
Characterization of the role of spatial proximity of DNA double-strand breaks in the formation of CRISPR-Cas9-induced large structural variations
- 1Department of Biomedicine, Aarhus University, 8000 Aarhus, Denmark;
- 2Department of Clinical Medicine, Aarhus University, 8200 Aarhus, Denmark;
- 3Department for Clinical Genetics, Aarhus University Hospital, 8200 Aarhus, Denmark;
- 4Aarhus Institute of Advanced Studies (AIAS), Aarhus University, 8000 Aarhus, Denmark
-
↵5 These authors contributed equally to this work.
Abstract
Structural variations (SVs) play important roles in genetic diversity, evolution, and carcinogenesis and are, as such, important for human health. However, it remains unclear how spatial proximity of double-strand breaks (DSBs) affects the formation of SVs. To investigate if spatial proximity between two DSBs affects DNA repair, we used data from 3C experiments (Hi-C, ChIA-PET, and ChIP-seq) to identify highly interacting loci on six different chromosomes. The target regions correlate with the borders of megabase-sized topologically associated domains (TADs), and we used CRISPR-Cas9 nuclease and pairs of single guide RNAs (sgRNAs) against these targets to generate DSBs in both K562 cells and H9 human embryonic stem cells (hESCs). Droplet digital PCR (ddPCR) was used to quantify the resulting recombination events, and high-throughput sequencing was used to analyze the chimeric junctions created between the two DSBs. We observe a significantly higher formation frequency of deletions and inversions with DSBs in proximity compared with deletions and inversions with DSBs not in proximity in K562 cells. Additionally, our results suggest that DSB proximity may affect the ligation of chimeric deletion junctions. Taken together, spatial proximity between DSBs is a significant predictor of large-scale deletion and inversion frequency induced by CRISPR-Cas9 in K562 cells. This finding has implications for understanding SVs in the human genome and for the future application of CRISPR-Cas9 in gene editing and the modeling of rare SVs.
Structural variations (SVs) encompass a variety of DNA alterations, including inversions, deletions, and insertions of DNA segments. Although many SVs are without clinical significance, some SVs that arise in germ cells lead to genetic disorders, and some that arise in somatic cells contribute to the development of cancer (Yoshioka et al. 2021). SVs can be created by CRISPR-Cas9 genome editing via two double-strand breaks (DSBs) that occur at the target sites of two different single guide RNAs (sgRNAs), which we refer to as an sgRNA pair, or at multiple target sites of an individual sgRNA (Cullot et al. 2019; Höijer et al. 2022; Wu et al. 2022).
Unequal crossing over is a common and well-described mechanism for the creation of duplications and deletions. However, SV formation could also be influenced by other factors such as the three-dimensional genomic structure, as breakpoint proximity has been associated with both recurrent translocation formation (Nikiforova et al. 2000), and nonrecurrent translocation formation (Rothkamm et al. 2001; Engreitz et al. 2012; Zhang et al. 2012; Balajee et al. 2018; Eidelman et al. 2021). Furthermore, SV breakpoints have been shown to correlate with interaction frequencies measured by Hi-C (Gandhi et al. 2006; Swenson and Blanchette 2019; Akdemir et al. 2020; Sidiropoulos et al. 2022). These findings support that DSB proximity may play a role in SV formation, although this understanding has not been verified with genome engineering in a controlled manner. DNA has also shown the ability to shift position in the three-dimensional nucleus upon induction of DSBs in a process called “DSB clustering” (Aten et al. 2004; Roukos et al. 2013; Aymard et al. 2017; Arnould et al. 2023). Thus, two opposing understandings of SV formation have emerged, the “contact first” versus “breakage first” model (Misteli and Soutoglou 2009).
The development of Hi-C and Hi-C-related techniques in recent years have led to the discovery of topologically associated domains (TADs), which are loop-like genomic structures characterized by extensive self-interaction. TADs are established through a dynamic interplay between CTCF and cohesin, influenced by the orientation of the CTCF motif. Accumulation of CTCF and cohesin occurs at specific sites, known as TAD borders (Fudenberg et al. 2016). TAD borders are excellent candidates to verify spatial proximity effects experimentally, because they exhibit consistent interaction across the cell cycle and cell types (Fudenberg et al. 2016; Schmitt et al. 2016; Krefting et al. 2018) and because advances in Hi-C and related techniques offer high-resolution TAD border capture (Lieberman-Aiden et al. 2009; Dixon et al. 2012).
SVs are generally formed through nonallelic homologous recombination, replicative mechanisms, or canonical nonhomologous end-joining (c-NHEJ) (Gu et al. 2008; Liu et al. 2012). In studies with irradiation-induced DSBs, c-NHEJ has been shown to repair DNA with biphasic kinetics involving a fast and a slow process depending on chromatin context (DiBiase et al. 2000; Riballo et al. 2004; Biehs et al. 2017). The fast c-NHEJ process occurs in the G1 and G2 phases of the cell cycle and is resection-independent (Biehs et al. 2017). The slow c-NHEJ process occurs in the G1 phase. This process is resection-dependent and requires the endonuclease Artemis. The slow process is characterized by microhomology usage and is more error-prone than fast c-NHEJ. It is primarily associated with repairing DSBs in heterochromatin (Biehs et al. 2017). Yet, it is unknown if DSB proximity could change SV formation kinetics and junction characteristics in a similar manner.
In this study, we quantify the impact of spatial proximity on SV formation by targeting borders of highly conserved TADs with DSBs induced by CRISPR-Cas9, comparing the results to size-matched noninteracting loci. Additionally, we investigate whether proximity between DSBs induced by CRISPR-Cas9 affects SV formation kinetics and repair characteristics of chimeric deletion junctions.
