
Estimating the size of long tandem repeat expansions from short reads with ScatTR
- Rashid Al-Abri1,2 and
- Gamze Gürsoy1,2,3
Abstract
Tandem repeats (TRs) are sequences of DNA in which ≥2 bp are repeated back-to-back at specific locations in the genome. TR expansions, in which the number of repeat units exceeds the normal range, have been implicated in more than 50 conditions. However, accurately measuring the copy number of TRs is challenging, especially when their expansions are larger than the fragment sizes used in standard short-read genome sequencing. Here, we introduce ScatTR, a novel computational method that leverages a maximum likelihood framework to estimate the copy number of large TR expansions from short-read sequencing data. ScatTR calculates the likelihood of different alignments between sequencing reads and reference sequences that represent various TR lengths and employs a Monte Carlo technique to find the best match. In simulated data, ScatTR outperforms state-of-the-art methods, particularly for TRs with longer motifs and those with lengths that greatly exceed typical sequencing fragment sizes. When applied to data from the 1000 Genomes Project, ScatTR detects potential large TR expansions that other methods missed, highlighting its ability to better characterize genome-wide TR variation.
Tandem repeats (TRs) are consecutively repeated nucleotide sequence motifs that are widespread throughout the human genome. Studying TR variation in the human population is important owing to the high mutation rate and polymorphic nature of TRs (Press et al. 2014). TRs influence key biological processes, including gene regulation, and are implicated in a range of diseases, including neurodegenerative conditions, cancer, and neurodevelopmental and psychiatric disorders (Gymrek and et al. 2016; Song et al. 2018; Trost et al. 2020; Zhou et al. 2022; Erwin et al. 2023). Pathogenic repeat expansions vary widely in size, with size often having a correlation with earlier onset and more severe disease phenotype. For example, a CGG expansion of more than 200 units located in the 5′ untranslated region of FMR1 is known to cause Fragile-X-associated tremor/ataxia syndrome (FXTAS) (Willemsen et al. 2011), and the number of repeats correlates with earlier onset of tremor and ataxia (Tassone et al. 2007; Leehey et al. 2008). Expansions in RFC1, which cause cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS), typically exceed 250 repeats, with larger expansions also linked to earlier onset and more severe symptoms (Cortese et al. 2019; Currò et al. 2024). Moreover, some repeat expansions can be much larger. In Huntington's disease, CAG expansions in HTT can grow somatically from 40 to more than 500 repeats, driving neurodegeneration once a critical threshold is surpassed (Aziz et al. 2012). Myotonic dystrophy type 1 (DM1), caused by DMPK expansions, can range from 50 to more than 3000 units, with disease severity increasing alongside expansion size (Brook et al. 1992). Moreover, an expansion in the intron of C9orf72 is known to cause amyotrophic lateral sclerosis (ALS) (Cruts et al. 2013). Its size can exceed 2100 repeat units, with size also correlating with DNA methylation status and age of onset (Gijselinck et al. 2016). Despite their critical role in biological processes and their link to various diseases, the precise lengths of large TR expansions remain underexplored owing to significant technical challenges.
Studying repetitive DNA sequences has historically been hindered by the lack of tools. Many genomic studies utilize short-read-based whole-genome sequencing (WGS) but exclude half of the data with RepeatMasker, a tool that identifies repetitive DNA sequences for removal. Currently, >56% of the human genome is removed with RepeatMasker (Nishimura 2000). Repeat expansions are extremely difficult to detect owing to various reasons (Martorell et al. 1997). They can be highly variable in size and location, making them difficult to detect through simple alignment-based methods. Their sizes can be much larger than a typical fragment length in short-read-based WGS data and usually occur in regions of the genome that are difficult to align. To overcome these challenges, various computational methods have been developed to identify repeat expansions from short-read sequencing data. Current tools, such as STRling (Dashnow et al. 2022), ExpansionHunter (Dolzhenko et al. 2019), and GangSTR (Mousavi et al. 2019), use likelihood-based approaches to estimate TR copy number from short-read sequencing data but struggle with accuracy when the TR length exceeds the lengths of the fragments used in short-read sequencing. Another tool, Dante, introduces an alternative alignment-free approach that applies hidden Markov models to model repeat sequences and corrects for polymerase-induced stutter artifacts. Although Dante provides robust genotyping for complex TR loci, its accuracy is similarly limited for TRs that extend beyond the read length (Budiš et al. 2019). However, many pathogenic TRs expand to hundreds of base pairs, well above the fragment length in typical short-read data (Hannan 2018). Moreover, these methods are constrained by the length of the TR unit (i.e., motif) they can analyze: STRling is restricted to motifs between 2 and 6 bp, whereas ExpansionHunter and GangSTR support repeat unit sizes from 2 to 20 bp. Although newer methods like TRGT overcome these limitations by leveraging long-read sequencing data (Dolzhenko et al. 2023), short-read sequencing data are still the overwhelming majority of currently available sequencing data owing to its financial feasibility. Thus, there is still a need for a computational method that can accurately estimate TR copy numbers that are larger than fragment lengths.
In this study, we introduce ScatTR, a novel computational method designed to accurately estimate the copy number of large TRs from short-read WGS data. ScatTR uses a maximum likelihood framework to evaluate alignments of reads to reference sequences with variable TR copy numbers and applies a combination of Monte Carlo–based simulated annealing and golden section search (GSS). The goal of this study is to provide a method that enables accurate copy number estimation of long TRs that exceed fragment length, addressing a key gap in the current short-read analysis toolkit.
Results
Overview of the ScatTR method
ScatTR estimates the copy number of TR regions in a genome by reformulating copy number estimation as an optimization problem that considers both mapped and unmapped reads from aligned paired-end short-read WGS data (Fig. 1A). It uses a predefined catalog of TR loci that specifies the chromosome, the start and end positions, and the repeat motif for each region of interest. For each TR locus, ScatTR identifies relevant read pairs by scanning the alignment file for (1) pairs in which at least one mate overlaps the flanking regions (defined as a window of fragment-length size immediately upstream or downstream of the repeat locus) or (2) read pairs, mapped or unmapped, whose sequences show high similarity to the repeat motif using a weighted-purity score (see Methods) (Dolzhenko et al. 2017). All rotations of the motif and its reverse complement are taken into account to ensure sensitivity, because certain repeats can appear differently depending on strand orientation and sequence context. For instance, for a CAG repeat, we check for the presence of the rotations CAG, AGC, and GCA in addition to the reverse complements of each rotation. These read pairs, collectively called the “bag of reads,” may include “anchored” in-repeat read (IRR) pairs (in which one mate maps to the flanking region whereas the other lies within the repeat), “fully IRR” pairs (in which both mates show high similarity to the repeat motif and are often unmapped owing to high repetitiveness), and spanning pairs (in which both mates are mapped within the flanking regions, surrounding but not overlapping the repeat). This approach differs from many existing methods by explicitly incorporating fully IRR pairs, which provide valuable information when the repeat region exceeds the sequencing fragment length.
