
Probing the eukaryotic microbes of ruminants with a deep-learning classifier and comprehensive protein databases
- 1Department of Animal Sciences, The Ohio State University, Columbus, Ohio 43210, USA;
- 2Center of Microbiome Science, The Ohio State University, Columbus, Ohio 43210, USA;
- 3Faculty of Chemistry, Biotechnology and Food Science, Norwegian University of Life Sciences, Ås NO-7491, Norway;
- 4Faculty of Biosciences, Norwegian University of Life Sciences, Ås NO-7491, Norway;
- 5Centre for Microbiome Research, School of Biomedical Sciences, Queensland University of Technology (QUT), Translational Research Institute, Woolloongabba 4102, Queensland, Australia
Abstract
Metagenomics, particularly genome-resolved metagenomics, have significantly deepened our understanding of microbes, illuminating their taxonomic and functional diversity and roles in ecology, physiology, and evolution. However, eukaryotic populations within various microbiomes, including those in the mammalian gastrointestinal (GI) tract, remain relatively underexplored in metagenomic studies owing to the lack of comprehensive reference genome databases and robust bioinformatic tools. The GI tract of ruminants, particularly the rumen, contains a high eukaryotic biomass but a relatively low diversity of ciliates and fungi, which significantly impacts feed digestion, methane emissions, and rumen microbial ecology. In the present study, we developed GutEuk, a bioinformatics tool that improves upon the currently available Tiara and EukRep in accurately identifying eukaryotic sequences from metagenomes. GutEuk is optimized for high precision across different sequence lengths. It can also distinguish fungal and protozoal sequences, further elucidating their unique ecological, physiological, and nutritional impacts. GutEuk was shown to facilitate comprehensive analyses of protozoa and fungi within more than 1000 rumen metagenomes, revealing a greater genomic diversity among protozoa than previously documented. We further curated several ruminant eukaryotic protein databases, significantly enhancing our ability to distinguish the functional roles of ruminant fungi and protozoa from those of prokaryotes. Overall, the newly developed package GutEuk and its associated databases create new opportunities for the in-depth study of GI tract eukaryotes.
The complex and multikingdom microbial ecosystem unique to the rumen supplies ruminants with up to 70% of their energy requirement through the digestion and fermentation of plant materials (Bergman 1990). However, this process also results in methane emissions, which account for ∼14% of the anthropogenic greenhouse gas emissions (Gerber et al. 2013). The rumen microbes also convert dietary nitrogen, including protein and nonprotein nitrogen, into high-quality microbial protein, contributing up to 80% of the protein metabolized in the small intestine (Wallace et al. 1997). Although the rumen microbiome is primarily linked to ruminant nutrition and production, the lower gastrointestinal (GI) microbiome is more associated with animal health and host physiology (Huws et al. 2018). The intricate and interdependent multikingdom ecosystem comprises anaerobic bacteria, archaea, fungi, protozoa, and viruses, each occupying a unique niche. Although the prokaryotic communities of the rumen and intestines have been extensively investigated through multiomic technologies, analysis of the eukaryotic communities still primarily relies on amplicon sequencing and analysis of a phylogenetic marker (metataxonomics), mainly the 18S rRNA gene for protozoa and internal transcribed spacer (ITS) for fungi, which provide limited taxonomic resolution and functional information. The lack of a comprehensive genome database for rumen protozoa and fungi further limits omics analyses of these eukaryotes. Our understanding of the functional importance of the eukaryotic communities within the rumen is still primarily based on culture-based and genomic studies of a small number of species.
Unlike the human gut, in which eukaryotes account for <1% of the gut microbiome (Huffnagle and Noverr 2013), fungi and protozoa constitute up to 20% and 50% of the rumen microbiome biomass, respectively (Edwards et al. 2017; Huws et al. 2018; Andersen et al. 2023). Rumen fungi are specialized fiber degraders capable of producing diverse carbohydrate-active enzymes (CAZymes), especially those hydrolyzing recalcitrant plant cell wall materials (Hagen et al. 2021), complementing the CAZymes repertoire of rumen bacteria (Hagen et al. 2021; Peng et al. 2021). In addition, gut fungi produce diverse secondary metabolites with potential antimicrobial and therapeutic properties (Swift et al. 2021). Except for Pecoramyces sp. F1, which was isolated from the rumen (Jin et al. 2011), all available genomes of gut fungi from ruminants, Anaeromyces robustus, Caecomyces churrovis, Neocallimastix californiae, Neocallimastix lanati, and Pecoramyces ruminantium, were obtained exclusively from fecal samples, therefore also representing hindgut fungi. Although the rumen and the hindgut share some of their fungi, they have distinctive fungal communities (Fliegerova et al. 2015), and it remains unknown whether the fungal enzymatic repertoire differs between the rumen and the hindgut. Although the contribution of fungi to fiber degradation in the rumen is well recognized, the functions of rumen protozoa are intricate and multifaceted, encompassing productive roles such as fiber degradation and rumen pH stabilization, alongside counterproductive roles such as promoting methane emissions and wasteful intraruminal recycling of microbial protein (Firkins et al. 2020; Yu et al. 2024). Indeed, rumen protozoa exhibit a linear relationship with methane emissions (Guyader et al. 2014). Because of their negative associations with feed efficiency and methane emissions, a few studies have attempted to specifically inhibit the dominant species of Entodinium (Park 2019a,b). Alternatively, analyses of single-cell amplified genomes (SAGs) and subsequent proteomic analysis of rumen protozoa revealed an unexpectedly extensive repertoire of various CAZymes (Li et al. 2022; Andersen et al. 2023). Furthermore, recent studies have demonstrated an association between rumen protozoa and feed efficiency in beef cattle (Clemmons et al. 2021) and between intestinal protozoa and intestinal health in claves (Fu et al. 2023). Therefore, it is imperative to transcend beyond metataxonomic analysis of rumen protozoa, characterizing the genomes and enzymatic capacity of the eukaryotic fraction in the GI tract of ruminants. Moreover, a comprehensive protein database is needed to enable robust and unbiased analyses of protozoal gene/protein expression profiles in relation to key rumen functions and animal production, such as methane emissions and feed efficiency.