Results
Spatial proximity between DSBs increases SV formation in K562 cells
To investigate the impact of spatial proximity on CRISPR-Cas9-induced SV formation, we first identified six distinct TADs using publicly available Hi-C and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) data (Fig. 1A,B). We also used chromatin immunoprecipitation (ChIP-seq) data for cohesin, and CTCF, including motif orientation, to show enrichment and conservation of TAD border markers (Supplemental Figs. S1–S6). We then designed sgRNAs to target the six TADs at seven to eight distinct positions (sgRNAs A-H; see Methods). By combining these in pairs, the sgRNA pairs would induce DSBs in spatial proximity inside (AB) and outside (CD) the CTCF motifs at TAD borders, at spatially distant positions (EF) or at nonrelated TAD borders (GX). The sgRNAs were designed to generate DSBs at a similar linear distance from each other (Fig. 1A,B). The frequency of SVs formed between the two DSBs was quantified by ddPCR assays designed to detect both deletions and inversions (Fig. 1C).
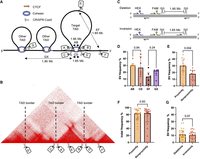
The effect of spatial proximity on SV frequency in K562 cells. (A) Six TADs were identified in K562 cells and named according to their position: Chr 2, Chr 6, Chr 10, Chr 17, Chr 21, and Chr X. Seven sgRNAs were designed for each TAD to investigate spatial proximity effects: sgRNAs A and B, which targeted sequences inside the CTCF motif at the left and right TAD border; sgRNAs C and D, which targeted sequences outside the CTCF motif at the left and right TAD border; sgRNA E, which targeted an intra-TAD sequence; sgRNA F, which targeted a sequence outside the TAD; and sgRNA G (or H, not shown), which targeted a sequence inside the CTCF motif of a neighboring TAD. Individual sgRNAs were electroporated into K562 cells in separate pairs as AB, CD, EF, and GX (X could be sgRNA A, B, or H). All pairs would have the same distance in base pairs between them, but AB and CD would be in spatial proximity, whereas EF and GX would be spatially distant. (B) An example Hi-C map of the Chr 6 TAD. (C) Illustration of the gain-of-signal ddPCR assay with primers surrounding the cut sites of the sgRNA pair and the binding site of the FAM probe. The HEX probe included to adjust for locus copy number is also illustrated. (D) SV frequencies (deletions + inversions) for AB, CD, EF, and GX sgRNA pairs (n = 6 sgRNA pairs in each group; six chromosome loci with one sgRNA pair); P-values from Mann–Whitney U test. (E) SV frequencies between proximity and nonproximity sgRNA pairs (n = 12 sgRNA pairs in each group; six chromosome loci with two sgRNA pairs); P-value from Mann–Whitney U test. (F) Individual sgRNA efficiencies measured by their ability to induce indels (n = 24 sgRNAs in each group; six chromosome loci with four sgRNAs); P-value from Mann–Whitney U test. (G) Frequencies of deletions and inversions for all sgRNA pairs (n = 24 sgRNA pairs in each group; six chromosome loci with four sgRNA pairs); P-value from Wilcoxon matched-pair signed-rank test. The observations are biological replicates, and the error bars show the median and IQR.
We hypothesized that a significant difference in SV frequency (defined as the sum of deletion and inversion frequencies) between sgRNA pairs AB and CD at TAD borders could result from steric hindrance of Cas9 caused by cohesin occupying the borders. Furthermore, we hypothesized that any difference between sgRNA pairs EF and GX would be because of DSB vulnerability at TAD borders (Canela et al. 2017). Therefore, we compared SV frequencies in sgRNA pairs AB with CD and EF with GX. There was no significant difference between AB (median 30%) and CD (median 27%) and thus no effect of the position relative to the binding site of CTCF/cohesin on SV frequency (P = 0.94) (Fig. 1D). Similarly, there was no significant difference between EF (median 13%) and GX (median 22%) and thus no significant effect from TAD DSB vulnerability with the caveat of a limited sample size (P = 0.24) (Fig. 1D). Thus, we grouped sgRNA pairs AB and CD as “proximity” and EF and GX as “nonproximity,” although we could not exclude that the pooled groups could differ according to additional factors that could influence the results despite no statistically significant difference in SV frequency.
Spatial proximity was associated with a significant increase in the formation of SVs, which increased from a median frequency of 18% in the nonproximity group to a median frequency of 30% in the proximity group (P = 0.004) (Fig. 1E). Because the formation of SVs could be influenced by the cutting efficiencies of the individual sgRNAs in the pairs, we performed Sanger sequencing across all cut sites and estimated the frequency of indels <30 bp as an indicator of sgRNA efficiency using the Inference of CRISPR Edits (ICE) tool. The individual sgRNA efficiencies, measured by their ability to facilitate indels at their target sites, also correlated with the frequency of SVs (Spearman's r = 0.45, P = 0.03) (Supplemental Fig. S11), but the sgRNA efficiencies did not differ significantly between the proximity and nonproximity groups (Fig. 1F).
The increase in SV frequency between proximity and nonproximity groups corresponded to a significant increase in deletion frequency (P = 0.002) but not significantly in inversion frequency (P = 0.06) (Supplemental Fig. S12A). We did not observe a significant difference when comparing the frequency of deletions and inversions (P = 0.07) (Fig. 1G). As shown later, resection of DNA may occur at the chimeric deletion junctions between the two DSBs owing to imprecise ligation. Such DNA resections could lead to disruption of ddPCR primers and/or probes, but high-throughput sequencing of the chimeric junctions revealed no significant difference in ddPCR-detectable deletions between the proximity and nonproximity groups (P = 0.53) (Supplemental Fig. S10C). Thus, the observed association between proximity and SV frequency was not influenced by differences in ddPCR sensitivity.
