
Kinetic measurement of gene-specific RNA polymerase II transcription elongation rates
Abstract
Transcription is regulated at multiple levels, including initiation, elongation, and termination. Whereas much research has focused on the initiation of transcription, regulation of elongation plays an important role not only in transcription dynamics but also in cotranscriptional RNA processing and genome stability. Despite advances in high-throughput approaches for global quantification of RNA polymerase II (RNAPII) speed, RNAPII elongation rate studies have been limited to a relatively small number of long genes or to velocity estimates inferred indirectly from RNAPII occupancy data. Here, we present DRB/TTchem-seq2, a modified version of the DRB/TTchem-seq method, to directly measure gene-specific elongation rates of more than 3000 genes. By combining short time point sampling after synchronized RNAPII release into the gene body and a new computational framework to track the distance traveled by RNAPII, we greatly increase the number of genes for which it is possible to obtain elongation rates. Our direct RNAPII elongation rate quantification reveals that elongation rates vary not only among genes but also within genes. Additionally, we describe how specific histone modifications and elongation factor occupancy correlate with subclasses of genes based on their elongation rates. Together, we present a robust and powerful method for RNAPII transcription elongation rate measurement.
Transcription elongation by RNA polymerase II (RNAPII) is one of the essential steps that give rise to precursor mRNAs in eukaryotes. Productive elongation begins when RNAPII is released from promoter-proximal pausing and proceeds along the gene body to transcribe RNA (Jonkers and Lis 2015; Core and Adelman 2019; Noe Gonzalez et al. 2021; Aoi and Shilatifard 2023). As with all steps in the transcription process, elongation is tightly regulated to ensure that RNAPII travels along the gene at a proper speed (i.e., elongation rate) and successfully transcribes the full-length RNA (i.e., processivity) (Muniz et al. 2021).
Advances in biochemical and high-throughput technologies have revealed that transcription elongation rates are highly regulated (Muniz et al. 2021). For example, depletion of elongation factor Paf1C in a mouse muscle myoblast line leads to transcription elongation rate defects and reduced H2B ubiquitylation early in gene bodies (Hou et al. 2019). In contrast, loss of elongation factor RECQL5 results in increased average elongation rates of RNAPII, which are associated with increased genomic instability (Saponaro et al. 2014). Factors involved in cotranscriptional RNA processing have also been shown to impact elongation rate. The splicing regulator SRSF2 (also known as SC35) promotes transcription elongation in a gene-specific manner (Lin et al. 2008), and U1 small nuclear ribonucleoprotein (U1 snRNP) complex involved in splicing and suppression of cryptic intronic polyadenylation similarly stimulates elongation and facilitates transcription of long genes (Mimoso and Adelman 2023). Furthermore, RNAPII elongation rates are modulated in response to certain stresses and stimuli such as heat shock and UV-induced DNA damage (Andrade-Lima et al. 2015; Williamson et al. 2017; Cugusi et al. 2022). In addition, dysregulated transcriptional elongation has been implicated in developmental defects as well as disease and aging (Aoi and Shilatifard 2023; Debès et al. 2023).
RNAPII elongation rates not only reflect transcription efficiency but also are known to affect cotranscriptional RNA processing (Muniz et al. 2021). This has been classically studied using point mutations in RPB1, the largest subunit in the RNAPII complex (Greenleaf 1983), which either increase or decrease global elongation rate. Such mutations have demonstrated that both slow and fast elongation can alter splice site selection of specific exons (De La Mata et al. 2003; Fong et al. 2014; Maslon et al. 2019). RNAPII elongation rates also influence polyadenylation and 3′ end mRNA processing (Fong et al. 2015; Geisberg et al. 2020). Moreover, RNAPII elongation rate changes can impact the chromatin environment, and, vice versa, changes in the chromatin state can influence RNAPII elongation (Veloso et al. 2014; Fong et al. 2017; Wang et al. 2023). For instance, lack of histone H3 lysine 4 trimethylation (H3K4me3) catalyzed by SET1/COMPASS methyltransferase leads to decreased transcription levels, increased promoter-proximal pausing, and slower elongation (Wang et al. 2023).
The influences on cotranscriptional RNA processing alter the composition and diversity of the transcriptome, which eventually affect gene regulation. Whereas studies using Rpb1 point mutations or factor perturbations followed by average RNAPII rate estimation have associated RNAPII speed with functionality, direct measurements of RNAPII transcription elongation rates for individual genes and their correlation with chromatin modifications, RNAPII-associated cofactors, or 3′end processing remain limited, partly due to technical challenges in obtaining elongation rates at single-gene resolution. Therefore, high quality measurement of elongation rates at a large scale of genes has the potential to advance our understanding of the connections between transcription elongation and processes such as cotranscriptional RNA processing and genome stability.
In recent years, high-throughput sequencing of nascent RNAs has been employed to estimate RNAPII elongation rates (Danko et al. 2013; Fuchs et al. 2014; Jonkers et al. 2014; Saponaro et al. 2014; Veloso et al. 2014; Liang et al. 2018; Baluapuri et al. 2019; Ardehali et al. 2021) (Supplemental Table S1). These methods adopted the same principle, which estimates the elongation rate by measuring the distance traveled by RNAPII as a function of time (Muniz et al. 2021). We recently presented DRB/TTchem-seq with a detailed protocol (Gregersen et al. 2019, 2020) for estimating average RNAPII elongation rates. Like the other methods mentioned, DRB/TTchem-seq tracks nascent transcripts at different time points after synchronized release from promoter-proximal pausing. RNAPII is first restricted to the area close to the transcription start site (TSS) by reversible inhibition effect with 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole (DRB) (Singh and Padgett 2009). Upon DRB washout, RNAPII molecules synchronously enter productive elongation genome-wide. The distances traveled into gene bodies are then measured at various time points, allowing elongation rate to be inferred as a function of time and distance traveled by RNAPII. The method estimates average RNAPII elongation rates from metagene profiles but also for individual genes with sufficient nascent RNA coverage.