Single-cell genomics has proven valuable in unraveling the fundamental biological characteristics of protozoa (Li et al. 2022). However, it is not cost effective for ecological studies of protozoal communities. Metagenomics has revolutionized the investigation of various microbiomes, including the GI microbiomes of ruminants, but most studies focus on bacteria and archaea while ignoring fungi and protozoa. Two machine learning–based tools (Tiara and EukRep) are available to identify eukaryotic sequences directly from metagenomic assemblies (West et al. 2018; Karlicki et al. 2022; Pronk and Medema 2022). Studies using these tools have revealed the ecological importance of protists in aquatic environments (Duncan et al. 2022; Alexander et al. 2023; Kavagutti et al. 2023; Saraiva et al. 2023). However, it remains unclear whether the above tools can be used to analyze eukaryotes abundant in host-associated ecosystems like the rumen. Moreover, the diversity and functional repertoire of the ruminant GI tracts, especially the less-studied hindgut and ruminant species, remains limited. Therefore, our first objective was to benchmark the existing tools for classifying ruminant gut eukaryotes while creating a new tool (referred to as GutEuk) to identify and classify eukaryotic microbes (fungi and protozoa) in metagenomes derived from the gut of ruminants. Using GutEuk, we also aimed to reanalyze more than 1000 metagenomes and metatranscriptomes from ruminant GI tracts and to create protein databases for ruminant gut eukaryotes. This analysis included samples from the foregut (mostly rumen but also including reticulum, omasum, and abomasum) and hindgut (cecum, colon, and rectum) across various ruminant species (mostly domestic species including Bos taurus and Ovis aries, as well as wild species including Capreolus capreolus and Hydropotes inermis). Our last objective was to benchmark the newly established protein databases by improving metaproteomic analysis using data from a recent study (Andersen et al. 2023). Overall, GutEuk and the new protein databases it creates open up new opportunities in analyzing and reanalyzing host-associated microbiomes for eukaryotic microbes, especially those within the rumen.
Results
The new GutEuk improves identification of eukaryotic microbes from metagenomes
Tiara (Karlicki et al. 2022) and EukRep (West et al. 2018) are tools for identifying eukaryotic sequences from metagenomic assemblies based on k-mer frequency. They were developed based on a limited selection of eukaryotic nuclear genomes (73 for Tiara and 70 for EukRep), encompassing those of algae and animals. The lack of representation of gut-associated eukaryotic microbes likely undermines their utility in analyzing gut-derived metagenomes. In addition, k-mer frequency merely represents metrics of simplified genomic contexts, whereas improved classification can be achieved using a convolutional neural network (CNN) with original DNA sequences as inputs, as illustrated by a recent study (Hou et al. 2024). We tested Tiara and EukRep for their ability to identify gut eukaryotic sequences using a data set from 52 SAGs of rumen protozoa (see Methods) (Li et al. 2022). Tiara and EukRep correctly classify only 30,081 and 44,395 out of 57,253 eukaryotic contigs, achieving accuracies of 52.5% and 77.5%, respectively, which are much lower than their reported performance.
The aforementioned accuracies of Tiara and EukRep indicate that these two existing tools cannot achieve a generalizable performance for less-represented genomes of eukaryotes. We thus developed a specialized deep-learning classifier built upon diverse protozoal, fungal, and prokaryotic genomes to improve the classification of eukaryotic microbes in the rumen and gut. Leveraging thousands of prokaryotic, fungal, and protozoal genomes, we developed GutEuk, an ensembled deep-learning model combining a CNN (one-hot encoded nucleotide bases as inputs) and a feedforward neural network (FNN; k-mer frequencies as inputs) (Fig. 1A,B). GutEuk uses a two-stage classification with user-defined confidence levels at each stage: The first stage differentiates between prokaryotic and eukaryotic (fungal and protozoal) sequences, and the second stage further distinguishes fungal from protozoal sequences.
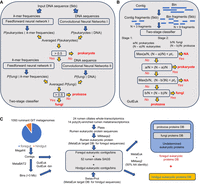
Illustrative schematic of the two-stage classification process used by GutEuk (A), the pipeline to classify individual contigs and bins from metagenomes (B), and the workflow to establish the ruminant foregut and hindgut eukaryotic protein databases (C).