Effect of DSB proximity in human embryonic stem cells
Having observed that spatial proximity affected the formation of CRISPR-Cas9-induced SVs in K562 cells, we subsequently investigated if spatial proximity also affected the formation of SVs in noncancer cells. Hence, we repeated the previous experiment for H9 human embryonic stem cells (hESCs) using the same sgRNA pairs, albeit without the Chr 10 TAD locus. Similar to K562 cells, we confirmed that neither cohesin nor TAD borders affected our results (see Fig. 2A). Hence, as previously, we grouped sgRNA pairs AB and CD as “proximity” and sgRNA pairs EF and GX as “nonproximity.”
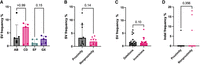
The effect of spatial proximity on SV frequency in H9 hESCs at the Chr 2, Chr 6, Chr 17, Chr 21, and Chr X loci. (A) SV frequencies for AB, CD, EF, and GX sgRNA pairs (n = 5 sgRNA pairs in each group; five chromosome loci with one sgRNA pair); P-values from Mann–Whitney U test. (B) SV frequencies between proximity and nonproximity sgRNA pairs (n = 10 sgRNA pairs in each group; five chromosome loci with two sgRNA pairs); P-value from Mann–Whitney U test. (C) Overall frequencies of deletions and inversions (n = 20 sgRNA pairs in each group; five chromosome loci with four sgRNA pairs); P-value from Wilcoxon matched-pair signed-rank test. (D) Individual sgRNA efficiencies by indel frequency (n = 20 sgRNAs in each group; five chromosome loci with four sgRNAs); P-value from Mann–Whitney U test. The observations are biological replicates, and the error bars show the median and IQR.
The median frequency of SVs in H9 hESCs was 1.8% in the nonproximity group and 3.8% in the proximity group, which was not statistically significantly different (P = 0.14) (Fig. 3C). Any increase in SV frequency was also not statistically significant for deletion (P = 0.17) or inversion frequency (P = 0.12) (Supplemental Fig. S12B). As for K562 cells, we did not observe a difference between the frequencies of deletions and inversions in H9 hESCs (P = 0.10) (Fig. 2C). Altogether, we could not confirm that DSB spatial proximity was associated with an increase in the formation of SVs in hESCs, with the caveat that the SV frequencies were an order of magnitude lower in H9 hESCs than in K562 cells.
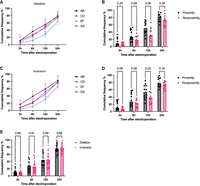
SV frequency at 3, 6, 12, and 24 h after electroporation in K562 cells and normalized to the end point SV frequency for each sgRNA pair as a measure of SV formation speed. (A) Formation speed of deletions for sgRNA pairs AB, CD, EF, and GX (n = 6 sgRNA pairs in each group; six chromosome loci with one sgRNA pair). (B) Formation speed of deletions for proximity versus nonproximity sgRNA pairs (n = 12 sgRNA pairs in each group; six chromosome loci with two sgRNA pairs); P-values from Mann–Whitney U test. (C) Formation speed of inversions for sgRNA pairs AB, CD, EF, and GX (n = 6 sgRNA pairs in each group; six chromosome loci with one sgRNA pair). (D) Formation speed of inversions for proximity versus nonproximity sgRNA pairs (n = 12 sgRNA pairs in each group; six chromosome loci with two sgRNA pairs); P-values from Mann–Whitney U test. (E) Formation speed of deletions versus inversions (n = 24 sgRNA pairs in each group; six chromosome loci with four sgRNA pairs); P-values from Wilcoxon matched-pair signed-rank test. The observations are biological replicates, and the error bars show the median and IQR.
We also assessed the capability of individual sgRNAs to induce indels at their target sites in H9 hESCs. In an analysis of indel frequencies at the sgRNA cut sites using the ICE tool, which has a reported lower sensitivity of 5% indel frequency, 32 out of 35 sgRNAs did not generate detectable indel frequencies, yet still facilitated the formation of SVs. Indel frequency at the cut sites of the sgRNAs measured by Sanger sequencing could thus be an unreliable indicator of sgRNA efficiency in H9 hESCs owing to the limited sensitivity of this analysis (Fig. 2D).
Notably, the CRISPR-Cas9 targets in the proximity group had significantly lower Hi-C scores in hESCs compared to in K562 cells (P = 0.004) (Supplemental Fig. S13), possibly suggesting that the target TADs were less conserved in hESCs than in K562 cells.
Effect of DSB proximity on SV formation speed
Having observed that spatial proximity of DSBs affected the formation frequency of SVs in K562 cells, we hypothesized that the distance between the DSBs might also affect the speed of SV formation.
To investigate the speed of SV formation, we compared the cumulative frequency of SVs at various time points to the end point frequency. This experiment did not confirm that formation of SVs occurred earlier in the proximity group compared with the nonproximity group. For example, at 12 h after electroporation, the median cumulative frequency of deletion formation was 51% in the proximity group and 31% in the nonproximity group, which was not statistically significantly different (P = 0.06) (Fig. 3A,B). By comparison, inversion formation 12 h after electroporation was 54% in the proximity group and 36% in the nonproximity group, showing a statistically significant difference (P = 0.03) (Fig. 3C,D). However, we noted that the kinetic profile of our GX sgRNA pairs resembled that of the proximity group rather than that of the sgRNA pairs EF (Fig. 3A,C).We did not observe any difference in the speed of formation between deletions and inversions (Fig. 3E).