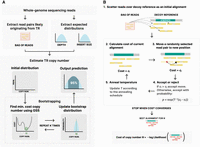
Overview of the ScatTR method. (A) Reads likely originating from the TR locus are collected from reference-aligned WGS data that include both mapped and unmapped reads to form the “bag of reads.” Additionally, the expected read depth and insert size distributions are extracted. These distributions are used as parameters in the likelihood function. TR copy number estimation is done with bootstrapping. ScatTR iteratively finds the copy number that minimizes a cost function using golden section search (GSS), and the result from each iteration is used to update a distribution of copy number estimates. After multiple iterations, this distribution is used to report a final estimate and a 95% confidence interval. (B) For a given copy number, ScatTR evaluates the cost by finding the best alignment to a decoy reference with the given number of repeat units. It starts with an initial alignment and then updates the alignment using Monte Carlo moves to reduce the cost and accept changes based on a probability function. This process is done via simulated annealing. It continues until convergence, yielding the best alignment for the given TR copy number. The best alignment is used to calculate the cost of the copy number, which is what GSS minimizes in A.
Once the relevant sequencing reads are collected, ScatTR estimates how many times a TR is repeated in a given region. To do this, it builds a series of test DNA sequences, called decoy references, for different possible repeat lengths. Each decoy reference combines the DNA sequence surrounding the repeat with a specific number of repeat units. The goal is to figure out which decoy reference most closely matches the observed sequencing data (Fig. 1B). ScatTR uses a maximum likelihood framework to do this, identifying the repeat length that makes the observed reads most likely. Although the most accurate method to calculate the likelihood would take into account every possible way the reads could align to each decoy, doing so would take far too much computing time. Instead, ScatTR approximates the likelihood by searching for the single best way the reads could align to each decoy and then using that to estimate how well the decoy fits. This search is done using a technique called simulated annealing (Kirkpatrick et al. 1983), which explores different possible alignments and occasionally accepts worse ones to avoid getting stuck in a suboptimal solution. The method aims to minimize a cost function, which reflects how well the proposed alignment explains the data. This function considers three things: how well the reads match the DNA sequence in the decoy, whether the spacing between read pairs looks typical for the sample, and whether the number of reads in the region is consistent with overall read depth across the genome. ScatTR learns what is typical for spacing and read depth by sampling high-quality regions of the genome from the same sequencing data.
Once ScatTR estimates how well the reads match a given repeat length, it compares that result to other possible repeat lengths to find the best fit, specifically, the one that gives the highest likelihood (or lowest cost). To speed up this process, ScatTR includes several optimizations. For instance, it avoids repeating calculations by storing and reusing alignment scores for reads that fall entirely within the repeat region. Instead of exhaustively testing every possible repeat length, which would be slow, ScatTR uses an efficient strategy called GSS (Kiefer 1953). This method assumes there is a single best answer within a given range and progressively narrows that range to find it faster. To kick off this search, ScatTR makes a rough estimate of the repeat length using the average sequencing depth and the number of reads that fall inside the repeat region. To improve reliability, ScatTR repeats the alignment and search process multiple times, a technique known as bootstrapping. This not only helps refine the final repeat length estimate but also provides a measure of confidence in the result. As a result, ScatTR reports both the most likely repeat count and a confidence interval showing how precise that estimate is (for details of the formulations and the algorithm, see Methods).
ScatTR outperforms existing methods on simulated data
We compared our method against STRling, ExpansionHunter, and GangSTR, in addition to a closed-form solution (see Methods) by calculating the root mean square error (RMSE) between the predicted and true TR copy numbers (Mousavi et al. 2019; Dolzhenko et al. 2020; Dashnow et al. 2022). Specifically, for each true copy number, we compute RMSE across all simulated samples with that same true copy number. A lower RMSE indicates that a method produces more accurate predictions. We sampled a subset of 30 TR loci identified by Tandem Repeats Finder (TRF) with motif lengths between 2 and 20 bp (Benson 1999) and simulated and reference-aligned short-read paired-end WGS samples with expansions for these loci (Supplemental Table S1). We simulated the samples with 30× coverage and a sequencing error profile based on the Illumina HiSeq X platform (see Methods). We simulated homozygous and heterozygous expansions in 30 TR loci with copy numbers ranging from 200 to 1000 for a total of 540 samples. We compared the predictions with respect to heterozygosity of TR and repeat motif length. We observe that ScatTR's predictions yield the lowest RMSE across all tested heterozygous TR copy numbers and eight out of nine homozygous TR copy numbers (Fig. 2A). Additionally, we observe that the copy number of heterozygous TRs is generally better predicted than those of homozygous TRs in all methods including ScatTR. Moreover, we show that the RMSE of ScatTR's predictions is consistent across different TR copy numbers.
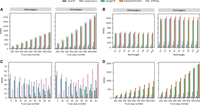
Benchmarking accuracy against the state-of-art with simulated data. We estimated the TR copy number using state-of-the-art methods (ExpansionHunter, GangSTR, and STRling), ScatTR, and a closed-form solution and calculated the root mean square error (RMSE) between the predicted and the true copy number of TRs on simulated data. (A) RMSE across samples compared with true copy numbers for large TRs with motif lengths between 2 and 20 bp. Heterozygous expansions on the left and homozygous on the right. These estimates were conducted on 540 simulated short-read WGS samples representing 30 TR loci and a range of known copy numbers (200–1000). (B) RMSE as a function of TR motif length for A. (C) RMSE across samples compared with true copy number of small TRs with motif lengths between 2 and 20 bp. Heterozygous expansions on the left and homozygous on the right. These estimates were conducted on 540 simulated short-read WGS samples representing 30 TR loci and a range of known copy numbers (five to 45). (D) RMSE across samples compared with true copy numbers for large TRs with motif lengths between 21 and 50 bp. These estimates were conducted on 540 simulated WGS samples with TR expansions, representing 30 TR loci and a range of known copy numbers (200–1000).
We further evaluated how motif length affects prediction accuracy for large TRs across all tested copy numbers (Fig. 2B). In both heterozygous and homozygous cases, ScatTR consistently achieves the lowest RMSE for all tested motif lengths, with only a small increase in error as motif length grows. In contrast, all other methods show higher RMSEs, particularly for homozygous expansions.
Although ScatTR is not designed to estimate the copy number of TRs shorter than the fragment length, we wanted to understand its accuracy relative to the state-of-the-art methods in such cases. Similar to our benchmarking method for large TRs, we simulated heterozygous and homozygous expansions in 30 TR loci with copy numbers ranging from five to 45 for a total of 540 samples. We observe that ScatTR underperforms compared with other methods for TRs shorter than the fragment length, whereas ExpansionHunter consistently maintains the lowest RMSE across different copy numbers for these short TRs (Fig. 2C).
Next, although ExpansionHunter and GangSTR were not specifically designed for large motifs, we wanted to test ScatTR's performance on long motifs compared with these tools. STRling was excluded from this analysis because it does not allow users to input motifs >6 bp. For this test, we selected 30 TR loci with repeat units ranging from 21 to 50 bp (see Supplemental Table S2). We again simulated homozygous and heterozygous expansions, this time with copy numbers ranging from 200 to 1000, yielding another 540 samples. In this large-motif setting, ScatTR achieved the lowest RMSE across the board (Fig. 2D).