In the original DRB/TTchem-seq method, distances traveled by RNAPII were measured from peak position in nascent transcription profiles, which approximates the maximum of the coverage (Gregersen et al. 2020). However, these peaks may not accurately reflect the exact distance traveled by RNAPII, as the signal likely comprises transcripts made from RNAPII complexes released at different times during the 4sU labeling period. Moreover, peak identification can be computationally challenging for genes with low read coverage or uneven nascent transcription profiles, limiting the number of analyzable genes. Additionally, the 10- to 40-min time windows used in DRB/TTchem-seq restrict analysis to longer genes because RNAPII moving at 2–3 kilobases per minute (kb/min) would travel 80–120 kb within that time frame. Accordingly, most studies using DRB/TTchem-seq or DRB/BrU-seq have typically measured RNAPII elongation rates for genes longer than 60 kb (Gregersen et al. 2019, 2020; Pappas et al. 2023), 75 kb (Danko et al. 2013), 100 kb (Saponaro et al. 2014; Veloso et al. 2014), or even 200 kb (Geijer et al. 2021; Cugusi et al. 2022). Addressing these limitations would enable more accurate and larger scale gene-specific RNAPII elongation rate measurements.
Results
Improved measurement of RNAPII elongation rates for individual genes
A major limitation of measuring RNAPII elongation rates using DRB/TTchem-seq, is that the elongation rates are restricted to long genes. This is due to the fact that, by 20 min of DRB release, the average length of nascent transcripts in human cells typically exceeds 60 kb (Fig. 1A, upper panel), making it difficult to resolve RNAPII elongation rates for shorter genes. To overcome this limitation and increase the number of measurable genes, we developed DRB/TTchem-seq2, in which we shortened the DRB release times to 5, 10, 15, and 20 min compared to the original 10, 20, 30, and 40 min used in DRB/TTchem-seq (Fig. 1B). This modification significantly expands the range of detectable nascent transcripts by enabling the inclusion of many shorter genes (Fig. 1A, lower panel). As a result, more than 5000 additional genes could be analyzed for RNAPII elongation rate (Fig. 1C).
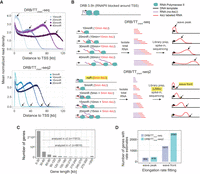
DRB/TTchem-seq2 enables estimation of RNAPII elongation rates for a larger number of individual genes. (A) Metagene plots showing the mean read density of nonoverlapping protein-coding and lncRNA genes from DRB/TTchem-seq and DRB/TTchem-seq2 data sets. Read depths were calculated from the transcription start site (TSS) to 120 kbdownstream. Black arrows indicate the positions of the wave peaks in DRB/TTchem-seq data and wave fronts in DRB/TTchem-seq2 data. (B) Overview of the DRB/TTchem-seq2 method with major modifications relative to DRB/TTchem-seq highlighted. A control sample without DRB release (noR) was included, where cells were treated with DRB for the same duration as other samples. The time durations post-DRB release were shortened to 0, 5, 10, 15, and 20 min. Correspondingly, 4sU incorporation times were reduced to 5 min, with 4sU added during the last 5 min of the experiments. Library preparation remains similar to DRB/TTchem-seq but incorporates Unique Molecular Identifier (UMI) as part of the sequencing adapter for PCR duplicates identification. Following sequencing, RNAPII elongation progression was identified by the transcription wave fronts instead of the wave peaks (see Methods). (C) Distribution of gene lengths for human protein-coding and lncRNA genes. The numbers of genes included in the RNAPII elongation rate analysis for both DRB/TTchem-seq and DRB/TTchem-seq2 are indicated. (D) Bar plots comparing the number of genes with RNAPII elongation rate estimates calculated from the same number of reads in DRB/TTchem-seq and DRB/TTchem-seq2.The DRB/TTchem-seq raw sequencing data were previously published (Pappas et al. 2023).
Consistent with the original DRB/TTchem-seq method, 4-thiouridine (4sU) was used to label and subsequently purify nascent transcripts following DRB release. To align with the shortened DRB release times, the 4sU labeling time was reduced to 5 min (Fig. 1B). Previous studies have shown that this short time period is sufficient for effective 4sU incorporation in K562 cells (Schwalb et al. 2016). Although shortening the labeling time could potentially lower 4sU incorporation and sequencing coverage, we confirmed through dot blot quantification that 4sU levels remained well above background for all release times tested (Supplemental Fig. S1A).
A second major improvement in the DRB/TTchem-seq2 protocol is the inclusion of a background control sample in which 4sU was added during the final 5 min of DRB treatment (Fig. 1B). This control enables the measurement of background transcription resulting either from nonspecific 4sU labeling or from nascent transcripts that escape DRB inhibition. As expected, transcript levels in the control sample were low but nonnegligible relative to the other nascent samples (Supplemental Fig. S1B). The control sample was subsequently used to correct for nonspecific signal and reduce noise in downstream analyses (see Methods).