Effect of DSB proximity on ligation of SV junctions
We hypothesized that utilization of fast c-NHEJ for SV formation would require proximity between DSBs. To assess this, we employed high-throughput amplicon sequencing on five chimeric deletion junctions for each of the sgRNA pairs AB, CD, EF, and GX. We centered our analysis on resection-independent end-joining and microhomology usage, as these are readily measurable metrics that are characteristic of either fast or slow c-NHEJ (Biehs et al. 2017). A chimeric deletion junction was defined as the ligation product of a deletion made with one of the sgRNA pairs (Fig. 4A). Resection-independent end-joining was measured in terms of precise ligation of the chimeric deletion junctions. Here, we defined precise ligation as a chimeric deletion junction with up to 2 bp insertions to account for templated insertions (Guo et al. 2018) and up to 2 bp resections to account for staggered cuts being generated by Cas9 (Xue and Greene 2021). Thus, chimeric deletion junctions could be ligated in four ways: (1) precise ligation (Guo et al. 2018; Xue and Greene 2021), (2) insertions >2 bp, (3) resections >2 bp, and (4) mixed resections and insertions >2 bp (Fig. 4B). The distribution of ligation outcomes in the proximity group and in the nonproximity group is illustrated (Fig. 4C).
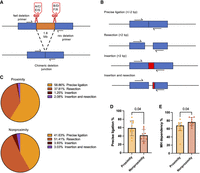
Five chimeric deletion junctions produced from each of the sgRNA pairs AB, CD, EF, and GX analyzed with Illumina MiSeq amplicon sequencing. (A) Definition of a chimeric deletion junction. (B) Characterization of sequencing reads. (C) Read distributions for the proximity and nonproximity sgRNA pairs (n = 10 sgRNA pairs in each group; six chromosome loci with one to two sgRNA pairs). (D) Precise ligation of chimeric deletion junctions for proximity and nonproximity sgRNA pairs (n = 10 sgRNA pairs in each group; six chromosome loci with one to two sgRNA pairs); P-value from Mann–Whitney U test. (E) Microhomology usage normalized to all reads with resection for proximity and nonproximity sgRNA pairs (n = 10 sgRNA pairs in each group; six chromosome loci with one to two sgRNA pairs); P-value from Mann–Whitney U test. The observations are biological replicates, and the error bars show the median and IQR.
The proximity group showed a significant increase in precise ligation of chimeric deletion junctions to 58.86% compared with 41.63% in the nonproximity group (P = 0.04), which suggested increased usage of the resection-independent fast c-NHEJ process (Fig. 4D). As a sensitivity analysis, we tested whether excluding 1–2 bp resections from the definition of precise ligation would significantly affect the results. Precise ligation differences were not significant with this definition (P = 0.089) (Supplemental Fig. S14A). There was no significant difference between precise ligation of the GX sgRNA pairs and the EF sgRNA pairs, which suggested no significant influence from TAD border nucleotide homology (P = 0.222) (Supplemental Fig. S14B).
Finally, the proximity group showed a signification reduction in microhomology usage at resected junctions compared to the nonproximity group (P = 0.04) (Fig. 4E). Taken together, the increase in precise ligation and the reduction in microhomology usage in the proximity group compared with the nonproximity group could suggest a difference in DSB repair with increased usage of the fast c-NHEJ pathway compared with the slow c-NHEJ pathway. However, the observed differences were only borderline significant, so we did not interpret the evidence to be conclusive.
Discussion
In this study, we investigated experimentally if spatial proximity between CRISPR-Cas9-generated DSBs would affect the frequency of deletion and inversion formation. The idea that spatial proximity of DSBs would affect the formation of SVs is not new, however, as multiple lines of prior evidence had established a connection between breakpoint proximity and SVs. Thus, FISH analysis was used to demonstrate that a known recurrent translocation occurred between loci in spatial proximity (Nikiforova et al. 2000). Another study observed that low-dose irradiation yielded fewer nonrecurrent SVs than high-dose irradiation, which the authors suggested could be owing to more spatiotemporal separation of DSBs in the low-dose group (Rothkamm et al. 2001). The development of Hi-C since then allowed the validation of these findings, revealing that nonrecurrent translocations induced by irradiation often involve loci with increased Hi-C interaction (Engreitz et al. 2012; Zhang et al. 2012; Balajee et al. 2018; Eidelman et al. 2021). Moreover, computational modeling linked nontranslocation SV breakpoints with increased Hi-C interaction (Swenson and Blanchette 2019). Although these studies showed a correlation between spatial proximity and both recurrent and nonrecurrent SV formation, they did not establish causality or quantify the impact of spatial proximity. In this study, we verify using genome engineering that spatial proximity between DSBs significantly increases SV frequency in K562 cells (Fig. 1E). Although we did not reach statistical significance when replicating the experiment in another cell line, H9 hESCs (Fig. 2B), it is possible that the observed effect in K562 cells also applies to other cell lines, as the replicate analysis was underpowered and limited by significantly lower Hi-C interaction strength between sgRNA pairs in the proximity group compared with K562 cells (Supplemental Fig. S13).
Our results align with the previous studies, reinforcing the idea that spatial proximity between DSBs has a significance in the generation of SVs. This idea, often referred to as the “contact first” hypothesis of SV formation, has been challenged by numerous studies showing that DSB loci cluster in space (Aten et al. 2004; Roukos et al. 2013; Aymard et al. 2017; Arnould et al. 2023), which has led to a contrasting “breakage first” hypothesis of SV formation (Misteli and Soutoglou 2009). Although we did not investigate DSB clustering specifically, we consider our work a relevant contribution to the debate.
Through the use of CRISPR-Cas9 in the generation of SVs, we also show that spatial proximity could be a relevant parameter to consider for CRISPR-Cas9 gene editing strategies, particularly those involving large DNA excisions using sgRNA pairs. A well-described example is the creation of microdystrophin through truncation of the DMD gene in Duchenne muscular dystrophy (Min et al. 2019). Additionally, DSB proximity could serve as a valuable parameter to consider, when assessing the feasibility of modeling large SVs within a cell line with CRISPR-Cas9 using sgRNA pairs. This approach is otherwise often restricted by low efficiency (Choi and Meyerson 2014).