ScatTR captures mismapped IRRs more effectively than existing methods
Accurately capturing and mapping IRRs is a key challenge in TR copy number estimation (Gymrek et al. 2016; Dolzhenko et al. 2017; Dashnow et al. 2022). IRRs originating from a TR of interest may be mismapped to other genomic regions or remain unmapped altogether. To assess ScatTR's ability to recover these reads, we identified the ground truth IRRs for the 30 repeats in the 540 simulated samples described earlier and compared them to the IRRs extracted by ScatTR. Across all samples, ScatTR achieved a mean precision of 0.999 (SD = 0.003) and a mean recall of 0.954 (SD = 0.0635), indicating that it captures nearly all true IRRs (Supplemental Fig. S1).
Some existing tools, such as ExpansionHunter and GangSTR, attempt to improve IRR retrieval and mapping by incorporating user-defined off-target regions, genomic regions where IRRs are likely to mismap (Gymrek and et al. 2016; Dolzhenko et al. 2020). These methods scan predefined off-target regions to recover additional IRRs before estimating the TR copy number. In contrast, ScatTR scans the entire WGS alignment file (CRAM/BAM/SAM) without requiring predefined off-target regions, allowing it to adaptively identify relevant reads. To evaluate how ScatTR performs relative to ExpansionHunter and GangSTR when off-target regions are used, we analyzed TR loci when each tool defines such regions. ExpansionHunter's catalog includes C9orf72 and FMR1, whereas GangSTR's catalog contains 12 TR loci with off-target regions, including SCA1–3, SCA6–8, SCA12, SCA17, HTT, DM1, FMR1, and C9orf72. We simulated WGS samples with varying copy numbers for each locus and estimated TR copy numbers using ExpansionHunter and GangSTR both with and without off-target regions. As shown in Figure 3A, ScatTR's RMSE was lower than 90.57 and 145.57 across all copy numbers for heterozygous and homozygous cases, respectively, whereas GangSTR's RMSE was much higher with off-target regions seeming to slightly increase RMSE. Additionally, in Figure 3B, we see that off-target regions help decrease ExpansionHunter's RMSE, but it is still much higher than ScatTR's RMSE, which is less than 180.08 and 249.90 across all copy numbers for heterozygous and homozygous cases, respectively. ScatTR consistently outperforms ExpansionHunter and GangSTR, even when off-target regions were incorporated.
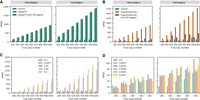
Impact of off-target regions, mutation rates, and sequencing errors on ScatTR accuracy. (A) RMSE of ScatTR compared with GangSTR, with and without off-target regions, in 108 simulated samples across different true copy numbers. Heterozygous cases are shown on the left and homozygous on the right. The simulated samples represent expansions in 12 TR loci for which GangSTR provided off-target regions, including SCA1–3, SCA6–8, SCA12, SCA17, HTT, DM1, FMR1, and C9orf72. (B) RMSE of ScatTR compared to ExpansionHunter, with and without off-target regions, in 18 simulated samples across different true copy numbers. Heterozygous cases are shown on the left and homozygous on the right. The simulated samples represent expansions in two TR loci for which ExpansionHunter provided off-target regions, including C9orf72 and FMR1. (C) RMSE of ScatTR across varying mutation rates in the repeat sequence. (D) RMSE of ScatTR across different sequencing error rates in whole-genome sequencing (WGS) samples.
ScatTR is robust against expected mutations and sequencing errors
To assess ScatTR's robustness to mutations within the repeat sequence, we selected five TR loci with motif lengths ranging from 2 to 20 bp (Supplemental Table S3). We generated WGS samples with homozygous and heterozygous repeat expansions, simulating copy numbers between 200 and 1000. For each locus, we systematically introduced varying levels of mutations within the repeat region to test the impact on performance. As expected, the accuracy of the ScatTR improved as the mutation rate decreased (Fig. 3C). We then calculated the expected mutation rate as 0.0047 at these loci using long-read sequencing data across five 1000 Genomes Project samples (see Methods) (Logsdon et al. 2025). At the expected mutation rate, we found that the highest RMSE for ScatTR was 193.32 across all tested copy numbers, which is comparable to 177.59 when there are no mutations present. This indicates that our method effectively estimates TR copy numbers even in the presence of naturally occurring sequence variation.
To assess the effect of sequencing errors on ScatTR's performance, we used the same 30 loci as introduced previously to simulate and align WGS samples with varying sequencing error rates. The read simulator we used, ART, allows for overriding the sequencing error rate with a user-specified value (Huang et al. 2012). The Illumina HiSeq X platform error profile, as provided by ART, assumes a sequencing error rate of approximately 0.0012. We simulated samples at multiples of this baseline, up to 16 times (i.e., 0.0192), and measured ScatTR's accuracy. As shown in Figure 3D, ScatTR's RMSE remains largely stable across increasing error rates. For example, at a homozygous copy number of 300 units, the largest observed difference in RMSE between error rates of zero and 0.0048 was only 54.35 bp, small relative to the overall size of the repeats. Furthermore, when examining absolute error, we observed substantial overlap in error bars across all error rates (Supplemental Fig. S2). This suggests that the variations are primarily owing to stochastic effects in read simulation and ScatTR's probabilistic inference rather than to a systematic decline in performance.
Genome-wide profiling of large, expanded TRs in the human population
To demonstrate the utility of ScatTR, we used 2495 PCR-free high-coverage WGS data from the 1000 Genomes Project (Byrska-Bishop et al. 2022). Because ScatTR and the other state-of-the-art methods require a catalog (except STRling), we first identified TR expansions larger than the read length with ExpansionHunter Denovo (Dolzhenko et al. 2020), which detects novel repeats from WGS data and reports the number of fully IRR pairs. Because it only provides approximate coordinates, we overlapped the identified expanded TR regions with repeat regions identified by TRF to find their exact coordinates. We retained loci that had at least one fully IRR pair in a minimum of 50 samples, resulting in eight TR loci. ScatTR estimated lengths larger than the fragment length (400 bp) for six out of the eight identified loci (Fig. 4A). In contrast, other methods consistently predict the lengths as shorter than or around the fragment length for the identified TR loci.
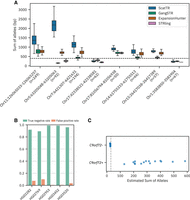
Evaluation of ScatTR on real WGS data. (A) Predicted sum of allele lengths (in base pairs) of TR expansions by ScatTR and state-of-the-art methods. The labels on the x-axis indicate the sample size for each locus. All loci have motif lengths >6 bp, except for the locus on Chromosome 5. The mean fragment length of 400 bp is shown as a horizontal line. (B) True-negative rate and false-positive rate of TR expansion calls for five samples with long-read ground truth across 171146 TR loci. Expansions are defined as exceeding the fragment length threshold of 400 bp. (C) Predicted sum of allele lengths (in base pairs) for 30 ALS patients based on short-read WGS data. The data set includes 15 samples with known C9orf72 expansions (C9orf72+) and 15 without (C9orf72−). The vertical line indicates the pathogenic cutoff of 30 repeat units. All samples were correctly classified.