A third key methodological improvement in DRB/TTchem-seq2 is the computational framework used to estimate RNAPII elongation rates. Instead of identifying transcriptional wave peaks, we implemented a new approach to detect the wave fronts of nascent RNA profiles following DRB release (Fig. 1A,B). Whereas wave peaks represent the positions with the highest read density, wave fronts reflect the distance traveled by the earliest RNAPII molecules released into productive elongation after DRB removal (Supplemental Fig. S1C). Therefore, wave fronts provide more accurate measurement of RNAPII elongation rates of individual genes. Moreover, wave peaks identified in DRB/TTchem-seq for the same gene at different DRB release times did not consistently progress in a clear, time-dependent manner (Supplemental Fig. S1D), which limited the number of genes that could be measured.
With the combined experimental and computational improvements introduced in DRB/TTchem-seq2, the method now enables robust elongation rate estimation for a substantially larger set of genes. Specifically, when elongation rates were calculated from the same sequencing depths, DRB/TTchem-seq2 yielded an at least seven-fold increase in the number of genes with reliable rate estimates compared to the original DRB/TTchem-seq (Fig. 1D). Taken together, the modifications in DRB/TTchem-seq2 greatly increase the resolution and scale at which RNAPII elongation rates can be measured.
Computational identification of gene-specific RNAPII elongation rates
To achieve accurate single-gene RNAPII elongation rate estimates, we implemented several improvements in our computational pipeline, including relying on wave front estimation as described above. Additionally, several other new method features, such as improved background correction and the removal of PCR duplicates, also played key roles in enhancing the RNAPII elongation rate calculations. An overview of our updated computational workflow is illustrated in Figure 2A and explained in detail in the Methods section.
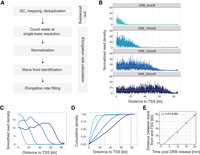
RNAPII elongation rates in DRB/TTchem-seq2 are calculated using transcription wave fronts. (A) Schematic illustration of the main data processing steps and RNAPII elongation rate estimation in DRB/TTchem-seq2. (B) Read coverage plots for an example gene CEMIP2 showing RNAPII elongation progression after DRB release. Read densities were calculated at single-base resolution from the annotated transcription start site (TSS) to transcription end site (TES), normalized to yeast spike-ins and the control sample (see Methods). (C) Smoothed splines of the read density profiles for gene CEMIP2 shown in panel B (see Methods). (D) Cumulative sums of the smoothed spline coverage from panel C for gene CEMIP2. Dashed lines mark the “elbow” point of the cumulative curves, indicating the transcription wave fronts (see Methods). (E) Linear regression of distance traveled by RNAPII versus DRB release time for gene CEMIP2 (see Methods). The slope corresponds to the RNAPII elongation rate.
First, sequencing reads were mapped to the reference genome and PCR duplicates were removed with the application of Unique Molecular Identifiers (UMIs) (Supplemental Fig. S2A). Reads were then assigned to gene features for each sample. Because both technical and biological replicates showed high correlations (Supplemental Fig. S2B–D), replicate samples were pooled, and average coverage values were used for subsequent analyses. To identify the distance traveled by RNAPII, we filtered out reads mapping to exon-exon junctions. These reads presented higher proportions in control samples (Supplemental Fig. S2E), suggesting that they originated mainly from contaminating mature mRNAs. In contrast, nascent RNA samples consistently contained low proportions (<2%) of exon-exon junction reads (Supplemental Fig. S2E). Given the fixed 5-min 4sU labeling time, the captured RNA segments primarily represent nascent transcripts adjacent to actively elongating RNAPII complexes. Therefore, the exon-exon junction reads in nascent samples likely represent more contaminating pre-existing spliced mRNAs than newly spliced nascent RNAs. Excluding exon-exon junction reads thus reduces background noise without sacrificing signal from genuine nascent transcripts.
The second stage of the analysis involved quantifying RNAPII elongation rates from noise-corrected read coverage profiles. For each individual gene, read coverage was quantified at every single base pair position from the transcription start site to the transcription end site (TES). Coverage values were normalized first using yeast spike-ins and then adjusted relative to the control sample (i.e., DRB-treated cells without release). The wave fronts in the coverage profiles represent the leading edge of the fastest RNAPII molecules following DRB release as illustrated by the example gene CEMIP2 (Fig. 2B; Supplemental Fig. S3A,B). To reduce local spikiness in the coverage profiles, we applied spline smoothing (Fig. 2C). We then calculated the cumulative sum of the smoothed coverage values, generating a continuously increasing curve between the TSS and TES (Fig. 2D). The “elbows” in these cumulative sum curves are thus representative of the wave fronts, as the coverage signal increase is notably slower after that point.
We identified these elbows computationally by locating the point on the curve that forms the largest angle with the start and end points (see Methods for details). After identifying the wave fronts, we performed linear regression on the distances between the TSS and the identified wave fronts (i.e., the distances traveled by RNAPII) across different DRB release times (Fig. 2E; Supplemental Fig. S3C,D). The slope of the regression line represents the RNAPII elongation rate within the gene body.
RNAPII elongation rates are calculated with high accuracy from simulated data
To evaluate the accuracy of our computational approach, we tested its performance using simulated data. Synthetic coverage profiles were generated to mimic coverage profiles obtained from DRB/TTchem-seq2 experiments. In the simulation, RNAPII molecules were modeled as being released from the promoter-proximal pause site at a constant frequency and transcribing through the gene body at a fixed elongation rate (Fig. 3A; also see Methods). Because 4sU is incorporated for only the final 5 min of each DRB release time point in DRB/TTchem-seq2, only RNA segments transcribed during the final 5 min were retained in the simulation (see Methods).