We benchmarked the performance of GutEuk against EukRep and Tiara using independent data sets that were not used to train these tools at the contig level (see Methods). GutEuk outperformed Tiara and EukRep consistently at different contig lengths (Fig. 2A). GutEuk and Tiara exhibited comparable precision for eukaryotic contigs, whereas EukRep displayed a slightly lower precision, especially for contigs <20 kb. However, for prokaryotic contigs, GutEuk achieved a consistent precision of ∼99%, even for contigs <15 kb, whereas Tiara and EukRep achieved a precision of 95% and 91%, respectively, for contigs <10 kb. The performance of Tiara and EukRep was poorer than that of GutEuk for contigs between 3 and 5 kb. In terms of recall rate, all three tools performed similarly with prokaryotic contigs. However, for eukaryotic contigs, GutEuk achieved a recall rate of ∼99% for contigs <15 kb and still maintained a recall rate of ∼98% for longer contigs. In comparison, EukRep and Tiara recorded lower recall rates, with EukRep achieving at best 95% and dropping below 90% with contigs <10 kb, whereas Tiara achieved only 82% at best with contigs between 5 and 10 kb, with lower recall rates with longer contigs. Regarding computing time, GutEuk processed the test data set of 965,741 sequences (totaling 4.6 Gb) in 9.3 h using 20 threads. In comparison, Tiara and EukRep completed the same task with the same resources in 0.3 h and 2 h, respectively.
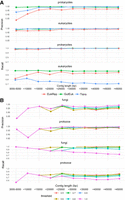
Benchmarking the performance of GutEuk, EukRep, and Tiara for contigs. (A) The performance of GutEuk, EukRep, and Tiara in differentiating prokaryotic and eukaryotic contigs of different lengths. EukRep and Tiara were used with their default settings. GutEuk was run at a pt1 of 0.5. (B) The performance of GutEuk in further differentiating fungal and protozoal contigs at different confidence levels (pt2) and contig lengths.
We evaluated GutEuk for its ability to further differentiate protozoal and fungal contigs (Fig. 2B). Because the maximum number of classifiable fragments is two for contigs <15 kb, the threshold used will not affect its performance (both fragments must provide the same prediction). For contigs >15 kb, we observed the expected increase in precision and decrease in recall as the confidence level (pt2) rose. Precision exceeded 90% even at a pt2 of 0.5 and reached 95% at a pt2 above 0.8. In terms of recall rate, GutEuk achieved >70% and >85% for protozoa and fungi, respectively, at a pt2 of 0.8. Therefore, pt2 can be adjusted to optimize GutEuk for precision and recall based on specific research objectives. Nevertheless, a balance between precision and recall was seen at a pt2 of 0.8, and the corresponding precision of >95% was considered conservative.
In addition to evaluating the performance of GutEuk in identifying microbial eukaryotic contigs, we further assessed whether it could achieve consistent results across genomes of diverse phylogenetic lineages, including aquatic fungi and protozoa included in the database (Fig. 3A–D). All three tools achieved >90% accuracy. However, GutEuk, leveraging an expanded training set and a more complex model, achieved >90% accuracy in classifying DNA from most eukaryotic microbes, especially the underrepresented rumen ciliate protozoa. The only eukaryotic genome whose fragments were identified with a <70% accuracy (∼45%) was that of Stentor coeruleus (accession ID GCA_001970955.1), a freshwater protozoan. In contrast, Tiara and EukRep each had 14 eukaryotic genomes classified with an accuracy <70%. These results indicate that at a pt1 of 0.5, GutEuk predicts eukaryotic microbial genomes/bins more accurately than Tiara and EukRep. More importantly, GutEuk can also further classify eukaryotic genomes as fungal or protozoal with high accuracy, except for a few fungi and protozoa not found in the mammalian GI tract. Specifically, GutEuk identified the following fungal genomes with <50% accuracy: Smittium mucronatum (GCA_001953115.1, an insect gut symbiont), Vavraia culicis (GCA_000192795.1, a microsporidian parasite), and Enterospora canceri (GCA_002087915.1, infecting mosquito and European shore crab). Similarly, protozoal genomes identified with <50% accuracy are not found in the GI tract either: Perkinsus marinus (GCA_000006405.1) and Perkinsus sp. BL_2016 (GCA_004369235.1), two parasitic flagellates inflecting mollusks, as well as S. coeruleus (GCA_001970955.1) and Lenisia limosa (GCA_001655205.1), two aquatic ciliates. Given the narrow phylogenetic range of gut eukaryotes, GutEuk is expected to perform well with gut metagenomic sequences (see the next section). A higher pt2 threshold (e.g., 0.8) can be used if a lower false-positive rate is required. Furthermore, GutEuk correctly identified 697 out of 708 (98.4%) newly sequenced human gut fungal genomes (Yan et al. 2024) as fungal (Supplemental Table S1).
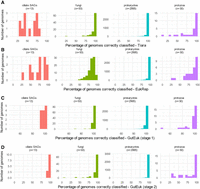
Benchmarking the performance of GutEuk, EukRep, and Tiara with genomic bins. The percentage of genomes being correctly classified as prokaryotic or eukaryotic by Tiara (A), EukRep (B), and GutEuk (C). The percentage of genomes being correctly classified as prokaryotic, protozoal, or fungal by GutEuk (D).
Diverse rumen protozoa are identified from underrepresented ruminant species
To date, 53 genomes of rumen ciliates have been sequenced, including 52 SAGs (Li et al. 2022) and one monospecies genome (Park et al. 2021). These genomes represent 19 species of rumen ciliates prevalent across various ruminant species. Using GutEuk, we systematically analyzed 15,000 bins (each exceeding 3 Mb) assembled from 1093 previously sequenced rumen metagenomes (see Methods) (Supplemental Table S2) for eukaryotic bins. These samples were collected from 13 different ruminant species, encompassing both domestic and wild species with varying feeding regimes. We identified hundreds of protozoal bins and one fungal bin. The scarcity of bins identified as fungal likely reflects their low abundance and low fungal DNA yield from rumen fluid samples because rumen fungi strongly adhere to digesta particles. We assessed the genome completeness of the identified eukaryotic bins based on the presence of lineage-resolved single-copy marker genes, finding that most bins were largely incomplete, consistent with findings from a recent study using EukRep for eukaryotic bin identification (Saraiva et al. 2023).