The fully IRR pairs originating from other TR loci might contaminate the bag of reads if there are multiple TRs with the same motif. Therefore, we also assessed the false-positive rate of expansions larger than the fragment length called by ScatTR. We used five 1000 Genomes Project samples that have both long-read (Logsdon et al. 2025) and short-read WGS available. We applied TRGT (Dolzhenko et al. 2023) to the long-read data to measure the lengths of 171,146 TR loci from the TRGT catalog (see Methods) and used ScatTR to estimate the same loci from short-read data. To evaluate performance, we classified each locus as expanded or not based on whether its estimated length exceeded the fragment length. We then compared these classifications between ScatTR and TRGT. Treating TRGT as a gold standard, the true-negative rate reflects the proportion of loci that both methods deemed as not expanded, whereas the false-positive rate reflects the proportion of loci classified as expanded by ScatTR but not by TRGT. As shown in Figure 4B, the mean false-positive rate was very low (0.047).
ScatTR correctly identifies samples with C9orf72 pathogenic repeat expansions
To further validate the utility of ScatTR, we assessed its ability to estimate the size of a well-characterized pathogenic repeat expansion in C9orf72 gene, a biomarker for familial ALS and frontotemporal dementia. The expansion consists of a hexanucleotide GGGGCC repeat (HGVS notation: NC_000009.12:g.27573529_27573546(GGGGCC)[>60]). We analyzed short-read WGS data from 30 ALS patients (Kenna et al. 2016), including 15 individuals with the known expansion (C9orf72+) and 15 without (C9orf72−). As shown in Figure 4C, ScatTR effectively distinguished C9orf72+ patients from C9orf72− patients with 100% accuracy. This demonstrates that ScatTR can reliably identify large pathogenic repeat expansions from short-read data.
To compare ScatTR's performance against existing tools in this clinical setting, we also analyzed the ALS cohort with ExpansionHunter, GangSTR, and STRling without using predefined off-target regions, as would be typical when analyzing a novel or uncharacterized locus. These methods consistently underestimated the repeat copy number in the C9orf72+ samples, with most estimates falling below the pathogenic cutoff of 30 repeats (Supplemental Fig. S3). In contrast, ScatTR estimated copy numbers well above the pathogenic threshold for all 15 positive samples, correctly classifying them as expanded.
Computational requirements
To evaluate ScatTR's runtime performance, we benchmarked its execution time against state-of-the-art methods on five WGS samples from the 1000 Genomes Project (Byrska-Bishop et al. 2022): HG03520, HG02282, HG02953, HG02769, and HG03452. Each tool was tested on randomly subsampled TR catalogs of varying sizes (one, 10, 100, 1000, and 10,000 loci) from the TRF catalog. All benchmarks were conducted on a Dell PowerEdge R6625 with 2× AMD EPYC 9254 24-core processors, operating in single-threaded mode. Memory usage was negligible across all methods.
As shown in Figure 5A, ScatTR required an average of 27.66 min per sample to estimate the copy number for one locus. Alternative tools such as GangSTR and ExpansionHunter completed the task significantly faster, whereas STRling performed more efficiently than ScatTR, requiring about half the execution time. However, the majority of ScatTR's runtime, an average of 26.94 min, was spent on read extraction.
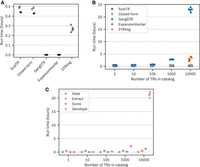
Runtime evaluation of ScatTR in simulated and real WGS samples (A) Measured runtime (hours) for estimating TR copy number at a single locus across five real WGS samples. Reported times are the mean of five runs. (B) Measured runtime (hours) for estimating TR copy numbers across varying catalog sizes in five WGS samples, comparing ScatTR, state-of-the-art methods, and a closed-form solution. (C) Breakdown of ScatTR runtime by processing step for B.
As TR catalog size increased, so did the runtime for all methods. For ScatTR, the read extraction and expected distribution extraction steps remained constant, averaging 25.20 min and 0.57 min per sample, respectively. However, the read scoring step (which calculates alignment scores for reads in the bag against decoy references; see Methods) and the genotyping step (which involves multiple bootstrap runs of GSS and Monte Carlo simulations; see Methods) varied with catalog size. At the largest catalog size (10,000 loci), ScatTR required an average of 22.94 h per sample, whereas the next slowest tool, ExpansionHunter, averaged 2.97 h (Fig. 5B). A breakdown of ScatTR's runtime by processing step (Fig. 5C) shows that most of the time was spent on scoring and genotyping. On average, ScatTR processed one locus in 8.08 sec compared with ExpansionHunter's 1.06 sec per locus.
Discussion
We present ScatTR, a novel method for estimating the copy number of TRs that exceed the fragment length in paired-end short-read WGS data. ScatTR introduces an innovative approach for estimating TR copy numbers. Unlike prior methods that classify reads into distinct categories such as flanking, enclosing, spanning, or anchored IRR pairs (Gymrek et al. 2012; Dolzhenko et al. 2017; Tang et al. 2017; Willems et al. 2017; Dashnow et al. 2018; Tankard et al. 2018; Mousavi et al. 2019), ScatTR uses a more flexible and comprehensive approach, overcoming key limitations of these classification-based strategies.
Existing methods typically exclude fully IRR pairs from copy number estimation unless the user specifies off-target regions where these pairs may have been incorrectly mapped. Our approach integrates fully IRR pairs directly into the estimation process. The closed-form solution, which incorporates fully IRR pairs, demonstrates significant improvements in accuracy for large TRs. However, incorporating these reads alone is insufficient for achieving high accuracy, necessitating the development of a more flexible likelihood function to further refine copy number estimates.
ScatTR adopts a likelihood model inspired by genome assembly methods. In genome assembly, the likelihood of an assembly is based on all read alignment combinations to the assumed true genome assembly (or reference). Recent methods approximate this likelihood by only using a few best read alignment combinations acquired using traditional read alignment methods such as BWA (Boža et al. 2015). However, ScatTR approximates the likelihood for a copy number using only the best alignment. Finding the best alignment to a decoy reference (containing a large repetitive region) is an ambiguous task because reads from within the repeat can perfectly align to multiple locations along the decoy reference. Thus, traditional read alignment methods are not helpful for finding the best alignment in TR copy number determination. This is also why it is difficult to assemble repetitive parts of genomes using short reads (Treangen and Salzberg 2012). ScatTR overcomes this challenge by using Monte Carlo sampling to efficiently find the best alignment to decoy references.
Our results show that our approach enables accurate estimation of TR lengths that exceed typical fragment lengths. ScatTR outperforms state-of-the-art methods for larger TRs and longer motifs up to 50 bp. Unlike classification-based approaches, ScatTR's model does not rely on directly counting repeat units within read sequences, allowing it to accommodate longer motifs with greater flexibility. Additionally, ScatTR offers the added advantage of generating a plausible alignment of reads to the predicted TR copy number. This alignment capability could facilitate deeper analysis of repeat mosaicism and interruptions. However, further testing is required to fully assess the utility of these alignment solutions.
Although ScatTR is computationally more demanding than existing approaches, this trade-off is a direct consequence of its design to maximize accuracy for large TRs. ScatTR processes all available reads spanning the repeat region, including IRRs that may be unmapped or mismapped elsewhere in the genome. This requires scanning the entire referenced-aligned NGS data, including both mapped and unmapped reads, which increases computational load. Additionally, ScatTR's Monte Carlo approach improves accuracy beyond that of closed-form solutions based solely on IRR counts. This exhaustive strategy is particularly valuable for large pathogenic TR expansions, in which precise repeat length determination might be critical for clinical interpretations. Given that no current method accurately genotypes very large TR expansions using short-read sequencing, we believe the trade-off between computational efficiency and accuracy is well justified, particularly for targeted analyses of TR loci.
