitochondrial shift, we asked whether gene expression of any other mitochondrial pathways is altered after birth. Therefore, we compared gene expression of the following mitochondrial-related pathways before and after birth: the mitochondrial ribosome, the tricarboxylic acid cycle (TCA), formation, and scavenging of reactive oxygen species (ROS), in addition to glycolysis.
First, the mitochondrial ribosome, which like the OXPHOS incorporates factors encoded by the nuclear and mitochondrial genomes, showed coordinated mitonuclear expression changes in heart of all organisms, except for the lizard (average 0.62, SD 0.24) (Supplemental Fig. S28; Supplemental Table S1), and in the kidney of humans, the rhesus monkey, and rabbit (levels of coordination = 0.64, 0.87, and 0.65, respectively). In addition, we observed strong CMNGE in the pig liver (Data set II, level of coordination = 0.78). The same trend, although to a lower extent, was observed in the mouse (Data set I, level of coordination = 0.28) but not in the liver of other tested mammals. In contrast, we observed a reduction of gene expression in the chicken liver as in OXPHOS genes (level of coordination = −0.52; e.g., downregulation). Finally, we found significantly decreased expression of the ribosomal genes in the cerebellum only in the chicken, similar to the OXPHOS genes (level of coordination = −0.5; e.g., downregulation) (Supplemental Fig. S28). These results suggest that the mitochondrial ribosomal genes undergo a similar expression change after birth as in OXPHOS, especially in the heart.
Second, we identified sharply elevated expression of genes related to the TCA cycle mainly in the heart and kidney after birth/hatching, as well as in several organisms in the rest of the tested tissues (Supplemental Fig. S29). Similar to the OXPHOS genes and the mitochondrial ribosome, the chicken liver displayed reduced TCA cycle genes’ expression. Taken together, these results support a conserved overall functional metabolic shift in the mitochondria after birth or hatching across tissues and across the evolution of vertebrates.
Our analysis of genes involved in glycolysis did not reveal any consistent overall expression pattern change while jointly considering a pathway in the comparison of prenatal and postnatal samples (Supplemental Fig. S30A). Nevertheless, lactate dehydrogenase (LDH), a key glycolysis enzyme, showed an interesting pattern: LDH has two subunits: LDHA and LDHB; the first is known to be upregulated mostly in glycolytic conditions, whereas LDHB is commonly upregulated in conditions that require OXPHOS (Read et al. 2001; Porporato et al. 2011). Although LDHA did not show any significantly conserved expression pattern across all tested tissues, it decreased in the heart after birth in four of six tested species (Supplemental Fig. S30B,C). LDHB was significantly upregulated in several tissues that displayed mitonuclear upregulation across species: the forebrain and hindbrain in three of six organisms, the kidney in four or six organisms, and the heart in five of six of the tested organisms (Supplemental Fig. S30B). It is worth noting that the expression of enzymes and regulators of the ROS scavenging pathway was not significantly altered in all tested mammals and in the chicken (Supplemental Fig. S31). These results suggest that by and large, OXPHOS likely replaced glycolysis as the dominant ATP source after birth at least in the mentioned tissues.
Nevertheless, the liver displayed significantly reduced LDHB expression after birth/hatching in four of seven species (human, rhesus, mouse, and chicken) (Supplemental Fig. S30B,C). Because the liver is known to have special metabolic activities, including gluconeogenesis, we asked whether the expression pattern we observed for OXPHOS and LDHB is also associated with similar expression changes of the key enzyme of this pathway, phosphoenolpyruvate carboxykinase (PCK2, also known as PEPCK) (Hanson and Reshef 1997). PEPCK (PCK1; the cytosolic form) also has a mitochondrial isoform, PCK2. Although PCK2 did not show any clear expression pattern change after birth/hatching, PCK1 was generally upregulated in all mammalian tissues and in the chicken heart. The expression of PCK1 was significantly elevated in the liver of four mammals (human, mouse, rat, and pig), yet did not significantly change in the chicken or in the chicken kidney after hatching, the main tissues in which gluconeogenesis takes place (Supplemental Fig. S32; Supplemental Table S1). In summary, the generally elevated expression of PCK1 in the mammalian liver and the lack of change in PCK1 in the chicken liver, along with only partial elevation of OXPHOS in this tissue in mammals and reduction of LDHB expression in mammals, may reflect a unique involvement of the OXPHOS in liver metabolism during evolution.
The lizard A. carolinensis did not show any consistent pattern in these pathways when comparing embryo to adult samples (Supplemental Figs. S33–S37; Supplemental Table S1). This suggests that although the lizard did show a trend toward elevated OXPHOS gene expression in postnatal periods, more detailed expression analysis in this species after hatching may shed light on the involvement of other additional metabolic pathways.
Taken together, the CMNGE-coordinated upregulation of mitonuclear gene expression (e.g., OXPHOS and the mitochondrial ribosome) is a widespread phenomenon across somatic tissues, is highly conserved among mammals and chicken, and is accompanied by significant elevation of TCA cycle enzymes. This supports an overall change in mitochondrial metabolism after birth/hatching that is regulated already at the transcript level.
The elevated expression and CMNGE after birth associates with altered expression of regulatory factors that modulate mitochondrial biogenesis
The discovery of elevated CMNGE in vertebrates during the transition to the neonate suggests an underlying regulatory mechanism. Accordingly, it is conceivable that certain regulators of mitonuclear transcripts modulate CMNGE elevation after birth. To identify such factors, we tested for two categories (transcription and post-transcription), while highlighting transcription factors involved in mitochondrial biogenesis. To this end, we analyzed bulk RNA-seq samples from Data sets I and II, including six mammals and chicken, for the expression pattern of about 70 known mitochondrial regulatory factors, including mitochondrial transcription, post-transcriptional activities, and biogenesis-related genes (Fig. 4; Supplemental Tables S1, S2; Supplemental Figs. S38–S40). Specifically, we compared the expression of these genes in tissue samples that preceded the time point at which the OXPHOS expression transition occurred to the samples right after the transition in each organism.