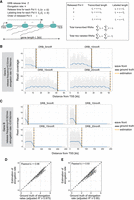
RNAPII elongation rates are estimated with high accuracy from simulated data. (A) Schematic illustration of the simulation model. (B) Example of a simulated gene with an RNAPII release frequency of 0.53/min and an elongation rate at 2.78 kb/min. Gray dots represent simulated read densities. Blue curves show the smoothed profiles. Vertical lines indicate the wave front positions from either the ground truth or computational estimation. (C) Example of another simulated gene with a different release frequency, elongation rate, and gene length. (D) Comparison of estimated RNAPII elongation rates with the ground truth values across 969 simulated genes. Genes were selected based on a linear regression adjusted R2 ≥0.975 (see Methods). (E) Same as shown in panel D but including 1061 genes selected using a slightly relaxed threshold (adjusted R2 ≥0.95) (see Methods).
By systematically varying the elongation rate, release frequency, and gene length, we generated a range of synthetic gene coverage profiles (see Methods). These profiles were then analyzed using our computational pipeline, including wave front identification and elongation rate estimation. As shown with two example genes, the wave fronts were correctly inferred by our approach (Fig. 3B,C). Furthermore, the estimated elongation rates were highly correlated with the ground truth values used in the simulations (Fig. 3D,E; also see Methods), demonstrating that our computational method reliably identifies wave fronts and accurately estimates RNAPII elongation rates.
RNAPII elongation rates vary both among and within protein-coding and lncRNA genes
As an application of DRB/TTchem-seq2, we estimated gene-specific RNAPII elongation rates in U2OS cells. Focusing on highly expressed genes across replicates, we obtained high-confidence elongation rate estimates (i.e., adjusted linear regression R2 ≥ 0.95) for 3009 genes. The substantial increase in the number of measurable genes was primarily achieved by the methodological improvements rather increased sequencing coverage, as downsizing of the sequencing depth only had a smaller impact on the number of genes with elongation rate estimates (Supplemental Fig. S4A). Importantly, downsampling of the sequencing depth did not affect the elongation rate, showing that DRB/TTchem-seq2 yields accurate elongation rate estimates even at substantially lower sequencing depth (Supplemental Fig. S4B). The mean RNAPII elongation rate across these genes was ∼4 kb/min, with the majority of rates ranging between 2 and 6 kb/min (Fig. 4A). As expected, this is higher than the rates derived from wave peak analyses (Gregersen et al. 2019, 2020), as wave peaks capture the majority of nascent reads transcribed throughout the entire DRB release interval, which can lead to overestimation of elongation time and consequently underestimate elongation rates.
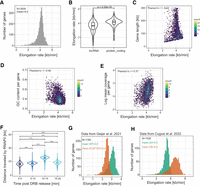
RNAPII transcription elongation rates are variable among and within genes. (A) Histogram showing the distribution of RNAPII elongation rates for 3009 genes estimated in U2OS cells using DRB/TTchem-seq2. Only genes with linear regression adjusted R2 ≥0.95 are included. (B) Violin plots comparing the distributions of the RNAPII elongation rates between protein-coding and lncRNA genes. Gene samples were matched in number for fair comparison (n = 300 in each group). (C) Correlation between RNAPII elongation rates and gene lengths. Genes are colored based on their biotypes. Only genes with linear regression adjusted R2 ≥0.975 are shown. (D) Correlation between RNAPII elongation rates and GC content of genomic sequences. GC content was calculated as the ratio of the number of G and C nucleotides to the total number of nucleotides per gene, based on the union of all transcripts. (E) Correlation between RNAPII elongation rates and mean read coverage in log scale of the DRB/TTchem-seq2 data. (F) Violin plots showing distances traveled by RNAPII during different time intervals after release from promoter-proximal pausing sites. Data included 1339 genes (see Methods). Wilcoxon rank-sum tests were used between each pair of samples. (***) P-value <1×10−6. (G) Distribution of RNAPII elongation rates for 1185 single genes in wild-type (WT) and ELOF1 knockout (KO) HCT116 cells. Rates were calculated using the DRB/TTchem-seq2 computational pipeline. Raw data from Geijer et al. (2021). (H) Distribution of RNAPII elongation rates for 1026 individual genes in both control and heat shock (HS) conditions in MRC5-VA cells. Rates were calculated using the DRB/TTchem-seq2 computational pipeline for data from Cugusi et al. (2022).
Notably, RNAPII elongation rates were generally higher in protein-coding genes than those in long noncoding RNA (lncRNA) genes (Fig. 4B). This difference was not attributed to gene length or GC contents (Supplemental Fig. S5A,B). Furthermore, no significant variation in elongation rates was observed among lncRNA subgroups classified according to their genomic locations relative to mRNAs (Supplemental Fig. S5C). Analysis of active enhancer-associated histone modifications, including H3K4me1 and H3K27ac, revealed greater enrichment of these marks in the promoter regions and reduced enrichment within gene bodies of lncRNAs compared to mRNAs (Supplemental Fig. S5D,E), potentially reflecting general differences of histone distribution patterns.