Although ScatTR excels in estimating simple TRs, it does not currently handle complex repeat structures that consist of multiple motifs or known interruptions. This limitation arises from the efficiency optimizations in ScatTR that assume a simple repeat structure. Notably, tools like STRling and GangSTR also do not support complex loci, whereas ExpansionHunter and Dante can handle such loci for short TRs, provided that the repeat structure is accurately defined. Future research could focus on algorithmic enhancements to extend ScatTR's applicability to complex loci, an important category of TR variation with significant implications for human genetic studies and disease research.
Methods
The ScatTR algorithm
In this section, we detail the steps of the ScatTR algorithm as shown in Figure 1. First, we describe how the expected distributions are extracted and then how the bag of reads is extracted, both of which are used as inputs to the copy number estimation process.
Next, we derive the likelihood function, which is optimized during the estimation process. To compute the likelihood of a copy number, the best alignment of the bag of reads needs to be found. We describe a Monte Carlo process to find the best alignment. Then, we describe an optimization process to efficiently search the space of all copy numbers to find the one with the highest likelihood value.
Expected depth and insert size distributions
To extract the expected read depth distribution from WGS short-read data, we sample aligned reads from 100 random regions, excluding those with an average mapping quality ≤60. Each position's depth within the sampled regions contributes to the distribution. Then, the distribution is trimmed to the 99th percentile to remove outlier depth values.
To extract the expected insert size distribution from WGS short-read data, we sample aligned reads from 100 random regions. A read's observed insert size, as determined by the aligner, is included in the distribution if the read meets specific criteria: It must be a primary read, must be part of a proper pair, must be unclipped, and have a mapping quality of at least 60.
Extracting the bag of reads
We extract paired-end reads that are likely to originate from the TR locus and refer to them collectively as the “bag of reads.” These reads fall into three categories: anchored IRR pairs, fully IRR pairs, and spanning pairs.
Anchored IRR pairs consist of one mate that originates from within the repeat region and the other from either the left or right flanking region, thereby “anchoring” the pair to the repeat locus. The flanking regions are the sequences immediately adjacent to the repeat. Anchored IRR pairs occur only when the repeat is longer than the read length; otherwise, it would be impossible to observe a read that originates entirely within the repeat.
Fully IRR pairs are those in which both mates originate entirely from within the repeat region. These occur only when the repeat is longer than the fragment length; otherwise, such pairs would not be observed.
Spanning pairs are those in which both mates originate entirely from the flanking regions, surrounding but not overlapping the repeat itself. These pairs provide additional information about the repeat's boundaries and are included in our analysis.
To extract these read pairs, we scan the alignment file (BAM/CRAM/SAM) for reads with at least one mate mapped to the flanking regions of the repeat (a window of read-length size around the reference repeat region). This process captures both anchored IRR pairs and spanning pairs. Fully IRR pairs are identified separately by testing all read pairs with a mapping quality ≤40. If both mates in a pair are classified as IRRs, the pair is also added to the bag of reads.
A read is classified as an IRR if its sequence matches with the expected repeat pattern closely, even after being rotated or reverse complemented. Dolzhenko et al. (2017) describe a weighted purity (WP) score to quantify how closely a read matches the expected repeat pattern. To compute the WP score, we compare the read to the nearest perfect repeat sequence across these transformations (e.g., a CAG repeat can manifest as CAG, AGC, or GCA in the forward direction or as CTG, TGC, or GCT in the reverse direction). The WP score accounts for both matching bases and mismatches, with scores of one for matches, 0.5 for low-quality mismatches, and −1 for high-quality mismatches. The resulting score is normalized by the read length to produce a WP value between −1 and one. Reads with a WP score above the threshold are deemed likely to originate entirely from the repeat region. Here we used a threshold of 0.9 (default in our tool).
Likelihood of a copy number
To determine the most probable TR copy number from a given bag of reads (i.e., set of sequencing reads), we derive the likelihood function that quantifies how well different candidate copy numbers explain the observed data. This process involves defining the probability of observing a set of reads given a proposed copy number and an alignment of those reads to a reference sequence (which we call a decoy reference). In this section, we introduce a general probability model that accounts for all possible alignments of the reads. Next, we refine this model by identifying the best alignment, which allows us to approximate the likelihood function more efficiently. By breaking down the likelihood into its component probabilities, we establish a structured framework for computing the likelihood in a way that is both theoretically sound and computationally feasible. Finally, we put together these building blocks to define the full form of the likelihood that ScatTR uses and adapt it to account for diploid genomes.
Here, we will derive Pr(R∣C), the probability (or likelihood) that a bag of reads R is observed assuming that C is the true TR copy number from which R was sequenced. In the ScatTR algorithm, we use this likelihood to compare proposed copy numbers for a given bag of reads with the goal of finding the copy number that maximizes the likelihood. The TR copy number that maximizes the likelihood is the most probable TR copy number given the bag of reads.
To show that maximizing the likelihood, Pr(R∣C), is equivalent to maximizing the probability of the TR copy number given the bag of reads, Pr(C∣R), we apply Bayes’ rule: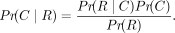 (1)
In the equation, Pr(C) is the prior probability of the TR copy number. We assume that this prior probability is constant across the set of reasonable
copy numbers for a given R. Pr(R) is the prior probability of observing the bag of reads. Because our primary goal is to compare the likelihood of various
copy numbers for the same bag of reads, we can assume Pr(R) is a constant. Therefore, for the purpose of comparing TR copy numbers, maximizing the values Pr(C∣R) and Pr(R∣C) over C is equivalent.
(1)
In the equation, Pr(C) is the prior probability of the TR copy number. We assume that this prior probability is constant across the set of reasonable
copy numbers for a given R. Pr(R) is the prior probability of observing the bag of reads. Because our primary goal is to compare the likelihood of various
copy numbers for the same bag of reads, we can assume Pr(R) is a constant. Therefore, for the purpose of comparing TR copy numbers, maximizing the values Pr(C∣R) and Pr(R∣C) over C is equivalent.
Previous work has typically modeled the likelihood Pr(R∣C) by categorizing reads into distinct classes like flanking, enclosing, spanning, or anchored IRR pairs (Gymrek et al. 2012; Dolzhenko and et al. 2017; Tang et al. 2017; Willems et al. 2017; Dashnow et al. 2018; Tankard et al. 2018; Mousavi et al. 2019). In these studies, the likelihood is determined by both the count of read pairs in each class and a characteristic specific to that class. In contrast, we do not define the likelihood by classifying reads into classes and modeling their characteristics. Instead, we use an approach similar to the one used in genome assembly problems, in which the likelihood is based on the alignments of the observed reads to the given genome assembly (i.e., TR copy number in our case).
The task of identifying the TR copy number that maximizes the likelihood of the observed reads can be reframed as a genome assembly problem. In genome assembly, estimating the most likely genome sequence inherently involves determining the correct number of repeat units. By searching for the genome sequence that best explains the read data, we implicitly evaluate all possible copy numbers. Therefore, solving the assembly problem also yields the most likely TR copy number.