Despite the majority of the eukaryotic bins identified from the rumen metagenomes being incomplete, we identified 21 protozoal bins with >50% completeness and <10% contamination. We further conducted a phylogenetic analysis on this set of protozoal bins and the rumen protozoa SAGs (Fig. 4). Examination of the phylogenetic tree, constructed from 30 concatenated single-copy marker proteins, reveals that the newly discovered rumen protozoal bins cluster with the SAGs within the Vestibuliferida order. In contrast, all Entodiniomorphida SAGs form a distinct cluster, suggesting a potentially greater diversity of Vestibuliferida species yet to be uncovered. Notably, the phylogenetic tree constructed with the 30 single-copy marker proteins exhibited the same topology as the original phylogenetic tree of the SAGs, which was based on 113 single-copy marker proteins. This consistency demonstrates the robustness of our phylogenetic analysis. It is also noteworthy that some of the newly identified protozoal bins were derived from metagenomes of ruminant species, including Bison bison and Bos indicus, from which no protozoal genomes have been previously sequenced. Therefore, future protozoal research should expand to include less-studied ruminant species to facilitate a more comprehensive genomic representation of rumen eukaryotes.
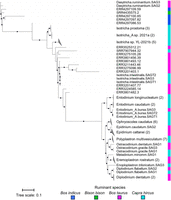
A phylogenomic tree of the identified rumen protozoal bins and 49 rumen protozoa single-cell-assembled genomes. All nodes with a >85% bootstrap support are indicated with dots in the branch.
Protein databases of ruminant gut eukaryotes and the repertoire of genes encoding glycoside hydrolases and peptidases
From the eukaryotic sequences (bins and contigs) identified from the gut metagenomes of various ruminants, predominantly cattle, sheep, and goats, we predicted and translated their protein sequences, subsequently constructing several protein databases (Fig. 1C). These gut eukaryotic microbial protein databases include one each for foregut protozoa (both newly identified and from the 52 SAGs), foregut fungi, and hindgut eukaryotes (Fig. 5A). The foregut protozoal database consisted of 25,498,129 proteins, which were clustered at 50% amino acid identity into 5,265,119 protein clusters, 54% of which are newly identified. Among these clusters, only 10% were annotated. The foregut fungal database is much smaller, containing 11,314 proteins, which formed 7293 protein clusters. Around half of the fungal protein clusters could be annotated. Notably, the proportion of the annotated hindgut eukaryotic protein clusters exceeded 92%. The markedly smaller proportion of annotated protozoan proteins relative to those of fungal and hindgut eukaryotes highlights the need for further genomic investigations into rumen protozoa. This need is also underscored by their complex functions and implications, such as predation on other rumen microbes, enzymatic breakdown of plant material, and symbiotic interactions with bacteria and archaea.
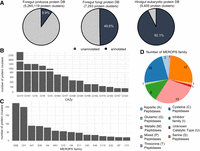
Diverse ruminant gut eukaryotic proteins identified in the databases. (A) The number of protein clusters identified in foregut protozoal, foregut fungal, and hindgut eukaryotic protein databases. (B) The most prevalent foregut protozoal glycoside hydrolases (GHs) identified. (C) The most prevalent foregut protozoal secretory (with identified signal peptides) peptidases and peptidase inhibitors identified. (D) The number of unique foregut protozoal secretory peptidases or peptidase inhibitors identified based on catalytic sites.
Previously, bacteria and fungi were credited for most of the cellulolytic activity within the rumen microbiome. However, a recent genomic study (Li et al. 2022) and a metaproteomic study (Andersen et al. 2023) have revealed a substantial proportion of genes encoding glycoside hydrolases (GHs), including cellulase and hemicellulases, within rumen protozoa (Andersen et al. 2023). In the present study, we annotated the foregut protozoal database for GHs. We found that amylases (e.g., GH13) and cellulases (e.g., GH5 and GH9) are among the most prevalent CAZymes identified (Fig. 5B), underscoring the significant fibrolytic and amylolytic capability of rumen protozoa.
Rumen protozoa are known for their ability to degrade protein, especially microbial protein. We thus also annotated the foregut protozoal database for peptidases. Among the peptidases identified, metallo, aspartic, cysteine, and serine peptidases were predominant, with M08, C01, A01, and S09 peptidases being among the most prevalent secretory peptidases (Fig. 5C). Prevotella, the most predominant genus of rumen bacteria with broad protease activity (Hartinger et al. 2018), exhibits 48 different families of secretory peptidases (Patra and Yu 2022). In comparison, 58 different secretory peptidase families were identified from the rumen protozoal proteins (Fig. 5D), highlighting their substantial proteolytic potential. Furthermore, the identification of ferredoxin hydrogenase of rumen protozoa (Supplemental Table S4) corroborates their ability to produce hydrogen.