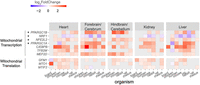
Expression of regulatory factors that modulate mitochondrial biogenesis associates with mitonuclear expression shift after birth/hatching across vertebrates. Heatmap presenting log2FC values of regulatory factors and genes related to mitochondrial biogenesis that displayed significant change in at least four tissues within any organism and in at least five organisms within any tissue (P-value < 0.05). Color bar indicates the log2FC. The x-axis indicates organisms divided by tissues; y-axis, genes. Only log2FC > 0.2 and log2FC < −0.2 are shown. (*) Genes related to mitochondrial biogenesis.
As we expected that only a few factors would be involved in the elevated CMNGE in the neonate, we counted for each factor the number of organisms and corresponding tissues that displayed significant value in the tested factors. Specifically, we considered factors as candidates if they consistently displayed significantly altered expression in the transition to neonatal life in at least four tissues within any organism and at least five organisms within any tissue (Fig. 4).
First, our analysis highlighted PPARG coactivator 1 alpha (PPARGC1A, PGC-1alpha), as a regulatory factor whose expression pattern best resembled the pattern of OXPHOS genes across organisms and tissues. PPARGC1A is regulated by environmental stimuli and is considered central in regulating mitochondrial biogenesis (Puigserver et al. 1998). Similar to mitonuclear OXPHOS genes, PPARGC1A was significantly upregulated in the liver of four of eight organisms—human (log2FC = 1.39), pig (log2FC = 2.64), rabbit (log2FC = 2.5), and rat (log2FC = 1.77)—whereas it was downregulated in the chicken liver (log2FC = −1.65). The elevated expression of PPARGC1A was also apparent in the forebrain/cerebrum (five of six organisms), hindbrain/cerebellum (three of six organisms), and kidney (three of seven), yet was not clearly evident in the heart (only in the mouse) (Fig. 4; Supplemental Fig. S38). This result supports the association of elevated CMNGE after birth with altered regulation of mitochondrial biogenesis in most tested tissues.
Second, NFE2L2 (also known as NRF2), a transcription factor that responds to oxidative stress and is involved in the regulation of mitochondrial biogenesis via regulation of PPARGC1A (Gureev et al. 2019; Deng et al. 2020), was significantly upregulated in the following tissues and organisms (Fig. 4; Supplemental Fig. S38; Supplemental Table S1): in the heart (all organisms, except for the lizard), in forebrain/cerebrum (four of six organisms), in hindbrain/cerebellum (five of six), in the kidney of five of seven organisms, and in liver of five of eight organisms. Unlike PPARGC1A, NFE2L2 expression was elevated in chicken liver.
Third, CCAAT enhancer binding protein beta (CEBPB) was significantly upregulated in the heart of five of seven organisms, in the forebrain/cerebrum of all organisms, in the hindbrain/cerebellum of three of six organisms, in the kidney of four of seven organisms, and in the liver of six of eight organisms (Fig. 4; Supplemental Fig. S38). Previous analysis of ChIP-seq experiments from the ENCODE consortium revealed that CEBPB binds upstream regulatory elements of OXPHOS genes more than expected by chance (Blumberg et al. 2014). This is the only transcription factor in the current analysis that was experimentally shown to both regulate nuclear gene transcription, bind the mtDNA in vivo, and localize to the mitochondria in human cells (Blumberg et al. 2014). CEBPB is a transcription factor involved in inflammation, metabolism, and differentiation as well as in fatty acid metabolism in collaboration with PPARGC1A (Takagi et al. 2022). Therefore, CEBPB is an attractive candidate to regulate the transcription of both nuclear and mtDNA-encoded OXPHOS genes in the transition to the neonate.
Fourth, G elongation factor mitochondrial 1 (GFM1) was upregulated in the heart (six of seven organisms), although it was downregulated in the lizard heart and upregulated in the forebrain/cerebrum of two of six organisms, in the hindbrain/cerebellum of four of six organisms, in the kidney of six of seven organisms, and in the liver of three of eight organisms (Fig. 4). The upregulation of GFM1 is of interest in the context of upregulated mitochondrial ribosomal proteins in the neonate. This raises the possibility that mitochondrial protein synthesis is also upregulated in the neonate.
The expression of NRF1, a known regulator of OXPHOS gene expression in the nucleus and of mitochondrial biogenesis (Gureev et al. 2019), was significantly downregulated in the liver of five of eight organisms (human, mouse, pig, rabbit, and rat) but was elevated in the chicken liver and downregulated in the kidney and forebrain/cerebrum of four of seven and four of six organisms, respectively (Fig. 4; Supplemental Fig. S38). This suggests that only a subset of the factors that regulate mitochondrial biogenesis form candidate regulatory factors to be involved in CMNGE after birth/hatching.