We next investigated whether gene architectures influence RNAPII elongation rates. Whereas previous studies reported corrections of RNAPII elongation rates with gene length, exon density, and first intron length (Jonkers et al. 2014; Veloso et al. 2014), we found no dependence of RNAPII elongation rates on gene length (Fig. 4C). Moreover, it is common to observe genes of varying lengths exhibiting similar RNAPII elongation rates (Supplemental Fig. S6A–D). No significant correlations were observed for exon density and first intron length either (Supplemental Fig S7A,B). Among the explored features, only GC content showed a significant negative correlation with RNAPII elongation rates (Fig. 4D), consistent with previous reports (Jonkers et al. 2014; Veloso et al. 2014) and recent indirect estimates via TT-seq and PRO-seq signal ratios (Mimoso and Adelman 2023).
We also explored the relationship between RNAPII elongation rate and transcriptional activity. A modest positive correlation with mean nascent transcript coverage was observed (Fig. 4E), indicating a nonlinear and partial relationship. This observation aligns with our theoretical model (Supplemental Fig. S1C), which suggests that transcriptional output is influenced more directly by the RNAPII release frequency than by the gene body elongation rate.
Because we sequenced the nascent transcripts at four incremental time points following DRB release, we were able to estimate RNAPII elongation rates within gene bodies over these intervals. Specifically, we calculated the distances traveled by RNAPII during each 5-min interval following DRB release (i.e., 0–5, 5–10, 10–15, and 15–20 min). Notably, RNAPII elongation rates varied substantially within individual genes. RNAPII elongation progressed markedly slower in the first 5 min after release from the promoter-proximal pausing site (Fig. 4F), a phenomenon also observed in previous studies (Danko et al. 2013; Saponaro et al. 2014). Our fine-scaled time point of nascent RNA labeling following DRB release provides a direct and quantitative view of this initial transcriptional slow phase. Between 5 and 10 min, elongation rates increased slightly and then accelerated in the 1015-min interval as RNAPII moved deeper into gene bodies (Fig. 4F; Supplemental Fig. S7C). Notably, elongation slowed down again significantly during the last 5-min interval (15–20 min) (Fig. 4F; Supplemental Fig. S7C). These measurements, derived simply by calculating the distances traveled by RNAPII between each of the two time points, were consistent across thousands of genes (Fig. 4F). Therefore, it is evident that dynamic change in RNAPII elongation rates within gene bodies is a common feature of transcription.
DRB/TTchem-seq2 is broadly applicable to various cell lines with enhanced single-gene RNAPII elongation rate estimation
To confirm that DRB/TTchem-seq2 is applicable across cell lines, we applied the same experimental and computational procedures to a DLD-1 derived cell line. Consistent with observations in U2OS cells, we obtained RNAPII elongation rate estimates for 3168 genes, with most rates varying between 2 and 5 kb/min (Supplemental Fig. S7D,E). The mean RNAPII elongation rate in DLD-1 cells was around 3.0 kb/min, slightly lower than that obtained from U2OS cells, likely reflecting cell line variations in RNAPII elongation rates.
Importantly, the improved computational method developed for DRB/TTchem-seq2 is also applicable to existing DRB/TTchem-seq data sets generated using longer nascent RNA labeling times. To illustrate this, we reanalyzed a previously published DRB/TTchem-seq data set from U2OS cells (Pappas et al. 2023). Using our wave front identification method, we estimated RNAPII elongation rates for 1549 genes, with an average elongation rate of ∼3.4 kb/min (Supplemental Fig. S7F). Similarly to our DRB/TTchem-seq2 data, RNAPII elongation rates varied between genes, in this case, ranging from 2 to 4 kb/min (Supplemental Fig. S7F). This means that the average RNAPII elongation rates in U2OS cells from our DRB/TTchem-seq2 data set is somewhat higher than that in the U2OS cells from this published data set, potentially reflecting differences in experimental conditions or cellular states.
We further applied our computational method to two additional published DRB/TTchem-seq data sets in which RNAPII elongation was perturbed. In the case of ELOF1 knockout (KO) in HCT116 cells (Geijer et al. 2021), we observed a significant decrease in RNAPII elongation rates compared to the wild-type (WT) controls (Fig. 4G). Although the original study reported a similar conclusion based on metagene profiles for genes longer than 200 kb (Geijer et al. 2021), it did not provide single-gene elongation rate estimates. In contrast, our analysis yielded estimates for 1855 genes without restricting the analysis to long genes, enabling a more detailed understanding of how specific genes have been affected by ELOF1 loss.
Similarly, we analyzed the effects of heat shock-induced stress in MRC5-VA cells (Cugusi et al. 2022). In agreement with previous metagene-based findings, we confirmed that heat shock substantially accelerates RNAPII elongation rates, which was evident in elongation rate estimates for 1026 individual genes (Fig. 4H). Notably, the magnitude of elongation rate increase varied among genes, indicating differential gene-specific transcriptional responses to heat shock.
Leveraging RNAPII elongation rates obtained across multiple cell lines using either the modified DRB/TTchem-seq2 method with short release time points or the computational pipeline alone, we explored inter-cell line variability. Comparison of RNAPII elongation rates with previously published data revealed considerable variability across studies employing different methodologies (Supplemental Fig. S8A). Nevertheless, variation within studies utilizing the same method was comparatively smaller, particularly when comparing RNAPII elongations rates processed through our computational pipeline (Supplemental Fig. S8A,B). Overall, RNAPII elongation rates obtained by the computational pipeline described here showed good consistency among different cell lines (Supplemental Fig. S8B). Specifically, we observed high correlations between rates derived from different cell lines using DRB/TTchem-seq2, as well as rates derived from the same cell line (i.e., U2OS cells) using either DRB/TTchem-seq or DRB/TTchem-seq2 (Supplemental Fig. S8B). Together, these findings underscore the reliability and broad applicability of our method and emphasize the importance of accounting for methodological differences when comparing RNAPII elongation rates across data sets.