The notable variability in our data, however, implies that spatial proximity should not be regarded as an absolute predictor of increased SV formation. Rather, it could be considered as one of several parameters, such as transcriptional activity, cell type, chromatin state, and cell cycle (Canoy et al. 2022). Specifically, for CRISPR-Cas9-induced SVs, sgRNA efficiency is a known predictor of SV frequency (Choi and Meyerson 2014). In our study, the ability of an sgRNA to generate indels was also independently associated with higher SV frequency in K562 (Supplemental Fig. S11), but not in H9 hESCs, in which most sgRNAs did not produce indels despite being capable of creating SVs (Supplemental Table S8). Similar to a recent finding in human primary cells (Selvaraj et al. 2024), we therefore caution that indel frequency might not reflect actual DSB frequency in all cell lines. A recent study showed that individual sgRNA efficiency could be affected by the number of spatial interactions at its DNA target (Bergman and Tuller 2024). Similar to our finding, this underlines the importance of evaluating 3D genomic context for CRISPR-Cas9 experiments.
Regarding the generation of large CRISPR-Cas9-induced SVs, it is also notable that deletion and inversion frequencies were similar in our experiments (Figs. 1G, 2C). This observation aligns with a prior study that generated a 2.5 Mbp deletion using an sgRNA pair (Miyata et al. 2023). However, it differs from another study that suggested a preference for deletions over inversions when inducing small SVs with sgRNA pairs (Watry et al. 2020).
We also investigated if spatial proximity impacted the underlying repair dynamics of SV formation, hypothesizing that utilization of fast c-NHEJ for SV formation would require proximity between DSBs. Our hypothesis was based on the knowledge that fast and slow c-NHEJs both share a similar first step in their repair pathways (Biehs et al. 2017). This step involves the rapid recruitment of the XRCC6/XRCC5 (also known as KU70/80) protein to DSBs, which, in turn, facilitates the recruitment of additional repair factors, such as DNA-PKcs. Together, these repair factors form a synapsis between DNA ends that guards against the formation of SVs (Frit et al. 2019; Watanabe and Lieber 2023).
Although we observed that deletions from proximity DSBs showed more precise ligation (Fig. 4D) and less microhomology usage (Fig. 4E) compared with deletions from nonproximity DSBs, which are characteristics associated with the fast c-NHEJ repair pathway (Biehs et al. 2017), we did not detect a statistically significant difference in SV formation speed, except at a single time point for inversions (Fig. 3D). Furthermore, the sgRNA pairs, which targeted nonproximity TAD borders, GX, had a kinetics and precise ligation profile similar to the proximity sgRNA pairs rather than the other nonproximity sgRNA pairs targeting inter-TAD loci (Fig. 3A,C; Supplemental Fig. S14B). Thus, the kinetics and precise ligation differences could be explained by other factors than proximity. Also, precise ligation differences were not significant in a sensitivity analysis with an alternate definition of precise ligation (Supplemental Fig. S14A). More research is therefore warranted to investigate if proximity changes DSB repair dynamics.
Nonetheless, it is interesting how SVs arise, when DSBs are not in proximity, as Ku-PKcs synapses are expected to keep the original free DNA ends together. However, studies have demonstrated that these synapses can break when XRCC6/XRCC5 or DNA-PKcs are phosphorylated (Uematsu et al. 2007; Lee et al. 2016), potentially allowing for DNA movement through space and the subsequent formation of SVs through slow c-NHEJ. SVs without DSB proximity could also emerge in a Ku-PKcs-independent manner, a process referred to as alternative-NHEJ (alt-NHEJ) (Chang et al. 2017), although this is less likely, as human cells do not typically utilize alt-NHEJ when Ku is present, neither when repairing DSBs induced by irradiation (Biehs et al. 2017) nor DSBs induced by designer nucleases (Ghezraoui et al. 2014).
Our study had some notable limitations. First, many parameters are likely to influence SV frequency as previously discussed. Thus, comparing SV frequency at different loci is an inherent limitation of our study design. Second, we assumed conservation of TAD borders across different cell lines (Schmitt et al. 2016; Krefting et al. 2018), as this allowed us to reuse the same sgRNAs for hESCs as for K562 cells. However, when we analyzed Hi-C interaction between the breakpoints in the proximity group in both hESCs and K562 cells, the interaction was significantly lower in hESCs, indicating less TAD conservation in hESCs than K562 cells (Supplemental Fig. S13). We also observed slight length variations in CTCF ChIA-PET interactions for each of the targeted TADs (Supplemental Figs. S1–S6). Because Hi-C and ChIA-PET data only represent the statistically most predominant average configurations within the cell population (Chang et al. 2020), this suggests that there were subtle differences in TAD extent within the K562 cell population. A recent review also challenged the notion of TAD conservation (Eres and Gilad 2021). Our assumption of TAD border conservation could therefore have led to a slight underestimation of the impact of spatial proximity.
Third, the quantification of SV frequency from sgRNA pairs targeting nonrelated TAD borders, GX, was not part of the original study design, and DNA was thus not extracted simultaneously with AB, CD, and EF for each TAD locus in K562 cells. Thus, it is important to note that inter-experimental variation could have affected our results despite the high transfection efficiencies we usually obtain in experiments with this immortal, fast-growing cell line. Moreover, we noticed that the sgRNA pairs GX appeared to have higher SV frequencies compared with the sgRNA pairs EF targeting inter-TAD loci (Figs. 1D, 2A). Although differences were not statistically significant, TAD borders are known to accumulate torsion from transcription-induced negative supercoiling of DNA (Racko et al. 2018), which can cause DSBs (Canela et al. 2017) and drive SV formation (Wu et al. 2017; Krefting et al. 2018). Potentially, this intrinsic propensity could have contributed to an overestimation of the true effect of spatial proximity in our study. Precise ligation at chimeric deletion junctions also seemed more prevalent for these sgRNA pairs compared with sgRNA pairs targeting inter-TAD loci (Supplemental Fig. S14B), indicating that nucleotide homology between TAD borders could have influenced the precise ligation analysis.