Finally, while considering the core mtDNA transcriptional regulatory factors, we noticed that TFB2M was significantly upregulated in the heart of six of seven organisms, in the forebrain/cerebrum of three of six organisms (chicken, rabbit, and rhesus), in the hindbrain/cerebellum of rhesus, in the kidney of three of seven organisms (mouse, rabbit, and rhesus), and in the liver of five of eight organisms (human, mouse, pig, rabbit, and rat) (Fig. 4; Supplemental Table S1). It is worth noting that TFB2M expression was significantly downregulated in chicken liver. Then, while considering mitochondrial transcription factor A (TFAM), we found that it was significantly downregulated after birth in the human heart (log2FC = −0.63); in forebrain/cerebrum samples from humans, rats, and rabbits (log2FC = −1.37, log2FC = −1.12, log2FC = −0.81, respectively); in the hindbrain/cerebellum of humans and rabbits (log2FC = −0.81, log2FC = −0.51, respectively); in the human and chicken kidney (log2FC = −0.5, log2FC = −0.34, respectively), and in the chicken liver (log2FC = −0.61, respectively) (Supplemental Fig. S38; Supplemental Table S1). Because in high cellular concentrations, TFAM coats the mtDNA and, in lower concentrations, it promotes mtDNA transcription and replication, it is conceivable that lower TFAM levels reduce mtDNA occupancy and increase mtDNA transcription and replication (Kukat et al. 2015). Consistent with this interpretation, the mitochondrial RNA polymerase (POLRMT) was significantly upregulated in the heart of humans, the mouse, and rat and in human kidney; these findings support the possibility that core mtDNA transcriptional machinery partially participates in the CMNGE upregulation after birth (Supplemental Table S1; Supplemental Fig. S38).
Taken together, the regulation of mitochondrial biogenesis seems the most prominent regulatory mechanism that responds similarly to the CMNGE shift in the neonate. Hence, it is conceivable that control of mitochondrial biogenesis is the main player that steers the mitochondrial–metabolic transition from fetus to neonate in mammals and in chicken.
Hypoxia and locomotion: assessment of altered pathways that likely require the upregulation of CMNGE after birth
Mammalian embryos develop in utero in hypoxic conditions (Webster and Abela 2007). Previous work revealed that animal exposure to hypoxia reduces mitochondrial metabolism and allows cardiomyocyte regeneration in the neonate and in the adult mouse heart (Nakada et al. 2017). Thus, we reasoned, that upregulation of CMNGE after birth mechanistically associates with the transition to an oxygen-rich environment and is important for the transition of embryonic to adult tissue. Therefore, as a first step, we tested for the expression of the top genes that are known to be upregulated in hypoxia in humans during development. Our results revealed a reduction in the expression of most of the genes that positively respond to hypoxia across tissues and species (Fig. 5; Supplemental Fig. S41). We also noticed that humans display a stronger reduction in the brain and liver compared with the heart and kidney.
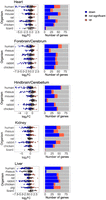
Hypoxia response gene expression is generally reduced after birth. (Left) Box plots representing the log2FC of hypoxia genes. The x-axis indicates log2FC values; y-axis, organisms. Color bar: blue dots indicate significant downregulation (log2FC < −0.2, P-value < 0.05); red dots, significant upregulation (log2FC > 0.2, P-value < 0.05); and black dots, nonsignificant. The dashed vertical red line corresponds to the value of zero log2FC on the x-axis. (Right) Bar plots representing the number of significant (upregulated in red, downregulated in blue) or insignificant genes (in gray). The x-axis indicates number of genes; y-axis, organisms tested.
Because the transition to the neonate is associated with an exit from a confined space (the womb, egg) to the external environment, we hypothesized that the expression of genes involved in the regulation of locomotion might be altered. While analyzing genes related to locomotion, we did not find a consistent overall change in expression (Supplemental Fig. S42). Because the forebrain harbors most neurons that direct locomotion (Ferreira-Pinto et al. 2018), our findings suggest that the upregulation of CMNGE in the forebrain may contribute to the emergence of locomotion. As the activity of proteins involved in locomotion is logically related to skeletal muscle, it would be of interest to study the expression of this pathway in muscle after birth when such data become available.
The lizard A. carolinensis did not show any consistent pattern in these pathways when comparing embryo to adult samples (Supplemental Figs. S33, S42–S44).
Together, the transition of the fetus from hypoxic conditions to breathing atmospheric oxygen might influence the OXPHOS gene expression after birth, whereas a shift in the mode of locomotion might not influence the OXPHOS gene expression in these tissues and organisms.
Elevation of mitonuclear gene expression before hatching associated with the initiation of spontaneous movement in Xenopus tropicalis and in Drosophila melanogaster
Although mammals and chicken displayed elevated CMNGE after birth, they did not show any clear CMNGE alteration before birth. We therefore asked whether putative changes in CMNGE during prenatal stages were masked by the major impact of postnatal samples. To address this possibility, we analyzed the same species, focusing only on prenatal samples before birth/hatching. This analysis did not reveal any consistent alteration in CMNGE among the tested species (mammals and chicken) during prenatal development (Supplemental Fig. S45). We next analyzed bulk RNA samples collected during embryonic and fetal development from two nonmammalian vertebrates—Xenopus tropicalis (tropical frog, with two experimental repeats) (Mitros et al. 2019) and Danio rerio (zebrafish) (Levin et al. 2016), as well as from two invertebrates—Drosophila melanogaster and C. elegans (whole organism) (Levin et al. 2016). X. tropicalis samples (whole organism) were collected from embryo cleavage stages until the tailbud free-swimming stage (Mitros et al. 2019), whereas D. rerio, D. melanogaster, and C. elegans were sampled from the cleavage stage until hatching (but not after). First, while analyzing mtDNA and nuclear DNA-encoded OXPHOS gene expression, we found that, unlike mammals and chicken, CMNGE elevation occurred in X. tropicalis before hatching (Fig. 6A; Supplemental Figs. S46–S48). In replicate one, both mtDNA and nuclear OXPHOS genes rose concurrently in timing and rate during developmental stages 24–26, whereas in replicate two, elevation occurred at stage 28 and aligned in timing, although with a distinct differential rate of elevated expression (Supplemental Figs. S47, S48). This early CMNGE coincided with initial motor neuron activity and spontaneous movement, respectively.