In summary, DRB/TTchem-seq2 enables accurate and gene-specific RNAPII elongation rate measurements for thousands of genes across multiple cell lines. Moreover, the improved computational pipeline can be used independently on previously published DRB/TTchem-seq data sets, which enables single-gene RNAPII elongation rate measurements beyond the original limitations to only very long genes (>200 kb) and to averaged rates from metagene analyses. Thus, our updated method not only improves the analysis of newly generated DRB/TTchem-seq2 data but also significantly enhances the utility of existing DRB/TTchem-seq data sets. By enabling robust and gene-specific RNAPII elongation rate estimates, our method provides deeper insights into how RNAPII-associated factors and cellular perturbations differentially affect transcription elongation of individual genes.
RNAPII elongation rates correlate with occupancy of RNAPII-associated factors and histone modifications
During the transcription cycle, elongating RNAPII complexes dynamically associate with multiple factors that are involved either directly in regulation of transcription dynamics or in cotranscription processing of nascent RNA. Whereas several of these factors have been individually implicated in modulating RNAPII elongation, these conclusions have typically been derived from measurements averaged across multiple genes or a limited number of gene examples. Our data set, which provides single-gene elongation rates, offers a unique opportunity to systematically investigate the relationship between RNAPII elongation rates and the occupancy of RNAPII-associated factors using publicly available ChIP-seq profiles.
To explore this, we categorized genes with RNAPII elongation rate estimates into “high,” “medium,” and “low” elongation rate groups (see Methods) and compared the enrichment of known elongation-associated factors across these groups. For canonical elongation factors such as PAF1, SSRP1, and SUPT5H (also known as SPT5), we observed higher ChIP-seq enrichment within gene bodies of genes exhibiting faster RNAPII elongation rates (Fig. 5A; Supplemental Fig. S9A). This positive association aligns with the established roles of these factors in promoting RNAPII elongation. Among the factors analyzed, SUPT5H showed the strongest correlation with elongation rate (Fig. 5A; Supplemental Fig. S9A).
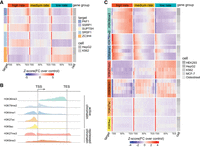
RNAPII transcription elongation rates are tightly associated with elongation factor binding and histone modifications. (A) Heat map displaying correlation between RNAPII elongation rates and RNAPII-associated elongation factor binding. Genes (n = 3009) with RNAPII elongation rate estimates were binned into three rate groups: high (4.28–7.02 kb/min); medium (3.93–4.28 kb/min); and low (0.14–3.93 kb/min). For each group, ENCODE ChIP-seq enrichment signals (see Methods) of elongation factor across gene bodies are displayed. The heat map colors represent Z-scores of fold change of ChIP-seq target signal relative to control. (B) Schematic illustration of the typical enrichment locations of various histone modifications based on ChIP-seq data relative to transcription start site (TSS) and transcription end site (TES). (C) Heat map displaying the correlation between RNAPII elongation rates and enrichment of histone modifications. The same 3009 genes were grouped equally by elongation rates as described in panel A. The histone modification enrichment of ENCODE ChIP-seq data sets across gene bodies is shown. Colors correspond to Z-scores of fold change of ChIP-seq target over control.
Whereas some RNAPII-bound factors are associated with increased RNAPII elongation rates, others serve to restrict RNAPII elongation, often in connection with early quality control of RNAPII complexes. Notably, ZC3H4, a component of the restrictor complex involved in transcription termination of early nonproductive elongation (Estell et al. 2023; Rouvière et al. 2023), showed a negative correlation between its enrichment along the gene body occupancy and RNAPII elongation rates (Fig. 5A; Supplemental Fig. S9A). Given that our RNAPII elongation rates reflect productive elongation across gene bodies, this finding indicates that higher ZC3H4 binding tends to associate with both earlier abrogation of elongation as well as slower gene body elongation.
Chromatin state is another key regulator of transcription. Histone posttranscriptional modifications (PTMs) are important markers of chromatin states. These modifications display characteristic distribution patterns across transcriptional units, with certain marks being most frequently localized upstream or around the TSS, whereas others show decreasing or increasing abundance across gene bodies (Fig. 5B; Kimura 2013). Whereas previous investigations have mostly relied on indirect velocity estimations or limited gene sets (Fuchs et al. 2014; Veloso et al. 2014; Shao et al. 2022), our extensive data set of gene-specific RNAPII elongation rates allowed for a more comprehensive analysis.
We found that active chromatin marks including H3K36me3, H3K79me2 and H3K4me1 positively correlated with RNAPII elongation rates, whereas histone modifications associated with inactive chromatin states such as H3K9me3 and H3K27me3 negatively correlated with RNAPII elongation rates (Fig. 5C; Supplemental Fig. S9B). No significant correlation was observed for histone acetylation modifications within gene bodies (Fig. 5C; Supplemental Fig. S9B).