Fourth, we decided to categorize 1–2 bp indels at chimeric deletion junctions as precise ligation to account for CRISPR-Cas9-induced staggered ends (Xue and Greene 2021), yet a study of induced SVs in Arabidopsis did not (Beying et al. 2020). Such classification differences make comparisons difficult and accentuate the need for a standardized approach.
Lastly, we did not adjust for multiple testing in this study. The CRISPR-Cas9 experiments were specifically designed to test the hypothesis that spatial proximity between DSBs might affect the frequency of CRISPR-Cas9-induced SVs, which meant that multiple testing was unlikely to affect the main conclusions. However, as our observations prompted us to initiate additional investigations to explore, for example, differential DSB repair mechanisms, we cannot exclude that some of the borderline statistically significant differences could be affected by chance, which underlines the need for future studies to explore this area.
Although our work offers evidence suggesting that DSB proximity increases SV frequency, the limitations of our study highlight the need for further experimental validation to definitively determine if and how DSB proximity influences SV formation.
Methods
Experimental design
To investigate the impact of spatial proximity on CRISPR-Cas9-induced SV formation (defined as deletions or inversions occurring between two DSBs), sgRNAs were designed for TADs on Chr 2, Chr 6, Chr 10, Chr 17, Chr 21, and Chr X. At each TAD locus, sgRNA targets were identified at the four edges surrounding the interacting TAD borders (targets A, B, C, and D), at two noninteracting sites distant from the TAD borders (targets E and F) and at a CTCF-binding region that did not spatially interact with the borders of the studied TAD (targets G and H). The sgRNAs were paired for targets A and B (sgRNA pairs AB) at the proximal edges, namely, inside, of the TAD borders, for targets C and D (CD) at the distal edges, namely, outside, of the TAD borders, for targets E and F (EF) away from the TAD borders and for targets G and either A, B or H (GX) at nonrelated (i.e., nonmutually interacting) TAD borders. The G and H targets were designed in a later phase of the study, owing to reports that TAD borders could be more vulnerable to DSBs than non-TAD loci (Canela et al. 2017), and therefore were not included in the initial experiments. The targets were designed such that the linear distance in nucleotides between the sgRNAs in each pair should be approximately the same for all sgRNA pairs at each TAD. To adhere to this rule, the additional target H was designed for Chr 10 because none of the sgRNAs A, B, C, or D could be paired with G to keep the linear distance approximately the same for GX as for the other sgRNA pairs. The sgRNA pairs were tested one pair by one with no multiplexing of the sgRNA pairs in transfections to prevent unwanted interference between multiple cut sites.
The primary aim of the study was to measure the frequency of large deletions and inversions generated by CRISPR-Cas9 depending on spatial proximity of the DSBs. A secondary aim was to explore for temporal trends in the development of these SVs. Thus, CRISPR-Cas9 experiments were performed in human myeloid K562 cells with the sgRNA pairs AB, CD, EF, and GX for all six TADs (Chr 2, Chr 6, Chr 10, Chr 17, Chr 21, and Chr X) and analyzed for deletions and inversions at several time points (3, 6, 12, 24, and 48–72 h). For feasibility, this time course experiment was divided over multiple experimental sessions, one TAD at a time, so that AB, CD, and EF were repeated in the same session. The later designed sgRNA pair GX was investigated for all TADs and time points in a single experimental session separate in time from the other sgRNA pairs. Additionally, the time course experiment was repeated in a later session for the sgRNA pair EF on Chr 10 after resynthesis of the nonfunctional sgRNA E for Chr 10, which had failed in the original experiments because of production error. Thus, the experiment was divided over a total of eight sessions of transfections (n = 6 sessions for AB, CD, and EF, one TAD at a time; n = 1 session for GX, all TADs; one session for repeating EF, only the Chr 10 TAD).
For biological replication, CRISPR-Cas9 was repeated in H9 hESCs in a replication experiment that included all four sgRNA pairs (AB, CD, EF, and GX) for five of the six TADs (Chr 2, Chr 6, Chr 17, Chr 21, and Chr X) with DNA extraction at a single time point (72 h) during the same experimental session. The TAD on Chr 10 was not included in the H9-ESC experiment owing to failure of sgRNAs to generate indels in K562 cells. In addition to these primary analyses, high-throughput sequencing of the chimeric deletion junctions in K562 cells was performed to explore the sequence composition.
Locating TAD borders in the human genome with high resolution
TAD borders are discernible as clustered inter-ligation paired-end tags (PETs) in ChIA-PET for CTCF (Tang et al. 2015) and as corner dots in Hi-C heat maps (Lieberman-Aiden et al. 2009; Beagan and Phillips-Cremins 2020). Furthermore, they are characterized by accumulation of CTCF and cohesin, which can be identified by ChIP-seq. These idiosyncratic features of TAD borders allow for their precise identification using Hi-C, ChIA-PET, and ChIP-seq data (Sadowski et al. 2019).
To locate TAD borders for CRISPR-Cas9 targeting, publicly available ChIA-PET data for CTCF in K562 cells (ENCODE: ENCFF000KYD) (Tang et al. 2015) were therefore stratified for size and confidence score, and the six largest, most-confident inter-ligation PETs (average size 1.85 megabases; range 0.9–3 megabases) were selected. The inter-ligation PETs were then visualized in the UCSC Genome Browser and compared with K562 Hi-C data (Rao et al. 2014) to assess whether the PETs corresponded with TAD border characteristic corner dots in heat maps (Beagan and Phillips-Cremins 2020). Furthermore, enrichment of evolutionarily conserved CTCF and cohesin (RAD21 and SMC3) traces were confirmed using publicly available ChIP-seq data (Supplemental Table S1; Davis et al. 2018) and CTCF motif orientation assessed using the JASPAR database (Supplemental Figs. S1–S6). In case of a TAD comprising multiple inter-ligation PETs, with cohesin enrichment and convergent CTCF orientation, only the PET with the highest confidence score was chosen.