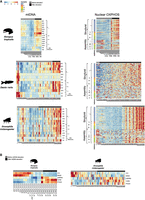
Mitonuclear gene expression is elevated during prenatal development. (A) Heatmaps of mtDNA and nuclear DNA–encoded OXPHOS gene expression from X. tropicalis, C. elegans, D. melanogaster, and D. rerio. The color bar indicates the scaled expression. Red scale indicates elevated expression; blue scale, reduced expression. The x-axis indicates prenatal samples; y-axis, mtDNA/OXPHOS genes. Notice division into OXPHOS complexes and into structural and assembly genes of the OXPHOS. (B) Heatmap representing log2FC values of regulatory factors and genes related to mitochondrial biogenesis. The color bar indicates the log2FC. The x-axis indicates organisms; y-axis, genes. Only log2FC > 0.2 and log2FC < −0.02 are shown.
In D. melanogaster, elevated mtDNA genes expression was observed at 585 min postfertilization (mpf), whereas elevated expression of nuclear DNA-encoded structural OXPHOS genes was observed shortly later, 645 mpf (Fig. 6A; Supplemental Figs. S49, S50); the elevated expression of both genomes preceded the onset of muscle twitching (840 mpf; i.e., early movement) (Halpern et al. 1991; Crisp et al. 2008).
For C. elegans, mtDNA genes expression was elevated at 60 mpf (Supplemental Figs. S46, S51, S52); however, only a small subset of nuclear DNA-encoded OXPHOS structural genes showed transient upregulation at this point, which was not sustained throughout the remainder of prenatal development, suggesting limited coordination in early stages.
In D. rerio (zebrafish), mtDNA genes reached peak of expression at 3520 mpf in the window w = 4 (Fig. 6A; Supplemental Figs. S53, S54). Conversely, nuclear DNA-encoded OXPHOS genes peaked earlier at 280 mpf. Additionally, analyses across varied window sizes (from w = 4 to w = 20) indicated different time frames for mtDNA expression peaks, with the highest FCs shifting between 2120 and 3520 mpf. This variability suggests biphasic mtDNA expression elevation that may be independent of nuclear OXPHOS genes, implying more complex regulatory dynamics in this species during prenatal development.
As mentioned above, our analysis suggests that in X. tropicalis and in D. melanogaster the timing of elevated CMNGE is associated with the beginning of embryonic movement, thus tempting us to speculate a causal connection. However, our analysis of the expression of genes related to the locomotion pathway in these two species (Supplemental Fig. S55) revealed that only a small subset of the locomotion pathway genes displayed altered expression in the same time frame of CMNGE. Therefore, the association of CMNGE with the locomotive phenotype during development (e.g., beginning of embryonic movements) cannot be easily explained by overall analysis of the locomotion pathway. This underlines the need for a future study that will focus on the involvement of mitochondrial function in the regulation of muscle movement, while taking advantage of single cells analysis.
We next asked whether the elevation of CMNGE toward hatching is unique to OXPHOS or could be found in other mitochondrial pathways of metabolism. Our analysis revealed that the expression of TCA cycle enzymes and the mitochondrial ribosome genes were also elevated toward hatching in X. tropicalis and in D. melanogaster (Supplemental Figs. S56–S59). Glycolysis genes did not show any altered expression pattern toward hatching.
Next, we asked whether CMNGE that was observed toward hatching in D. melanogaster and X. tropicalis is modulated by mitochondrial transcription factors, RNA binding factors, or biogenesis regulatory factors (Fig. 6B; Supplemental Fig. S46). The results highlighted three candidate regulatory genes that were shared between D. melanogaster and X. tropicalis (in two independent data sets): mitochondrial translation elongation factor (TUFM), PTCD3 (binds mtDNA-encoded rRNAs and regulator of mitochondrial translation), and LRPPRC (regulator of mitochondrial RNA stability). YY1, a known regulator of OXPHOS nuclear gene expression (Nandi et al. 2020), showed reduced expression in D. melanogaster and X. tropicalis, thus excluding it from positive involvement in regulating the elevated CMNGE during development in these species. An additional gene that displayed reduced expression in these organisms is SIN3A, a transcriptional repressor protein known to regulate mitochondrial components. Consistent with this finding, a previous study showed that loss of SIN3 in D. melanogaster cultured cells resulted in upregulation of both nuclear DNA–encoded and mtDNA-encoded genes (Barnes et al. 2010). Hence, D. melanogaster and X. tropicalis likely share a mechanism controlling CMNGE during prenatal development.