Previous studies employing direct RNAPII elongation rate estimates reported positive correlations for H3K79me2 and H4K20me1 but did not find a significant association with H3K36me3 (Fuchs et al. 2014; Veloso et al. 2014). In contrast, our data show that H3K36me3 exhibits the most pronounced differences among genes with varying RNAPII elongation rates (Fig. 5C; Supplemental Fig. S9B), consistent with recent elongation velocity estimates (Shao et al. 2022). To investigate this discrepancy, we reanalyzed the subset of genes characterized by Fuchs et al. (2014), using our elongation rate estimates alongside the ENCODE data sets utilized in this study. Within this common gene set, we again observed greater enrichment of H3K36me3 in genes with higher elongation rates (Supplemental Fig. S10A), suggesting that the different findings likely stem from differences in methodology.
In summary, our extensive data set of gene-specific elongation rates reveals clear correlation with both RNAPII-associated factors and histone modifications. Specifically, faster RNAPII elongation is associated with increased gene body occupancy of SUPT5H and enrichment of active histone marks such as H3K36me3, H3K79me2, and H3K20me1. Conversely, high ZC3H4 binding is associated with reduced RNAPII elongation rates.
Discussion
Here, we present an improved methodology and computational framework to directly quantify RNAPII elongation rates across more than 3000 individual human genes. Key modifications to the original method include shortening the RNA metabolic labeling pulse, incorporating a no-DRB background normalization control, and refining the computational strategy for detecting elongation progression. Together, these improvements significantly enhance the resolution and reliability of RNAPII elongation rate calculations at the single-gene level.
Unlike previous methods that relied on data from only one (Veloso et al. 2014; Baluapuri et al. 2019) or two (Fuchs et al. 2014) time points following DRB release, restricting measurement to data obtained immediately following promoter proximal release, our method utilizes four time points to capture RNAPII progression across gene bodies. This enables direct measurement of absolute elongation rates in kb/min.
Alternative indirect approaches, such as the elongation indexes derived from the ratio of TT-seq to RNAPII occupancy (e.g., from mNET-seq or PRO-seq), have been used to infer elongation velocity along the gene body (Gressel et al. 2017; Caizzi et al. 2021; Mimoso and Adelman 2023). However, these indirect approaches come with certain assumptions, such as that all RNAPII engagement is resulting in productive elongation, which does not hold true for all genes or in all conditions. Moreover, the elongation index can be influenced by technical differences in how TT-seq and RNAPII occupancy data are generated (e.g., fragmentation size, metabolic labeling times, immunoprecipitation conditions, etc.) as well as by coverage variability for the separate TT-seq and RNAPII occupancy data. As a result, elongation index values offer only relative estimates and cannot provide actual elongation rates in kb/min based on progression of initiated RNAPII complexes and levels of nascent transcriptions.
Accurate and high-resolution RNAPII elongation rate measurement is crucial as elongation speed is not only a readout of RNAPII progression dynamics but also influences transcriptional output and cotranscriptional processes such as splicing (Fong et al. 2014) and polyadenylation site usage through regulation of RNAPII termination (Pinto et al. 2011; Yague-Sanz et al. 2020), both impacting mRNA isoform expression. In addition, cotranscriptional deposition of RNA modifications such as N6-methyladenosine (m6A), known to impact splicing, mRNA stability, and translation, is also influenced by RNAPII elongation rates (Slobodin et al. 2017) Thus, it must be assumed that RNAPII elongation rates are carefully tuned in accordance with cotranscriptional processing. Our direct measurements furthermore reveal a positive correlation between RNAPII elongation rates and nascent RNA levels, suggesting that elongation speed can affect transcriptional activity. This agrees with previous observations both in human cell lines (Danko et al. 2013; Jonkers et al. 2014) and in plants (Leng et al. 2020). The application of multiple time points in our analysis allowed us to examine dynamic changes of RNAPII elongation rates across gene bodies. Notably, RNAPII elongation is slower near the promoter-proximal pause site in the first 5 min, which agrees with early stalling and increased regulation of RNAPII during this stage of the transcription cycle (Jonkers and Lis 2015). As RNAPII proceeds downstream, elongation speeds up before slowing down again near the gene end. These changes in RNAPII elongation rates within gene bodies have previously only been directly measured for a very small number of genes (Danko et al. 2013; Jonkers et al. 2014) or inferred indirectly from occupancy data (Gressel et al. 2017; Caizzi et al. 2021; Mimoso and Adelman 2023) or modeled probabilistically (Liu et al. 2025). Our direct measurements, therefore, provide new insights into RNAPII elongation dynamics within genes. Importantly, this variability can have significant functional implications as elongation rate affects the timing of cotranscriptional processing events such as RNA splicing, polyadenylation, and 3′ end formation. Additionally, these changes may influence chromatin modifications that depend on RNAPII progression speed.
A major advantage of having high-confidence single-gene RNAPII elongation rates for thousands of genes is the ability to correlate RNAPII elongation rates with specific genomic or epigenetic features. As an example, we find that H3K36me3 levels are significantly correlated with high RNAPII elongation rates, in line with recent indirect measurement of elongation velocity (Shao et al. 2022). H3K36me3 is a well-known active chromatin mark deposited along gene bodies by methyltransferase SETD2 (Set2 in yeast) (Fig. 5B; Strahl et al. 2002; Krogan et al. 2003; Morris et al. 2005). Recent structural data showed that SETD2 interacts closely with SUPT6H (also known as SPT6) within the RNAPII elongation complex, facilitating H3K36me3 deposition during nucleosome reassembly downstream of the RNAPII complex (Markert et al. 2025). In our data, genes with faster elongation rates show strong H3K36me3 enrichment towards the 3′ end, suggesting a functional link between RNAPII elongation speed and the positional deposition of H3K36me3. In support of this, earlier observations have shown that slowing down RNAPII (by expression of a Rpb1 mutant) shifts H3K36me3 enrichment towards the 5′ end of genes (Fong et al. 2017).