No new targets for H9 hESCs were designed owing to the assumption of TAD border conservation across cell types (Krefting et al. 2018). To validate this assumption, embryonic stem cell and K562 interaction frequencies were compared in Juicebox (Robinson et al. 2018) using publicly available data from Hi-C experiments of H1 hESCs (Dekker et al. 2017) and K562 (Rao et al. 2014). H1 hESC Hi-C data was used as a proxy for H9 hESCs. Both Hi-C maps had high read depths (>1 × 109 reads) and used the same restriction enzyme. A Hi-C score, defined as the log10 of the observed/expected ratio (Engreitz et al. 2012), was then used to compare H1 hESC and K562 interaction. Narrower bins were used for the proximity groups (5 kb) compared with the nonproximity groups (25–50 kb) to obtain enough observations for a ratio (Supplemental Fig. S13).
sgRNA design
All sgRNAs with predicted high efficiencies based on the Doench score (Doench et al. 2016) were designed using the CRISPOR online tool (Concordet and Haeussler 2018). FAIRE-seq data from ENCODE (ENCSR000DCM) (Davis et al. 2018) were used to ensure targeting of loci with similar chromatin accessibility for Cas9 (Jensen et al. 2017). The binding of Cas9 to DNA is directed by the position of the PAM on either the Watson (W) or the Crick (C) strand. Directing one sgRNA in an sgRNA pair to the W strand and the other sgRNA to the C strand (W/C orientation) has been shown to reduce potentially confounding +1 templated insertions (Guo et al. 2018). The sgRNA pairs AB, CD, and EF were therefore designed in this orientation. However, this was not possible for the sgRNA pairs GX owing to the reuse of the A or B sgRNAs.
sgRNAs were purchased from Synthego and purified Cas9-nuclease from IDT. The sequences, orientation, MIT specificity scores, and predicted efficiencies are provided in Supplemental Table S2. All sgRNA efficiencies were evaluated by indel frequencies using the ICE tool from Synthego (Conant et al. 2022) in both K562 cells (Supplemental Table S7) and H9 hESCs (Supplemental Table S8). For this experiment, amplicons were obtained from both edited and nonedited DNA extracted 48 to 72 h post electroporation (see primer sequences in Supplemental Table S5). To purify amplicons, Thermo Fisher Scientific GeneJET gel extraction or Thermo Fisher Scientific GeneJET PCR purification kits were used. Sanger sequencing was performed by Eurofins Genomics. Individual sgRNA efficiencies were then evaluated for differences between groups, and their correlation with SV frequency was assessed.
Electroporation of Cas9 RNPs
An electroporation protocol was used to transfect cells with individual sgRNA pairs (Laustsen and Bak 2019). To assemble RNPs with the sgRNA pairs, 6 µg Cas9 enzyme (0.6 µL of 10 µg/µL) was mixed with 1.6 µg (0.5 µL of 3.2 µg/µL) of each of the two sgRNAs in PCR tubes in no predefined order. RNPs were then stored at −20°C or used immediately. Before electroporation, K562 cells were grown in RPMI (+L-glutamine) with 10% fetal bovine serum (FBS) and 100 U/µg per mL of penicillin/streptomycin (P/S) in T25 flasks and passaged when confluent. H9 hESCs were grown on vitronectin XF in TeSR-E8 with 50 U/µg per milliliter of P/S, passaged weekly using PBS‐‐/EDTA (0.5 mM) buffer, and supplemented with TeSR-E8 with 10 µM ROCKi for 24 h after each passage. The morphology of the hESC colonies was evaluated by phase-contrast microscopy during maintenance. Expression of pluripotency markers POU5F1 (also known as OCT4) and TRA-1-60 was confirmed by immunocytochemistry (Supplemental Fig. S7).
For nucleofection of K562 cells, 20 µL of cells in Opti-MEM (37,500 cells/µL) with RNPs were electroporated using program CM138 on the Lonza 4D-Nucleofector and then incubated in T25 flasks with RPMI complete medium (+FBS + P/S) at 200,000 cells/mL. The sgRNA pairs were electroporated individually but simultaneously for all six different chromosomes except the sgRNA pairs GX targeting nonrelated TAD borders, as it was not part of the original design, and the sgRNA pair EF for Chr 10 (see Experimental Design). For nucleofection of H9 hESCs, areas of differentiation were aspirated before single-cell dissociation of the colonies with Accutase. Twenty microliters of cells was resuspended in P3 complete buffer (15,000 cells/µL), electroporated with RNPs using program CB-150, and split into two separate culture wells with E8 medium supplemented with 10 µM ROCKi.
DNA extraction
Genomic DNA from approximately 200,000 cells was extracted at 3, 6, 12, 24, and 72 h after electroporation to assess SV formation over time using Thermo Fisher Scientific PureLink genomic mini DNA kit. DNA was quantified on NanoDrop, normalized to 25 ng/uL, and stored at −20°C.
Quantification of SV events
A gain-of-signal ddPCR protocol was used to quantify SV formation (Watry et al. 2020). HEX reference probes were ordered from Bio-Rad, and FAM probes from IDT with 5′6-FAM/ZEN/3′IBFQ modifications (Supplemental Table S4). Primers were designed using Primer3Plus with the settings recommended for ddPCR by Bio-Rad Laboratories. Amplicon and primer specificities were evaluated with the NCBI BLAST tool (Ye et al. 2012), and Mfold was used to verify the absence of strong secondary structures at the primer binding sites (Zuker 2003). Additionally, the primer target sequences were analyzed for SNPs in K562 cells (Zhou et al. 2019). All ddPCR primer pairs were validated beforehand with regular PCR using ddPCR settings and enzyme (for primer sequences, see Supplemental Table S3).