Discussion
In the current study, we assessed the cross talk between prenatal development and regulation of mitochondrial function at the RNA level during metazoan evolution across multiple tissues (Fig. 7). Our analysis showed a concomitant increase of both mtDNA and nuclear DNA–encoded OXPHOS gene expression either immediately or shortly after birth (mammals) or after hatching (G. gallus and A. carolinensis). It was previously shown that the transition from the fetus to the neonate in the mouse heart requires a postnatal metabolic switch to OXPHOS (Talman et al. 2018; Zhao et al. 2019; Secco and Giacca 2023), as well as profound alterations of mitochondrial protein functions in the early postnatal period (Talman et al. 2018). Our findings argue for a general, cross-tissue, and evolutionarily conserved elevation in mitochondrial function after birth; our results imply that the transition of the embryo to the neonate most probably relies on CMNGE. While considering OXPHOS, most of the elevated CMNGE was observed in structural OXPHOS subunits rather than in the assembly genes. This suggests that CMNGE involves the former, implying that OXPHOS assembly genes are regulated differently from the structural OXPHOS genes during development. Second, along with the elevated mitonuclear gene expression after birth, we observed elevated expression of major mitochondrial metabolic pathways, such as the mitochondrial ribosome and TCA cycle enzymes. Moreover, the mtDNA copy number was reported to have significantly increased in postnatal human liver and muscle, supporting a general increase in mitochondrial mass after birth (Pejznochova et al. 2010). As we did not observe consistent change in the expression of mtDNA replication factors after birth (Supplemental Table S1), the reported change in mtDNA copy number might only reflect change in mitochondrial mass rather than mtDNA replication. Finally, we noticed that the CMNGE alteration after birth/hatching was evident in somatic tissues but less in the reproductive organs (testis and ovary), apart from certain organisms (Supplemental Fig. S7). Because reproductive organs become more active upon puberty, it would be of interest to assess mitochondrial gene expression in reproductive organs before and after puberty. Additionally, because mitonuclear gene expression is likely coordinated also at the level of single cells (Medini et al. 2021a), it would be of interest to assess the mitonuclear gene expression in different cell types and to perform scRNA-seq analysis before and after birth from different tissues, hopefully from a variety of organisms. Finally, it would also be of interest to assess in future experiments whether elevated CMNGE after birth is also reflected at the protein level, especially in light of the claim for a strong impact of mitochondrial translation on the coregulation of the two genomes (McShane et al. 2024). Taken together, our results strongly support a regulatory basis underlying the coordinated response of mitochondrial and nuclear OXPHOS gene expression at the RNA level to the transition from fetal to neonatal life. Furthermore, although the association between the elevated gene expression in both genomes during development is correlative, its conservation across species and prevalence in different tissues strongly suggests a mechanistic basis for coordination.
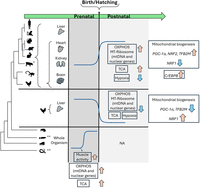
The landscape of CMNGE during development across metazoan evolution. OXPHOS, mitochondrial ribosome, and TCA genes show elevated expression right after birth in mammals chicken (after hatching, apart from the liver), and in the lizard across tissues. This pattern is associated with elevated expression of regulators of mitochondrial biogenesis and with transition from hypoxia to normoxia. The frog and Drosophila show elevated CMNGE in association with the start of locomotion in the embryos. Blue arrows indicate downregulation; orange arrows, upregulation; and gray areas, elevated or reduced mitonuclear gene expression. (*) S. scroffa (pig) samples were available only from the liver. (**) D. rerio (zebrafish) and C. elegans did not show clear CMNGE.
One clear exception from the coordinated upregulation is the chicken liver: Unlike the elevated CMNGE in mammalian liver samples, the chicken liver showed reduced CMNGE. After hatching, the chick's main source of energy is derived from the conversion of feed-based carbohydrates into fatty acids, which mainly occur in the liver, as in humans (Noble and Cocchi 1990; Hicks et al. 2022). However, unlike humans and other mammals that harbor adipocytes in a variety of body sites, much of the fat of the chick is stored in the liver (Leveille 1969). It is thus conceivable that the hepatic molecular metabolic response to posthatching in the chicken involves an increase in lipolysis-related gene expression (Latour et al. 1995). This is different from the mammalian neonatal liver, which performs uptake of fatty acids from maternal milk, esterification of fatty acids into triglycerides, and storage in lipid droplets. These differences may impact the usage and underlying regulatory difference of CMNGE in the chick versus neonatal liver upon birth/hatching. Regardless, mitonuclear gene expression is coordinately increased (mammals) or reduced (chicken), again supporting an underlying regulatory mechanism.
Although OXPHOS genes are upregulated in the A. carolinensis heart, this pattern was not consistently observed in other mitochondrial pathways. This suggests a species-specific regulation of mitochondrial gene expression in lizards, which calls for further exploration of metabolic gene expression across the development of other additional lizard species. Once gene expression data sets become publicly available from several lizard species, such study may shed light on the dynamic of mitochondrial gene expression and on the underlying regulation of metabolism, across vertebrates.
To identify the best candidate factors to regulate the altered CMNGE after birth/hatching, we screened a compendium of known regulators of transcription, post-transcription, and mitochondrial biogenesis that displayed expression changes after birth across tissues and across evolution. Our findings highlighted an elevation of PPARGC1A expression, a major regulator of mitochondrial biogenesis (see more below), right after birth in many tissues and most organisms (apart from the human heart). In contrast, the chicken liver displayed a decrease in PPARGC1A expression, the very same pattern observed for mitonuclear gene expression. With this in mind, we also found that the expression pattern of NRF2, a known transcriptional regulator of nuclear DNA–encoded OXPHOS genes, which regulates PPARGC1A transcription and numerous respiratory chain genes, including cytochrome c oxidase (COX) components (Scarpulla 2002; Gureev et al. 2019), also followed the mitonuclear gene expression pattern across tissues and tested organisms. Whereas NRF2 expression was elevated in the heart of most tested organisms, PPARGC1A expression was not, suggesting that the upregulation of NRF2 in the heart after birth might be regulated by other factors. This interpretation should be tested in the future.