There exist several definitions of the likelihood of reads given a genome assembly (Clark et al. 2013; Ghodsi et al. 2013; Rahman and Pachter 2013). Previous studies have shown that the likelihood is typically maximized for correct genome assemblies. Therefore, we can
adapt these likelihood functions with the expectation that finding their maximum will result in finding the correct TR copy
numbers. However, several adaptations need to be made. Based on the method of Ghodsi et al. (2013), we can define the likelihood Pr(R∣C) by considering all possible alignment positions of the reads in R to the decoy reference DC. The decoy reference DC is constructed by concatenating the left flanking region of the repeat, C repeat units, and the right flanking region of the motif. Note that in the context of finding the TR copy number that maximizes
the likelihood of the reads, we are constructing multiple decoy references with varying values of C.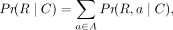 (2)
where A is the set of all alignments of the reads. A single alignment a is a mapping of each read to its positions and orientation (forward or reverse) along the decoy reference DC. The alignment refers to the positions only and does not include pairwise alignment information. To compute the exact value
of Pr(R∣C), Ghodsi et al. (2013) developed a dynamic programming algorithm that marginalizes over all possible alignments of the reads in R to the reference. However, this algorithm is resource-intensive and is mainly used to compute the quality of assembled genomes
and not to find a high likelihood assembly. In practice, only several best alignments contribute significantly to the overall
probability (Boža et al. 2015). We approximate the likelihood with the best alignment
(2)
where A is the set of all alignments of the reads. A single alignment a is a mapping of each read to its positions and orientation (forward or reverse) along the decoy reference DC. The alignment refers to the positions only and does not include pairwise alignment information. To compute the exact value
of Pr(R∣C), Ghodsi et al. (2013) developed a dynamic programming algorithm that marginalizes over all possible alignments of the reads in R to the reference. However, this algorithm is resource-intensive and is mainly used to compute the quality of assembled genomes
and not to find a high likelihood assembly. In practice, only several best alignments contribute significantly to the overall
probability (Boža et al. 2015). We approximate the likelihood with the best alignment 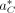 of the reads to decoy reference DC. So,
of the reads to decoy reference DC. So, 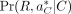 is used to approximate the overall likelihood Pr(R|C) because it contributes the largest value. Later, we describe how we find this best alignment. This likelihood is defined
as
is used to approximate the overall likelihood Pr(R|C) because it contributes the largest value. Later, we describe how we find this best alignment. This likelihood is defined
as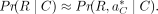 (3)
Now, we define P(R, a|C), the probability of a fixed alignment a of R given the copy number C. Using the chain rule, the joint probability P(R, a|G) can be expressed as the product of a conditional probability and a marginal probability:
(3)
Now, we define P(R, a|C), the probability of a fixed alignment a of R given the copy number C. Using the chain rule, the joint probability P(R, a|G) can be expressed as the product of a conditional probability and a marginal probability: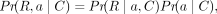 (4)
where Pr(R∣a, C) is the probability of the reads given that the true alignment is a and the TR copy number is C, and
(4)
where Pr(R∣a, C) is the probability of the reads given that the true alignment is a and the TR copy number is C, and 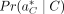 is the probability of the alignment positions given that the true copy number is C. In the following two sections, we define the two probability terms that make up Pr(R, a∣C) in general form, independent of
is the probability of the alignment positions given that the true copy number is C. In the following two sections, we define the two probability terms that make up Pr(R, a∣C) in general form, independent of 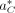 . This allows us to model alignment probability in a general sense before incorporating the best alignment in the overall
likelihood formulation.
. This allows us to model alignment probability in a general sense before incorporating the best alignment in the overall
likelihood formulation.
Probability of observing a set of reads given their true alignment and TR copy number
The model assumes that individual reads are independently sampled, thus the overall probability Pr(R∣a, C) is the product of the individual read mapping probabilities. To model the read mapping probability, we incorporate a simple
sequencing model that accounts for substitution errors (Boža et al. 2015). Then, the probability for a single read r aligned to position j is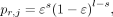 (5)
where ε is the sequencing error rate, s is the number of substitutions relative to DC at position j, and l is the length of the read r.
(5)
where ε is the sequencing error rate, s is the number of substitutions relative to DC at position j, and l is the length of the read r.
Then, the overall probability becomes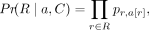 (6)
where a[r] is the position of r in the alignment a.
(6)
where a[r] is the position of r in the alignment a.
Probability of observing an alignment given the true TR copy number
The alignment a, regardless of the exact sequences of the reads, corresponds to an observed insert size and read depth distribution. The read depth is the number of reads that overlap a position along the decoy reference DC. We expect both these observed distributions to be close to the expected distributions of the sample.
We model the probability of the alignment a given the true TR copy number, Pr(a∣C), as the product of the observed insert size and depth distribution probabilities.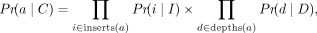 (7)
where I and D are the expected insert size and read depth distributions of the sample, respectively. inserts(a) and depths(a) are the sets of insert sizes and read depths implied by the alignment, respectively.
(7)
where I and D are the expected insert size and read depth distributions of the sample, respectively. inserts(a) and depths(a) are the sets of insert sizes and read depths implied by the alignment, respectively.
Overall likelihood of the bag of reads given a true TR copy number for a diploid sample
We combine the Equations 3, 6, and 7 into the overall approximate definition of the likelihood: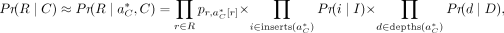 (8)
where
(8)
where 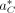 is the best alignment of R to the decoy reference DC.
is the best alignment of R to the decoy reference DC.
This derivation assumes a single haplotype. In practice, human genomes are diploid and may have different copy numbers (also
known as alleles) on each haplotype. To overcome this, ScatTR aligns reads to two decoy haplotypes (maternal and paternal)
and assumes that read depth is evenly distributed between them. To adapt our derivation, let 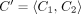 , where C1 and C2 are the corresponding copy numbers on each haplotype. We redefine the likelihood as such:
, where C1 and C2 are the corresponding copy numbers on each haplotype. We redefine the likelihood as such: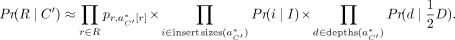 (9)
The alignment,
(9)
The alignment, 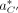 is now a mapping of each read to its position, orientation, and the haplotype. A read can only be aligned to one haplotype.
An alignment is only valid if all pairs are aligned to the same haplotype and in opposing orientations. The likelihood is
not defined for invalid alignments in which pairs are aligned to different haplotypes or have the same orientation.
is now a mapping of each read to its position, orientation, and the haplotype. A read can only be aligned to one haplotype.
An alignment is only valid if all pairs are aligned to the same haplotype and in opposing orientations. The likelihood is
not defined for invalid alignments in which pairs are aligned to different haplotypes or have the same orientation. 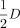 represents the halved depth distribution, assuming that reads are equally likely to originate from either haplotype.
represents the halved depth distribution, assuming that reads are equally likely to originate from either haplotype.