To avoid inaccurate DNA normalization owing to K562 cells’ aneuploidy (Zhou et al. 2019), different reference HEX assays were placed in physical proximity of the FAM probe for each chromosome. This normalization approach was validated with ddPCR before proceeding to quantification of SVs (see Supplemental Fig. S8).
ddPCR protocol
For each assay, sterile PCR tubes with 1.68 µL of 25 ng/µL DNA (40 ng + 5%) from each extraction point were prepared in addition to a negative control and a no template control (NTC) with ddH2O. Then, 19.32 µL of the master mix containing all other components was added to each PCR tube, yielding a final volume of 21 µL of ddPCR mixture. Droplets containing 20 µL of ddPCR mixture were generated, transferred to a 96-well ddPCR plate, and heat-sealed. Samples were run in a Bio-Rad C1000 thermal cycler at ramp rate 2°C/sec for the following steps: (1) 10 min at 95°C, (2) 30 sec at 94°C, (3) 1 min at 60°C, and (4) 10 min at 98°C (steps 2–3 ×40). The QX200 droplet reader was used to analyze droplets, and QX manager 1.2 was used to process results. The negative control was used to gate the FAM channel (the SV detecting probe), and the NTC to gate the HEX channel (the reference).
Validation of ddPCR quantification with single-cell clones
One sample was randomly selected for single cell cloning. After the 72 h extraction, leftover cells were sorted into a 96-well V plate with 100 µL of RPMI complete medium using the BD FACSAria III and transferred to a 96-well U-plate after 2–3 weeks. Once colonies reached confluency, DNA was extracted using QuickExtract from Lucigen. PCR was conducted to detect deletions by band separation using the Phusion polymerase from Thermo Fisher Scientific (Mullis et al. 1986). The following conditions were used for PCR: (1) 30 sec at 98°C, (2) 10 sec at 98°C, (3) 30 sec at X°C, (4) 25 sec at 72°C, and (5) 10 min at 72°C (steps 2–4 repeated ×35). X was approximated using the Tm calculator from Thermo Fisher Scientific. Results can be seen in Supplemental Figure S9.
High-throughput sequencing
Twenty chimeric deletion-junctions (five from each of the AB, CD, EF, and GX sgRNA pairs) were successfully sequenced using Eurofin Genomics’ NGSelect service on the Illumina MiSeq platform. Primer sequences can be found in Supplemental Table S6. FASTQ files were analyzed using the Cas-Analyzer online tool (Park et al. 2017) with the following parameters: R: 113, n: 0, r: 5. R = 113 would set the analysis range using linker sequences of 13 bp, allowing 100 bp upstream of and downstream from the ligation site to be analyzed for resection and insertion events; n = 0 would set the minimum frequency for an event, whereas r = 5 would define a wild-type sequence of 10 bp around the ligation site that would mark a read as perfect repair if present. These parameters were used to homogenize readouts across differences in amplicon lengths. Adjusting the position of the linker sequences allowed us to quantify ddPCR sensitivity (Supplemental Fig. S10).
All chimeric deletion junctions were assessed for precise ligation and microhomology usage using an R script from Beying et al. (2020). Precise ligation was defined as the proportion of reads with no resections or insertions at the predicted ligation site, normalized to the total number of reads. Reads with 1–2 bp insertions were included in this precise ligation definition to account for templated insertions (Guo et al. 2018) and so were reads with 1–2 bp resections to account for Cas9-generated staggered ends (Xue and Greene 2021). The R script analyzed microhomology usage by comparing sequences before and after a resection for homology. A read was classified as using microhomology if one or more matching bases were found at the resection junction. Reads identified as using microhomology for ligation were then normalized to the subset of reads with resection.
Statistical analysis
The results were reported as group medians with error bars showing the interquartile range owing to the nonnormal distribution of the data. Statistical comparisons were made with nonparametric tests. Comparisons between groups were made with the two-sided Mann–Whitney U test. Comparisons within the same samples were made with the two-sided Wilcoxon matched-pair signed-rank test. Correlation was assessed using Spearman's rank correlation coefficient. Statistical significance was considered at a significance level of 0.05. Statistical tests were performed in GraphPad Prism.
Data access
The high-throughput sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1117804.
Competing interest statement
R.O.B. is a cofounder of and consultant to UNIKUM Tx and is coinventor on patents and patent applications related to gene editing. UNIKUM Tx was not involved in the present study.
Acknowledgments
We thank Holger Puchta's laboratory at the Karlsruhe Institute of Technology for help with analyzing high-throughput sequencing data. We thank Mark Denham's laboratory at Aarhus University for H9 hESC cells. This work was funded (U.B.J.) by the Independent Research Fund Denmark (grant no. 1149-00024B and 9039-00337B).
Author contributions: Conceptualization was by U.B.J. Methodology was by M.D.-J., T.T., and R.O.B. Formal analysis was by M.D.-J., T.T., R.O.B., and U.B.J. Investigation was by M.D.-J., T.T., R.O.B., and U.B.J. Resources were by R.O.B. and U.B.J. Writing of the original draft was by M.D.-J., U.B.J., and T.T. Review and editing were by M.D.-J., T.T., R.O.B., and U.B.J. Visualization was by M.D.-J., T.T., and U.B.J. Supervision was by T.T., R.O.B., and U.B.J. Funding acquisition was by U.B.J. and R.O.B. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278575.123.
- Received September 28, 2023.
- Accepted January 8, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