PPARGC1A acts as a transcriptional activator via protein–protein interactions with DNA-bound transcription factors (Tavares et al. 2020). Previous study showed that impaired PPARGC1A in mouse hearts led to reduced growth, arrested mitochondrial development late in gestation, and high lethality shortly after birth (Lai et al. 2008). As current data support the limitation of PPARGC1A activity to the nucleus, it is plausible that its involvement in regulation of mitonuclear gene expression occurs via regulating the regulators of both mitochondrial and nuclear OXPHOS genes. This thought is partially supported by our observed elevated expression of the mitochondrial RNA polymerase, POLRMT, in certain tissues and species; POLRMT is indirectly regulated by PPARGC1A (Wu et al. 1999; Gureev et al. 2019). The expression of TFB2M, a mtDNA transcription initiation factor that interacts with POLRMT (Falkenberg et al. 2002), was elevated after birth/hatching, and its transcription is regulated by NRF2 (Gleyzer et al. 2005). In addition to PPARGC1A, PPARGC1B (PGC-1beta), a known transcriptional regulator of mitochondrial functions (Shao et al. 2010), was significantly upregulated across organs and species. These pieces of evidence suggest that the modulators of mitochondrial biogenesis form strong candidates to regulate CMNGE during the transition to postnatal life.
Nevertheless, this interpretation is limited by the following: Although PPARGC1A is known to increase the expression of both NRF1 and NRF2 mRNAs (Wu et al. 1999; Gureev et al. 2019; Popov 2020), we found that the expression of NRF1 was decreased after birth across tissues and organisms. Therefore, only some regulators of mitochondrial biogenesis are candidates to associate with the regulation of CMNGE after birth.
The expression of CEBPB was consistently upregulated after birth in most tissues and organisms tested, apart from the chicken liver. Unlike PPARGC1A and PPARGC1B, CEBPB has been in vivo localized both to the nucleus and to the mitochondria in cells, thus serving as an attractive candidate to directly participate in CMNGE. This interpretation awaits support from future silencing and overexpression experiments of CEBPB in cells, which will enable direct assessment of the impact of CEBPB on CMNGE.
Because CMNGE after birth and hatching is common to all mammals, the chicken, and the lizard, we reasoned that there should be a common condition in the transition to the neonate, or neonatal phenotype, that probably requires such a sharp and conserved shift in mitochondrial function. The first condition to be considered is the transition from hypoxia to breathing atmospheric oxygen. With this in mind, it is important to mention that hypoxia suppressed the activity of PPARGC1A and reduced its mRNA levels in cell lines (LaGory et al. 2015). Furthermore, hypoxia-inducible factor (HIF) is essential for mediating PPARGC1A suppression in hypoxia (LaGory et al. 2015), which is consistent with upregulation of PPARGC1A upon birth. However, this explanation is likely tissue specific in some organisms, as we found a tissue-dependent response of hypoxia regulatory factors after birth: Although, as expected, most of the hypoxia genes were downregulated in the human brain, liver, and kidney, the human heart displayed a weaker effect in the expression of hypoxia-responsive genes after birth. As the heart experiences a sudden elevation in oxygen levels after birth, one would expect a hypoxic response in the heart after birth owing to oxidative stress (Secco and Giacca 2023) and in response to hypoxia, an elevated mitochondrial energy production in cardiac cells (Essop 2007). Therefore, despite the weaker reduction in the hypoxia pathway in the human heart, our findings suggest that hypoxia may explain the need for elevated CMNGE after birth. With this in mind, it is conceivable that species-specific regulatory mechanisms are also important for regulating mitochondrial function during development, especially while considering the differences in the physiological requirements of different organisms. As more gene expression data sets become available for additional species, future analyses will allow us to better delineate these unique patterns and their potential biological significance.
Although we did not identify a clearly consistent change in CMNGE during prenatal development of mammals and chicken, we asked whether such changes could be identified in more distant metazoans, including vertebrates and invertebrates. In X. tropicalis, synchronized mtDNA and nuclear OXPHOS gene elevated expression aligns with the onset of motor activity. Similarly, in D. melanogaster, the coregulation between these gene sets precedes muscle twitches (Crisp et al. 2008), suggesting an associated need for muscle movement with the regulation of energy production in both the fly and the frog. Nevertheless, this interpretation is limited by the ambiguous mitonuclear gene expression coordination in D. rerio and in C. elegans. Thus, the connection between CMNGE and the emergence of movement during development emphasizes the need to study mitonuclear gene expression in a larger set of metazoans. Association between the upregulation of CMNGE and the initiation of locomotion is especially attractive because many mitochondrial disorders display impaired movement phenotypes (Tranchant and Anheim 2016; Ticci et al. 2021), further emphasizing the importance of the study of CMNGE in such disorders, an interpretation that should be tested in future experiments.
In summary, our study revealed coordination of gene expression between the mitochondria and the nucleus in the transition from fetal to neonatal life and, for the first time, identified its conservation in vertebrates. Such conservation implies similar selective constraints across evolution and, hence, functional importance. Unexpectedly, our analysis revealed elevated CMNGE, which reflected a conserved metabolic shift postbirth (mammals) or hatching (bird and lizard). Key regulatory factors, for example, PPARGC1A, PPARGC1B, NRF2, TFB2M, and CEBPB, emerge as potential regulators of such CMNGE shifts. The identification of these factors as candidates to regulate CMNGE across species forms a testable hypothesis, which can be experimentally addressed while manipulating the expression of these factors in cells and organisms. Second, we found that elevated CMNGE preceded the initiation of locomotion in the frog and Drosophila. Our findings pave the path toward future identification of the underlying mechanism of CMNGE during development, as well as in particular tissues, during the course of evolution.
Methods
Data availability
Data source details and number of samples per analyzed data set are indicated in Table 1.