Ruminant gut eukaryotic protein databases improve protein identification in proteomics
Recently, a metaproteomic study demonstrated the utility of protozoal SAGs and genomes as a reference search database in annotating rumen protozoal proteins and assessing the significance of rumen eukaryotes in rumen functions (Andersen et al. 2023). Given that reference search databases are a major bottleneck in metaproteomic studies of complex microbiomes, and considering the previously demonstrated high genomic and functional diversity of the rumen eukaryotes, we evaluated the potential of expanding protozoal genome databases with ruminant eukaryotic proteins identified with GutEuk to enhance protein detection from rumen metaproteomic data. Using the same pipeline and threshold as the original study (Andersen et al. 2023), we compared the number of protein groups identified in each sample using (1) the original genome database (consisting of one Entodinium caudatum genome and 18 SAGs of rumen protozoa) and (2) an expanded database combining the original genome database with the GutEuk-identified rumen eukaryotic proteins. The expanded database enabled substantially more protozoal protein groups to be identified in both cow and goat rumen samples, with an average increase of 64% for cow samples and 20% for goat samples (Fig. 6). In some samples, an increase of ∼200% was seen for protozoal protein groups, suggesting that previously unknown protozoa are likely dominant in certain samples. Similarly, the expanded database greatly increased the number of fungal proteins identified, with a 118% increase in cow samples and 70% increase in goat samples. Notably, on average, only 10 fungal protein groups were identified in the original study. It is also important to note that rumen samples were obtained for this particular metaproteomic study via esophageal tubing, which primarily collects rumen fluid and significantly lowers fungal biomass.
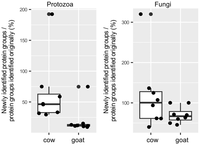
Increases in the number of rumen protozoal and fungal protein groups identified with the new protein databases compared with the previous rumen proteomic study. The numbers represent the percentage ratio between newly identified protein groups and those identified in the original study. Box plots indicate the median (middle line), the 25th and 75th percentiles (box), as well as individual observations (dots).
Discussion
Historically, gut eukaryotes have been studied primarily from a parasitological perspective. However, eukaryotic populations are also ubiquitous components of the gut microbiome (Parfrey et al. 2014; Ramayo-Caldas et al. 2020), and they contribute significantly to the overall microbial assembly, host nutrition, and physiology (Hoffmann et al. 2013; Laforest-Lapointe and Arrieta 2018; Gerrick et al. 2024). Their role is particularly significant in the rumen, the foregut of ruminants, where they can constitute up to 50% of the microbial biomass, a proportion much higher than in other ecosystems. These microbes play direct roles in rumen fermentation and fiber degradation (Huws et al. 2018) and directly influence several important host characteristics, including feed efficiency and methane emissions (Firkins et al. 2020). Despite recent efforts in the cultivation and genomic sequencing of rumen protozoa and fungi, which is a highly labor-intensive process, we still lack a robust bioinformatic tool to analyze the eukaryotic microbes represented in shotgun metagenomes generated from rumen samples. Therefore, it is crucial to develop bioinformatic tools tailored to gut eukaryotic microbes, particularly those in the rumen, to obtain a more comprehensive representation of their genomes and proteomes. Such new tools will enable a shift from taxonomic profiling to functional profiling, allowing for a deeper understanding of gut eukaryotic microbes as well as whole-gut microbiomes.
Tiara and EukRep have been used for eukaryotic sequence identification; however, they fail to accurately identify rumen eukaryotes. To facilitate metagenomic and functional analysis of gut eukaryotic microbes, especially those in the rumen, we developed GutEuk. This tool outperforms Tiara and EukRep in both precision and recall when identifying microbial eukaryotic genome fragments. The ensemble model implemented in GutEuk (Fig. 1), which combines CNN and FNN, enables consistent performance for both short sequences (<5 kb) and long sequences (>50 kb). The ability of GutEuk to further differentiate fungal and protozoal sequences is especially valuable for analyses of metagenomes derived from microbiomes that contain both types of eukaryotic microbes, such as the rumen microbiome. Although GutEuk was designed for metagenomes from the GI ecosystem, it also demonstrates consistent performance across genomes of diverse taxonomic lineages by accurately differentiating fungi and protozoa from other ecosystems. Because eukaryotic communities are less diverse in the gut than in aquatic and terrestrial environments, and there is minimal overlap in eukaryotic microbial diversity among these ecosystems (Parfrey et al. 2014), GutEuk is well suited for gut metagenomic studies, as demonstrated by its use with the newly sequenced human gut fungi genomes (Supplemental Table S1). In addition to prokaryotes and eukaryotes, metagenomes can contain significant amounts of mobile genetic elements, such as plasmids and viruses. If these elements are of particular interest, we recommend using specialized tools like geNomad (Camargo et al. 2024) to filter the input sequences.