Finding the best alignment for a copy number
Here we describe a Monte Carlo optimization routine that finds the best alignment 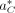 of reads R to a decoy reference DC among all possible alignments AC. The alignment is a mapping of each read to its position and orientation on the decoy reference. In the previous sections,
we described how this best alignment is used to approximate the likelihood of R given the true copy number. The likelihood of the alignment Pr(R, a∣C) is used to approximate the overall likelihood Pr(R∣C) because it contributes the largest value to the overall likelihood. So, we seek to find
of reads R to a decoy reference DC among all possible alignments AC. The alignment is a mapping of each read to its position and orientation on the decoy reference. In the previous sections,
we described how this best alignment is used to approximate the likelihood of R given the true copy number. The likelihood of the alignment Pr(R, a∣C) is used to approximate the overall likelihood Pr(R∣C) because it contributes the largest value to the overall likelihood. So, we seek to find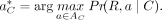 (10)
Finding
(10)
Finding 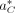 is difficult because the search space, AC, is combinatorial in size with respect to C and |R|. Therefore, we use simulated annealing (SA), which is an iterative probabilistic technique for approximating the global
minimum of a cost function (Kirkpatrick et al. 1983). It is especially useful when the search space is large, and it is often used when the search space is discrete.
is difficult because the search space, AC, is combinatorial in size with respect to C and |R|. Therefore, we use simulated annealing (SA), which is an iterative probabilistic technique for approximating the global
minimum of a cost function (Kirkpatrick et al. 1983). It is especially useful when the search space is large, and it is often used when the search space is discrete.
In our implementation, we seek to minimize the following cost function, which is the negative log likelihood of observing
the reads R given the true alignment a and copy number C.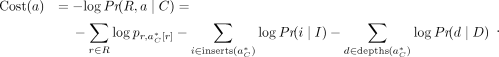 (11)
We start from a randomly initialized alignment a and an initial temperature t0 (a parameter of the SA algorithm). In each iteration i, we choose a number of read pairs to move, and we randomly sample new positions on the decoy reference to align them. This
forms a new alignment a′. The number of read pairs that are moved is proportional to the temperature ti. The next step depends on the costs of the alignments a and a′:
(11)
We start from a randomly initialized alignment a and an initial temperature t0 (a parameter of the SA algorithm). In each iteration i, we choose a number of read pairs to move, and we randomly sample new positions on the decoy reference to align them. This
forms a new alignment a′. The number of read pairs that are moved is proportional to the temperature ti. The next step depends on the costs of the alignments a and a′:
-
If Cost(a′) ≤ Cost(a), then the new alignment a′ is accepted, and the algorithm continues with the new alignment.
-
If Cost(a′) > Cost(a), then the new alignment a′ is accepted with probability proportional to the difference in cost. If a′ is rejected, the algorithm keeps the old alignment a for the next step.
The acceptance probability is 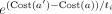 . When the temperature ti is high, changes to the alignment that increase the cost substantially (decrease the likelihood) are more likely to be accepted.
We use a standard cooling schedule in which
. When the temperature ti is high, changes to the alignment that increase the cost substantially (decrease the likelihood) are more likely to be accepted.
We use a standard cooling schedule in which 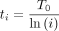 at iteration i of the algorithm.
at iteration i of the algorithm.
The algorithm stops when no new best solutions are found for 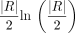 iterations, where
iterations, where 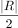 is the number of read pairs. If no new best solutions are found for
is the number of read pairs. If no new best solutions are found for 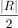 iterations, the algorithm resets the temperature to the initial temperature t0. When the algorithm terminates, the alignment with the lowest observed cost is considered the optimal alignment, denoted
as
iterations, the algorithm resets the temperature to the initial temperature t0. When the algorithm terminates, the alignment with the lowest observed cost is considered the optimal alignment, denoted
as 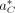 .
.
Finding the copy number that maximizes the likelihood of the bag of reads
To find the most likely copy number C*, we find the copy number that maximizes the likelihood of the observed bag of reads Pr(R∣C). We explained earlier how maximizing the former is equivalent to the latter and show how its value can be approximated using
the best alignment for the given C. Thus, our goal is to find C*: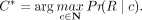 (12)
Every evaluation of Pr(R∣c) requires finding the corresponding best alignment
(12)
Every evaluation of Pr(R∣c) requires finding the corresponding best alignment 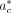 , which is used to calculate the value of Pr(R∣c). Because the search space is the set of natural numbers, it is inefficient to compute the likelihood for every copy number
to find the maximum likelihood. Optimization techniques such as gradient descent are not appropriate because the likelihood
function is nondifferentiable.
, which is used to calculate the value of Pr(R∣c). Because the search space is the set of natural numbers, it is inefficient to compute the likelihood for every copy number
to find the maximum likelihood. Optimization techniques such as gradient descent are not appropriate because the likelihood
function is nondifferentiable.
An applicable technique is GSS (Kiefer 1953). It is an iterative optimization technique used to find a local extremum of a unimodal function within a specified interval. It works by successively narrowing the interval in which the extremum lies, reducing the search space in each iteration. It significantly reduces the number of evaluations that are needed to find the copy number that maximizes the likelihood. Because it requires defining an initial interval, we calculate a reasonable range of copy numbers using a closed-form solution that relies on the mean depth of the sample and the number of IRRs present in the bag. For the minimum value of the range, we compute the closed-form estimate using a depth value of μdepth + 3σdepth and for the maximum using μdepth − 3σdepth. The closed-form estimate takes as input the number of IRRs observed and the expected mean depth, which is described in a later section.
In practice, to maintain numerical stability, we use the negative log likelihood as the minimization objective of GSS. Additionally, because GSS is designed to converge to any local maximum within an interval, it can sometimes converge to saddle points in our likelihood function. To improve our estimate of C*, we use bootstrapping to perform GSS multiple times. We start with an initial bootstrap distribution with the probability set to zero for all copy numbers. For every bootstrap iteration, the copy number returned by GSS gets its probability incremented by the inverse of its the negative log likelihood. After bootstrapping is finished, the bootstrap distribution is normalized to sum to one, and the median is reported as the estimate. This allows us to report a 95% confidence interval.
Efficiently scoring reads against repeat decoy references
When trying to find the best alignment for a set of reads to a decoy reference genome, we need to efficiently compute a set of permitted alignment positions for each read and the associated alignment error. Here, “alignment positions” refer to specific positions along the decoy reference. By default, we compute the alignment error as the Hamming distance between the read sequence and the corresponding reference sequence, separately for both the forward and reverse directions. Additionally, we provide an option for users to compute the alignment error using the Levenshtein distance, which accounts for insertions and deletions. This flexibility allows users to choose the appropriate metric based on the characteristics of the locus being analyzed, particularly for loci at which indels are expected to play a significant role.
For a given read, we compute the alignment error across three regions of the decoy reference: the left flank, the repeat region, and the right flank. The alignment error for positions on the left and right flanks is computed directly. Within the repeat region, although the total number of possible alignment positions increases with the number of repeat units, the repetitive structure of the sequence allows us to simplify the computation. Because the repeat is composed of copies of the same motif, we only need to consider one position per motif offset, specifically, M positions, where M is the motif length. For each of these positions, we compute the alignment score once and replicate it across the repeat by shifting in steps of the motif length. This takes advantage of the periodic nature of the repeat sequence and eliminates redundant computation. As a result, we can efficiently identify the best-scoring alignment positions for a read across all regions of the decoy reference, regardless of the TR's copy number.