For Data set I, bulk RNA-seq FASTQ files were downloaded from ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) with the following accession codes: E-MTAB-6769 (G. gallus), E-MTAB-6782 (O. cuniculus) E-MTAB-6798 (M. musculus), E-MTAB-6811 (R. norvegicus), E-MTAB-6813 (M. mulatta), and E-MTAB-6814 (H. sapiens) (Cardoso-Moreira et al. 2019). For data analysis, see the RNA-seq Analyses section. Data set I included 1346 RNA-seq libraries, encompassing embryo, fetal, and neonatal development in seven organs, nine to 23 developmental stages, depending on the analyzed organism. Samples that were collected during the same developmental week were considered biological replicates. For instance, samples that were generated from Carnegie stages 13 and 14 in a given organism were considered as replicates (4 weeks postconception [wpc]), as previously reported (Cardoso-Moreira et al. 2019). Accordingly, the data included one to four replicates per stage. Briefly, human prenatal samples were available at 4 wpc and for each week until 20 wpc (except for 14, 15, and 17 wpc). It is worth noting that there were no human samples available between 20 and 38 wpc. Postnatal human samples included “infants” (6–9 months old), “toddlers” (2–4 years old), “school” (7–9 years old), “teenagers” (13–19 years old), and adults from each decade until 63 years of age. Human ovary samples were collected only from prenatal samples (until 18 wpc), and therefore did not present insight into the transition to the neonate. Finally, the human kidney was sampled during development up until 8 years of age (hereby termed “school”). Samples from rhesus monkey (M. mulatta) were collected while taking into account that gestation lasts ∼167 days. These samples were collected starting from fetal stage e93 in addition to four subsequent stages prior to birth (until e130). Notice that samples for the rhesus monkey were not available for e130 and e160. When considering the mouse (M. musculus), samples were collected from e10.5 and during subsequent days, each day, until birth (i.e., until e18.5). Postnatal mouse samples included P0, P3, P14, P28, and P63. For the rabbit, O. cuniculus (outbred New Zealand breed) samples were collected starting from e12 and the subsequent 11 time points up until (including) e27, while taking into account that gestation length is 29–32 days. Postnatal rabbit samples included P0, P14, and P84 and between P186 and P548. For the rat, R. norvegicus (outbred strain Holtzman SD) samples were collected starting from e11 and at subsequent daily time points until birth (until e20). Postnatal samples include P0, P3, P7, P14, P42, and P112. For the chicken, G. gallus (red junglefowl) egg incubation lasts ∼21 days. Thus, collection time series started at e10 and at subsequent three additional stages until e17. Postnatal samples include P0, P7, P35, P70, and P155.
Data set II included RNA-seq experimental data collected during S. scroffa (pig) liver development. The data comprised a normalized count matrix of processed transcripts per million (TPM), which was downloaded from the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) accession number GSE176387. We analyzed five developmental stages from porcine liver: E38, E80, the day of birth (0d), weaning at 28 days, sexual maturity at 110 days, with six replicates in each stage.
For Data set III, the fragments per kilobase million normalized count matrix was downloaded from GEO accession number GSE121019. We analyzed three developmental stages available for chicken liver: e13 and postnatal stages at 5 weeks and 42 weeks of age, with each having three replicates.
For Data set IV, the raw read count matrix was downloaded from GEO accession number GSE58455. We analyzed seven independently collected e13.5 samples and 12 adult samples (six at 6 weeks of age, six at 16 weeks of age) from mouse heart.
For Data set V (A. carolinensis), we took into account that A. carolinensis development typically lasts 30–33 days and includes 19 stages (Sanger et al. 2008). Raw bulk RNA-seq FASTQ files were downloaded from GEO accession number GSE97367. We analyzed 22 embryonic samples (from stages 15–16) and 30 adult samples from four organs (brain, heart, kidney, liver) and 26 whole embryonic samples.
For Data set VI (X. tropicalis), raw data FASTQ files were downloaded from GEO accession number GSE37452. The samples included two clutches: (1) from two cells to stage 45, and (2) from stage 9 to stage 42. These stages span from the beginning of cleavage to the tailbud free swimming stage. In all cases, the whole organism was sampled.
For Data set VII, raw read count matrices for the zebrafish (D. rerio), D. melanogaster, and C. elegans were downloaded from GEO accession number GSE70185 (Levin et al. 2016). Time series included samples from the cleavage stage up to the time point prior to hatching.
RNA-seq analyses: mapping, read count and control for nuclear mitochondrial sequences
For Data sets I, V, and VI, FASTQ files were mapped, and read counts were generated for further analyses (see below). For Data sets II, III, IV, and VII, we downloaded normalized postmapping read count files for subsequent analyses.
Specifically, for further analyses of Data sets I, V, and VI, sequenced reads were trimmed using Trim Galore! (version 0.4.5; https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Prior to read count analyses, we controlled for possible noise originating from nuclear mitochondrial fragments (NUMTs), for example, mtDNA sequence fragments that were transferred from the mitochondria to the nucleus during the course of time (Mishmar et al. 2004). NUMTs potentially pose an obstacle to mtDNA gene expression assessment, as a subset of RNA reads, particularly those that are relatively recent (Mishmar et al. 2004), might be more similar to the active mtDNA and sequencing reads from mtDNA genes; such fragments might be erroneously filtered out while applying the unique mapping protocol. To overcome such a problem, reads were first mapped solely against the mtDNA genome using BWA using the aln parameter (BWA-backtrack algorithm) (Li and Durbin 2009); then, the reads that did not map to the mtDNA were uniquely mapped against the entire reference genome of the relevant organisms using STAR (version 2.5.3), while employing default parameters and the parameter [—outFilterMultimapNmax 1] (Dobin et al. 2013). Expression levels of all genes were counted using HTSeq-count v0.11.2 (Anders et al. 2015) using default parameters while employing the [-f bam] parameters. High-quality samples were used as listed in the original study (Cardoso-Moreira et al. 2019).