Using GutEuk, we screened thousands of metagenomic bins assembled from the rumen and identified hundreds of individual eukaryotic bins. Our phylogenetic analysis of the dozens of identified protozoal bins with at least medium quality demonstrated a great genomic diversity of rumen protozoa, particularly those in less-studied ruminant species. The relative sparsity of the recovered eukaryotic bins with high quality may be attributable to multiple factors, including the low abundance of eukaryotes in the rumen, their large and complex genomes, high genetic heterogeneity, and the presence of highly repetitive genomic sequences (Cho et al. 2023). Additionally, there is a risk of chimeric bins owing to the presence of mobile elements and paralogous genes (West et al. 2018). Annotation of eukaryotic bins should, therefore, be performed with caution. Binning of contigs into high-quality MAGs of eukaryotic microbes from metagenomic sequences, especially short sequencing reads, is probably challenging. Sequencing repetitive regions contiguously (Cho et al. 2023), long-read sequencing, such as Pacific BioSciences (PacBio) HiFi and Oxford Nanopore Technologies (ONT) sequencing, may facilitate assembling and binning of eukaryotic sequences from metagenomes, especially for protozoa that have highly fragmented small chromosomes, such as the Entodinium genus (Park et al. 2021). With the growing use of single-cell sequencing, GutEuk can be applied to filter out symbiotic prokaryotic sequences prior to assembly, enhancing genome quality owing to its high accuracy in distinguishing between prokaryotic and eukaryotic sequences. Additionally, the recently developed bin refinement tool ACR (Seong et al. 2023) can enhance genome-centric analyses of eukaryotes through metagenomic approaches. Furthermore, similar to the assessment of prokaryotic genomes and MAGs, the completeness and contamination rates of eukaryotic bins should be determined based on the presence of lineage-specific marker genes and corresponding databases as implemented in BUSCO (Simão et al. 2015) and EukCC (Saary et al. 2020). However, both tools show limited performance when applied to eukaryotic clades with sparse reference genomes (Saary et al. 2020; Jauhal and Newcomb 2021). With the expansion of reference genomes and advanced quality assessment using newly developed tools like ContScout (Bálint et al. 2024), we can better assess the quality of the obtained eukaryotic microbial genomes.
Several genomic structural features of eukaryotes, such as the exon–intron architecture and the lack of conserved splicing signatures, complicate the gene-calling process in eukaryotic genome annotations. To overcome these challenges, a reference- and homology-based pipeline, MetaEuk (Levy Karin et al. 2020), with high precision (>99.9%) has been developed (Levy Karin et al. 2020). Using this pipeline and a database combining single-cell protozoal transcriptomes and poly(A)-enriched rumen metatranscriptomes, we screened the contigs assembled from around 1000 ruminant GI metagenomes (see Methods). This analysis led to the identification of millions of ruminant eukaryotic proteins, further highlighting the great metabolic potential of rumen protozoa, consistent with the previous studies (Li et al. 2022; Andersen et al. 2023).
Leveraging the databases we compiled, we tested their resource applicability by reanalyzing the detected proteomic profiles of the rumen of cows and goats (Andersen et al. 2023). We substantially increased the number of identified protein groups from both fungi and protozoa. The additional proteins identified are primarily from the ruminant eukaryotic sequences identified by GutEuk, validating the genomic identification and protein prediction pipeline we used. The potential for chimeric binning is unlikely to confound the protein prediction as all the genomic sequences are predicted to be either fungal or protozoal. With the assistance of GutEuk and the derived protein database, we could better understand the distinct metabolic contributions of fungi and protozoa in the rumen, particularly under varying dietary and microbial interventions. However, protein prediction will require further genome curation, as described previously (West et al. 2018), to attribute expressed protein to specific species.
In summary, the new GutEuk tool, in conjunction with its associated databases, enables comprehensive analyses of GI eukaryotes in metagenomes. The integration of the proteins identified from rumen metagenomes into reference genome databases markedly enhances metaproteomic studies of the rumen microbiome. Additionally, the expanded databases enhance read-based ecological analyses of gut eukaryotic microbes with tools such as EukDetect (Lind and Pollard 2021), offering finer taxonomic resolution than marker-based metataxonomic approaches. Collectively, GutEuk and the associated databases can help provide new insights into the functional contributions of ruminant fungi and protozoa to the rumen ecosystem, as well as their interactions with diets and other microbes.
Methods
Evaluating existing tools in identifying and classifying rumen protozoa
Currently, Tiara (Karlicki et al. 2022) and EukRep (West et al. 2018) are tools available for identifying eukaryotic contigs assembled from metagenomes. To evaluate their performance in identifying sequences of eukaryotes, we used the 52 rumen protozoa SAGs reported recently by Li et al. (2022). The telomere sequences were trimmed off from the telomere-capped contigs with a custom Python script (Supplemental Code). Because both tools were trained with contigs of 5 kb length, the contigs of each SAG were chopped into 5 kb fragments with a 1 kb sliding window and used for benchmarking (see below).
Genome data set preparation
To specifically identify eukaryotic sequences in metagenomes derived from the gut, we compiled a data set comprising genomes of prokaryotes, fungi, and protozoa. Briefly, 85,205 representative genomes of prokaryotes were downloaded from GTDB release 214.0 (https://data.gtdb.ecogenomic.org/releases/release214/214.0/). These genomes represent 80,789 species of bacteria and 4416 species of archaea. Fungal genomes were downloaded from the MycoCosm genome portal (https://mycocosm.jgi.doe.gov/mycocosm/home). When multiple genomes were available for a fungal species, we only selected one genome with the highest quality. We also filtered out the genomes of mushrooms and soil fungi, reducing the number of fungal genomes not relevant to gut microbiomes. A total of 679 fungal genomes were retained, representing 639 genera. Protozoal genomes were downloaded from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) on September 2, 2023, and we selected the most complete genomes per species. We combined these protozoal genomes with the 52 rumen protozoal SAGs representing 19 species across 13 genera (Li et al. 2022). To remove potential sequence contamination from symbiotic prokaryotes, we retained only the contigs flanked by the identified telomere sequences of protozoa. Additionally, we included only one representative species per genus from the fungal and prokaryotic genomes to minimize classification bias toward overrepresented lineages and to prevent data leakage. The data set includes 20,739 prokaryotic genomes/contigs, 639 fungal genomes/contigs, and 380 protozoal genomes/contigs.