Once alignment errors are computed for all positions on the decoy reference, we allow a read to align only to the position(s) with the lowest error, which may include multiple equally optimal positions.
Simulating samples with TR expansions
We obtained a set of TR loci from the Simple Repeats catalog (hg38 coordinates) published on the UCSC Genome Browser (Kent et al. 2002). The catalog was generated by TRF (Benson 1999). First, we removed loci with a motif length <2 bp, >20 bp. Additionally, we selected for loci that perfectly lift over to the T2T-v2 assembly (Kent et al. 2002; Nurk et al. 2022). A locus passes the filter only if the region ±550 bp around the repeat locus is identical in both the GRCh38 and the T2T-v2 assembly. We then randomly selected 30 loci for evaluating the performance of our method (Supplemental Table S1).
We used the T2T-v2 assembly (Nurk et al. 2022) as the reference genome from which WGS with paired short-read sequencing was simulated. We used ART (Huang et al. 2012), which simulates reads with customized error profiles. We generated our samples with the HiSeq X PCR-free profile, with 30× coverage, 150 bp read length, 450 bp mean fragment size, and 50 bp fragment size standard deviation. All other parameters were kept to the default. Specifically, the substitution error rate is approximately 0.0012. The insertion error rates are 0.00009 and 0.00015 for the first and second reads, respectively. The deletion error rates are 0.00011 and 0.00023, respectively. For each locus, we simulated a set of samples corresponding to copy numbers of 200 to 1000, with a step size of 100, as well as heterozygous and homozygous status. So, for each locus, we simulated 18 samples. In total, we simulated 540 samples.
Baseline closed-form solution
As a baseline, we derived a closed-form solution to predict the copy number. The solution is equivalent to the copy number
that maximizes the likelihood of the number of IRRs observed. We counted a read as an IRR if it has a weighted-purity score
≥0.9.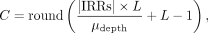 (13)
where L is the read length, and μdepth is the mean read depth of the sample. In the diploid case, we multiply the above value by two to get the sum of the copy
numbers on both haplotypes.
(13)
where L is the read length, and μdepth is the mean read depth of the sample. In the diploid case, we multiply the above value by two to get the sum of the copy
numbers on both haplotypes.
Comparison with GangSTR
We obtained the source code for GangSTR version 2.5.0 from its GitHub repository (https://github.com/gymreklab/GangSTR). The binary was built using htslib-1.11 bindings. We ran the simulated samples with the ‐‐targeted option. Because we are only interested in the genotype and estimated length, we used ‐‐skip-qscore. The ‐‐readlength option was set to 150, ‐‐coverage to 30, ‐‐insertmean to 300, and ‐‐insertsdev to 50. We also set ‐‐bam-samps to the name of the sample and ‐‐samp-sex to M because Chromosome Y was included in the reference from which the reads were simulated. The predicted repeat copy number is extracted from the output JSON file selected as the allele with the least absolute error to the true copy number.
Comparison with ExpansionHunter
We obtained the source code for ExpansionHunter version 5.0.0 (https://github.com/Illumina/ExpansionHunter). We ran the simulated samples using default parameters and set the ‐‐sex option to male. The predicted repeat copy number is extracted from the output JSON file selected as the allele with the least absolute error to the true copy number.
Comparison with STRling
We obtained the source code for STRling version 0.5.2 (https://github.com/quinlan-lab/STRling). We ran the simulated samples using default parameters. Because STRling is not capable of analyzing TRs with motif lengths larger than six, samples with such motifs were excluded from STRling predictions.
Analysis of short-read data from the 1000 Genomes Project
We used PCR-free high-coverage short-read WGS data from 2495 individuals in the 1000 Genomes Project (Byrska-Bishop et al. 2022) to evaluate ScatTR on a population scale. The data are publicly available from the NIH 1000 Genomes mirror at https://ftp-trace.ncbi.nih.gov/1000genomes/ftp/1000G_2504_high_coverage/.
Analysis of long-read data from the 1000 Genomes Project
We obtained Pacific Biosciences (PacBio) HiFi long-read sequencing data for five individuals from the 1000 Genomes Project (HG02282, HG02769, HG02953, HG03452, and HG03520) from https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC3 (Logsdon et al. 2025). These data were used in two separate analyses: to estimate the natural mutation rate of TRs and to assess the false-positive rate of large TR expansions called by ScatTR.
For both analyses, reads were aligned to the GRCh38 reference genome using pbmm2 align ‐‐sort -j 48 <reference> <reads> <output> with pbmm2 version 1.13.1. TR loci were then genotyped using TRGT version 1.5.0 (Dolzhenko and et al. 2023) with the following command: trgt genotype -t 6 ‐‐genome <reference> ‐‐repeats <repeats> ‐‐reads <reads> ‐‐output-prefix <output>.
To estimate the mutation rate of TRs, we extracted the allele purity (AP) values from the FORMAT column of the TRGT VCF output. We averaged these values across all five samples and subtracted the result from one to obtain the mean mutation rate.
Analysis of ALS cohort WGS data
We analyzed short-read WGS data from 30 ALS patients (Supplemental Table S4) obtained from the New York Genome Center (NYGC) ALS Consortium (Kenna et al. 2016). The individual-level data are available to authorized investigators through The database of Genotypes and Phenotypes (dbGaP; https://dbgap.ncbi.nlm.nih.gov/) under accession number phs003067.
Software availability
ScatTR is an open-source software package available for download on GitHub (https://github.com/g2lab/scattr). The source code, installation instructions, and documentation are included in the GitHub repository and as Supplemental Code. The software is implemented in Rust and supports both Linux and Mac operating systems.
Prebuilt binaries are available for download on GitHub for macOS (both Intel x86_64 and Apple Silicon arm64), compiled on GitHub-hosted macOS runners (macOS 12 or later). For Linux, binaries are provided for x86_64 systems, built natively on Ubuntu 22.04 LTS runners, and for arm64 systems, which are cross-compiled on Ubuntu. These binaries are intended to be compatible with systems similar to the build environments. Users running other distributions or architectures are encouraged to build ScatTR from the source to ensure compatibility.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work has been supported by National Institutes of Health (NIH) grant R03OD036491 to G.G., and by the NIH National Institute of General Medical Sciences grant R35GM147004. This work was also made possible by the MacMillan Family Foundation as part of the MacMillan Center for the Study of the Non-Coding Cancer Genome at the New York Genome Center. We acknowledge ALS patients and their families. Samples and associated phenotype data used in this study were provided by the ALS Consortium members such as, but not limited to, the University of California San Diego, Johns Hopkins University, Columbia University Medical Center, Barrow Neurological Institute, the University of Edinburgh, Georgetown University, University College London, Massachusetts General Hospital, Academic Medical Center, Mount Sinai, Tel-Aviv Sourasky Medical Center, the University of Pennsylvania, Penn State University, Washington University, Henry Ford Health Systems, Cedars-Sinai Medical Center, the University of Thessaly, the University of Athens, Hadassah Medical Center. We acknowledge the ALS Association (ALSA, 19-SI-459) and the Tow Foundation for providing financial support to carry out the sequencing.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280563.125.
-
Freely available online through the Genome Research Open Access option.
- Received February 15, 2025.
- Accepted August 15, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