Libraries with nonzero read counts of mtDNA genes were used for subsequent analysis to avoid technical noise. Reference genomes that were used are listed in Supplemental Table S2.
Quantification of gene expression and statistical analysis
Read count data matrices were normalized using DESeq2 normalization method available from the DESeq2 R package (R Core Team 2013; Love et al. 2014). For Data set I, we used a median of two to four replicates for each stage for statistics. Heatmaps were created using the pheatmap R package (https://github.com/raivokolde/pheatmap; R Core Team 2013). To assess mtDNA gene expression across developmental stages, a sliding window algorithm was employed with varying window sizes: w = 2, w = 4, and w = 6. FC values for each mtDNA gene were computed over specific time point intervals. For w = 2, the FC was calculated between consecutive time points (e.g., from time point 1 to time point 2, followed by time point 2 to time point 3, etc.). For w = 4, the FC was determined by the median of the first two time points and the median of the subsequent two time points to capture broader temporal shifts. Similarly, for w = 6, the FC was calculated between the median of three subsequent time points to the following ones, providing a wider context for temporal changes. The smaller window size allows for more sensitive detection of rapid changes in expression (with higher signal-to-noise), whereas the larger window size emphasizes longer-term trends.
During data analysis, the average FC for mtDNA genes was computed for each time point to identify those exhibiting the highest mean FC values, thereby highlighting significant expression changes.
Window sizes w = 4 and w = 6 showed similar trends; however, given the limited number of time points in G. gallus (usually n = 9), using w = 6 would reduce resolution and potentially obscure key shifts in gene expression. Window w = 2, on the other hand, introduced more noise. Therefore, for the sake of consistency, w = 4 was chosen for the analysis across species, offering sufficient resolution in all tested organisms.
In nonmammalian species, including X. tropicalis, D. melanogaster, D. rerio, and C. elegans, the sliding window range was extended to incorporate sizes from w = 2 to w = 20, utilizing only even-numbered windows to ensure an equal number of samples in each iteration. This strategy accommodated variations in the number of sampled developmental stages across these species compared to mammals and birds.
The overall “topography” of mtDNA gene expression graphs remained consistent across the different window sizes used, suggesting that the chosen window size did not significantly impact the analysis outcomes. Again, window w = 4 was chosen to ensure a consistent analysis across species. To assess whether the change in FC was coordinated with nuclear DNA–encoded OXPHOS gene expression, we conducted the same analysis to this set of genes.
Supplemental Figures S1–S6, S13–S18, S21–S26, and S47–S54 present the results of the analysis across all window sizes, enabling a detailed and accurate representation of mtDNA, nuclear OXPHOS, and structural OXPHOS gene expression patterns over time, respectively, while maintaining consistency in the analysis across species.
Differential expression of mitochondrial genes in a given time point was calculated (DESeq2) separately for each data set,
while considering samples from the previous and subsequent time points. Correction for multiple testing for each specified
set of genes was performed using an adjusted P-value of 0.05. Box plot and bar plot graphs were generated using “ggplot2” (Wickham 2016) and RColorBrewer R packages (R Core Team 2013). Tables were generated using the R packages openxlsx, tidyr, and reshape2 (R Core Team 2013; Zhang 2016). The level of coordination of mitonuclear genes was calculated as follows: the number of significantly upregulated OXPHOS
genes (Xup) minus the number of significantly downregulated OXPHOS genes (Xdown), divided by the number of total available OXPHOS genes, as in the following formula: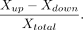
Expression of genes was considered significantly altered if their adjusted P-value was smaller than 0.05 and the absolute value of log2FC was greater than 0.2.
GO terms used in the analysis
The following Gene Ontology (GO) Terms were used to download and perform differential expression analysis: genes belonging to “TCA” (GO:0006099), “glycolytic process” (GO:0006096), ROS formation (GO:1903409), “locomotion” (GO:0040011), and “positive regulation of cellular response to hypoxia” (GO:1900039). We assessed the expression of candidate mtDNA regulatory factors of transcription, regulatory factors of mtDNA replication, and nuclear DNA-encoded factors with known mitochondrial RNA-binding activity (Wolf and Mootha 2014; Cohen et al. 2016), in addition to RNA- and DNA-binding proteins that were recently identified in human mitochondria (Ardail et al. 1993; Fernández-Vizarra et al. 2008; She et al. 2011; Blumberg et al. 2014; Lambertini et al. 2015; Chatterjee et al. 2016). The hypoxia gene set was downloaded from a previous study (Bono and Hirota 2020). Accordingly, the top 100 responding genes to hypoxia with the highest score in humans were used for the analyses.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was funded by grants from the Israeli Science Foundation (ISF 404/21) and from the life sciences division of the US Army Research Office (grant no. 80581-BB) awarded to D.M. We thank the Myles Thaler Genetics and Genomics Research cathedra, BGU, for supporting this research. We thank Ben-Gurion University for awarding H.M. the Negev scholarship for excellent PhD students and a short-term postdoc training grant.
Author contributions: H.M. performed all analyses and participated in all stages of writing the paper. D.M. conceived the idea of the study, supervised the analyses, and wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279700.124.
- Received June 25, 2024.
- Accepted February 4, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