The genomes/contigs from each class were randomly assigned to one of the following data sets: training (70%), validation (15%), and testing (15%) to be used to train the classifier (see below). Within each data set, genomes or contigs were randomly fragmented into 5 kb fragments with a sliding window of 5 kb. Then, we randomly selected 28.5% of the prokaryotic and fungal fragments and 75% of the protozoal fragments. This selection ensured a data set size balanced with prokaryotic and gut eukaryotic sequences. The genomes/contigs used for training, validation, and testing are detailed in Supplemental Table S3.
Development of GutEuk and training
We developed GutEuk, a two-stage classifier designed to differentiate prokaryotic and eukaryotic (fungal and protozoal) sequences in the first stage and then to further differentiate fungal and protozoal sequences in the second stage. GutEuk uses an ensemble model comprising a FNN and a CNN in each stage (Fig. 1A) to classify individual input sequences after they are fragmented into 5 kb length. For each input sequence, its 4-, 5-, and 6-mer frequencies were calculated, and its nucleotide bases (A, C, G, T) were one-hot encoded as [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0], and [0, 0, 0, 1], respectively. The k-mer frequencies and the one-hot encoded DNA sequences served as the input of FNN and CNN, respectively. In the first stage, the probability of a sequence being eukaryotic origin is calculated by FNN and CNN. The probabilities calculated by the two networks are then averaged. If the average probability of an input sequence exceeded 0.5, it was subjected to another round of FNN and CNN classification to be fungal or protozoal. Input sequences identified with a probability of less than 0.5 were considered prokaryotic. For input sequences <5 kb but longer than the user-defined minimal length, they were zero-padded to the right to reach 5 kb, and their k-mer frequencies are calculated based on the original input sequences (Fig. 1B). For sequences >5 kb, the remainder fragments <5 kb after the 5 kb fragmentation were also zero-padded on the right and processed as described above. The origin of the input sequences (before fragmentation) was determined based on the predicted origin of their fragmented sequences. A user-defined confidence level was preset at the first and second stages (pt1 and pt2, respectively). If the percentage of fragments of the input sequence classified as either prokaryote or eukaryote was less than the threshold pt1, it was classified as not available (NA). Otherwise, they were classified as either prokaryotic or eukaryotic based on the majority rule. Similarly, input sequences were further classified in the second stage if the percentage of their fragments classified as either fungal or protozoal exceeded the threshold pt2.
The FNN architecture consists of four sequential layers with decreasing dimensionality, accepting an initial 5376-dimensional input vector of k-mer frequencies. The first layer is a fully connected layer with 5376 input units and 1024 output units, followed by ReLU activation and a dropout rate of 0.5. The second layer has 1024 input units and 256 output units, with similar activation and dropout. The third layer features 256 input units and 48 output units, again followed by ReLU and a dropout rate of 0.5. The final output layer has 48 input units and two output units. The CNN architecture begins with an input layer accepting a one-hot encoded DNA as input. The first convolutional layer has one input channel and produces 32 output channels with a kernel size of (6, 4), followed by a ReLU activation. The output is then flattened starting from dimension 2 for subsequent 1D convolutions. The second layer transforms 32 input channels into 64 output channels with a kernel size of three, also followed by ReLU activation. This is followed by max pooling with a pool size of three and batch normalization for 64 channels. The architecture continues with two more convolutional layers that increase the output channels to 128 and then 256, each followed by ReLU activation. A second max pooling layer is applied with ceil mode enabled, along with batch normalization for 256 channels. The feature maps are processed through an adaptive average pooling layer, which is then flattened. The output from the convolutional layers feeds into fully connected layers, beginning with a layer that transforms 256 input features to 64 output features, followed by ReLU activation and dropout with a probability of 0.5. The second fully connected layer reduces this to eight output features, again with ReLU activation and dropout. Finally, the output layer consists of eight input features and two output features. The CNN and FNN models at each stage were trained separately in PyTorch (Paszke et al. 2019) with the cross-entropy loss and AdamW optimizer applied. Hyperparameter optimization involved manually adjusting and training the models across various configurations until the validation loss no longer improved over ten consecutive epochs. The model with the best performance (in terms of validation loss) was used to predict sequence origins.
Benchmarking EukRep, Tiara, and GutEuk
To benchmark the performance of GutEuk against Tiara and EukRep, we filtered out the genomes originally used for training Tiara and EukRep in the testing set (2995, 93, and 43 genomes for prokaryotes, fungi, and protozoa, respectively, of diverse taxonomic origins). We first evaluated the performance of the three classifiers at the contig level. Because contigs vary substantially in length, the first 50 kb of those >50 kb was chopped into fragments of 3, 4, 5, 8, 10, and 20 kb. Then, the performance of each tool was compared with the different lengths. For GutEuk, performance was also determined at different confidence levels. We further evaluated the performance of each tool at the genome level, with the criterion being the percentage of 5 kb fragments of the genomes correctly classified. A recent compendium of 706 genomes of cultured human gut fungi, encompassing 206 species across 48 families (Yan et al. 2024), was downloaded from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA835516 and used to benchmark GutEuk's performance with human gut eukaryotes at a confidence level of 0.5.
Identifying and functionally annotating microbial eukaryotic sequences from rumen metagenomes with GutEuk
We systematically analyzed 1093 previously sequenced rumen metagenomes (Supplemental Table S2) to identify microbial eukaryotic sequences (Fig. 1C). Specifically, the raw reads downloaded from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) were quality-filtered using fastp (Chen et al. 2018) and assembled (individually for each metagenome) with MEGAHIT (v.1.2.1) (Li et al. 2015) with the default setting. The quality-filtered reads were mapped to the resultant contigs with Bowtie 2 (Langmead and Salzberg 2012), and the mapping results were used for binning with MetaBAT2 (Kang et al. 2019). Then, GutEuk was used to identify eukaryotic sequences from the resultant contigs. The bins >3 Mb were also screened with GutEuk with the parameter “-bin.” Based on the benchmarking result, we determined conservative thresholds to classify the contigs and bins. Specifically, in the first stage, bins were classified as either prokaryotes or eukaryotes if 80% of the individual contigs within a bin were consistently classified as such and if the contigs were >20 kb. In the second stage, eukaryotic bins were classified as fungi or protozoa if >95% of the genomes were classified into the same category.
For functional annotations, the protein sequences from the previously sequenced polyadenylated-enriched rumen metatranscriptomics (Li et al. 2022) were first assembled using Plass (Steinegger et al. 2019). The resultant protein sequences were clustered at 50% amino acid identity with MMseqs2 (Steinegger and Söding 2017) to compile a rumen eukaryote protein sequence database. UniProtKB/Swiss-Prot release 2024_01 (The UniProt Consortium 2023) was downloaded and used as the target database to analyze the sequences derived from ruminant hindgut metagenomes. Then, genes were predicted from the identified eukaryotic contigs and bins from the foregut and hindgut using MetaEuk (Levy Karin et al. 2020) with the clustered rumen eukaryotic protein sequences and UniProtKB/Swiss-Prot target database, respectively. The obtained protein sequences were functionally annotated using eggNOG-mapper v2 (Cantalapiedra et al. 2021). Peptidases were also annotated from the identified protein sequences using DIAMOND (Buchfink et al. 2015) with the e-value of 1 × 10−5 as the threshold against the MEROPS database (Rawlings et al. 2010). Signal peptides were identified from the protozoal peptidases using SignalP 6.0 (Teufel et al. 2022), and those with a signal peptide were considered secretory.
Phylogenomic analysis of recovered protozoa genomes
The completeness of the identified protozoa genomes/bins was assessed with BUSCO v5.6.1 by identifying lineage-specific single copy marker genes with the parameter “-l alveolata_odb10” applied. Only those with completeness >50% were subjected to further phylogenomic analysis. Briefly, the multiple sequence alignment of 30 BUSCO proteins of the identified protozoal genomes/bins and the 52 rumen SAGs (Li et al. 2022) were concatenated. Genomes/SAGs with <70% of these markers were excluded to ensure robust phylogenomic analysis. Specifically, the amino acid sequences of individual BUSCO proteins of the collected genomes were aligned with MAFFT v7.505 (Katoh and Standley 2013) and trimmed with trimAl v1.4 (Capella-Gutiérrez et al. 2009). The trimmed alignments of all the BUSCO proteins were then concatenated together with catfasta2phyml.pl (https://github.com/nylander/catfasta2phyml). The concatenated alignment was used to build a phylogenetic tree using IQ-TREE (Nguyen et al. 2015) with the parameters “-redo -bb 1000 -m MFP -mrate E,I,G,I + G -mfreq FU -wbtl -nt AUTO” applied. The obtained tree was visualized with iTOL (Letunic and Bork 2019).
Evaluation of the new protein databases by reanalyzing the proteomics profiles from a recent rumen proteomic study
To benchmark the performance of the new protein databases in improving rumen eukaryotic protein identification and their applicability as a database resource for future analyses, we reanalyzed the metaproteomic data generated from both goat and cattle under two different dietary treatments using the same method described previously (Andersen et al. 2023). Briefly, FragPipe version 19 (Yu et al. 2023) was used to analyze the raw data obtained from mass spectrometry (MS). The raw data were then analyzed with (1) the original sample-specific databases used in the study or (2) the original databases supplemented with the newly established protozoal (no additional SAGs included) and fungal protein databases (described in the previous section) using MSFragger (Kong et al. 2017). To estimate false-discovery rates (FDRs), the databases were supplemented with contaminant protein entries such as human keratin, trypsin, and bovine serum albumin, alongside the reversed sequences of all protein entries. Variable modifications, including methionine oxidation and protein N-terminal acetylation, were accounted for, whereas carbamidomethylation of cysteine residues was held as a fixed modification. The choice of trypsin as the digestive enzyme allowed for a single missed cleavage, with matching tolerance levels set at 20 ppm for both MS and MS/MS. Filtering was executed to ensure a ≤1% FDR threshold. Quantitative analysis was conducted using IonQuant (Yu et al. 2021). Only the proteins identified in the majority of the replicates in at least one of the treatments were considered present, as described in the original study.
Data access
The established ruminant eukaryotic protein databases and identified eukaryotic bins have been submitted to Figshare (https://doi.org/10.6084/m9.figshare.c.7457755.v1). GutEuk, along with the codes used for visualization, is available at GitHub (https://github.com/yan1365/rumen_eukaryotes) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work is supported in part by the USDA National Institute of Food and Agriculture (award no. 2021-67015-33393). We also thank the Ohio Supercomputer Center for providing the computing resources.
Author contributions: M.Y. conceived this work, built the package, and performed bioinformatics analysis. T.O.A. performed the proteomics analysis. M.Y. and Z.Y. wrote the manuscript. All authors revised and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279825.124.
- Received July 17, 2024.
- Accepted December 19, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