In addition to histone marks, we also analyzed occupancy of RNAPII-associated factors known to globally modulate RNAPII elongation rates. We found that higher gene body binding of ZC3H4 correlates with slower RNAPII elongation rates. ZC3H4, together with WDR82, ARS2, and CKII, forms the restrictor complex that promotes early transcription termination and degradation of abortive transcripts through its interaction with the NEXT complex. Depletion of ZC3H4 leads to increased pervasive transcription from enhancer regions and noncoding RNAs including PROMTs, and also certain mRNAs (Austenaa et al. 2021; Estell et al. 2023; Rouvière et al. 2023). Whereas ZC3H4 has mainly been associated with early transcriptional regulation (Estell et al. 2023; Rouvière et al. 2023), our data raise the question of whether ZC3H4 and the restrictor complex might also serve to “restrict” RNAPII elongation during the productive elongation phase. As illustrated above, our data enable broad correlation studies with any RNAPII-associated factor, opening avenues to discover novel regulators of elongation, including factors whose effects may be gene-specific or context-dependent. We expect our method will be a powerful tool for identifying novel regulatory factors influencing RNAPII transcription elongation.
Finally, our methodology combines an improved experimental setup with advances in computational identification of RNAPII progression. When applied to existing DRB/TTchem-seq data sets, our computational pipeline substantially increases the number of genes with reliable elongation rate estimates compared to previous studies relying on metagene profiles of very long genes (Geijer et al. 2021; Cugusi et al. 2022). This provides flexibility to adopt either the full experimental-computational approach or apply the computational pipeline to reanalyze existing data sets to expand the resolution of elongation rate analysis.
In summary, our enhanced method provides a robust, high-resolution tool to accurately measure RNAPII elongation rates at the individual gene level, facilitating discovery of global or novel gene-specific regulators of RNAPII elongation dynamics.
Methods
Cell treatments, RNA extractions, and 4sU-RNA biotinylation
U2OS (gift from Jiri Bartek's group) or a DLD-1 derived cell line (gift from Brian Strahl's group) were cultured at 37°C with 5% CO2 in high glucose DMEM (Biowest, #L0104) supplemented with 10% v/v FBS (Gibco, #10270106), 100 U/mL penicillin, 100 µg/mL streptomycin (Gibco, #10378016), and 2 mM L-glutamine. Cells were routinely passaged 2–3 times a week. All cell lines were confirmed to be mycoplasma-free.
DRB/TTchem-seq2 was developed based on the previous protocol from Gregersen et al. (2020) with the major modifications being: (1) the RNA labeling time; (2) shorter DRB release time points; (3) inclusion of UMI adapters; and (4) addition of a no-DRB released sample for background subtraction. For RNAPII synchronization close to the TSS, cells were incubated with 100 uM DRB (5,6-dichlorobenzimidazole 1-β-D-ribofuranoside; Sigma-Aldrich, #D1916) for 3.5 h. DRB inhibition was released for 5, 10, 15, and 20 min by three washes with pre-warmed PBS. For each time point, newly synthesized RNA was labeled with 1 mM 4-thiourudine (4sU; Biosynth Carbosynth, #NT06186) for 5 min immediately prior to harvest, which was done by addition of TRIzol (Thermo Fisher Scientific, #15596026) directly on top of the cell monolayer. For the US2O experiment, samples were prepared in duplicate for all time points (each sample originating from a 10-cm dish). For the DLD-1 experiment, one replicate was prepared for all DRB release time points (each sample originating from a 10-cm dish).
Total RNA was extracted using TRIzol according to the manufacturer's instructions with an additional chloroform cleanup prior to isopropanol precipitation. For U2OS samples, 20 μg of total RNA were spiked in with 0.2 μg of yeast spike-ins (Saccharomyces cerevisiae W303a cells labeled with 5 mM 4-thiouracil (4tU; Sigma-Aldrich, #440736) for 5 min) (see Gregersen et al. 2020 for detailed procedure). For the DLD-1 experiment, 40 μg of total RNA were spiked in with 0.4 μg of yeast spike-ins and used as starting material for the protocol. RNA was fragmented for 20 min on ice by base hydrolysis and returned to a neutral pH buffer prior to biotinylation as previously described (Gregersen et al. 2020). For addition of biotin to 4sU residues, we used the MTSEA Biotin-XX linker (Biotium, #BT90066). Reactions were set up in 250 µL, containing 10 mM Tris-HCl [pH 7.4], 1 mM EDTA, and 5 mg MTSEA biotin-XX linker and incubated in the dark at room temperature for 30 min. Biotinylated RNA was purified by phenol:chloroform and isopropanol precipitation, heat-denatured for 10 min at 65 degrees, and used for purification with µMACS Streptavidin microbeads (Miltenyi, #130-074-101). RNA was incubated with beads for 15 min at room temperature on a rotating wheel. µColumns (Miltenyi) were equilibrated with the nucleic acid buffer supplied as part of the streptavidin kit while placed in the magnetic field of a µMACS magnetic separator. Bead samples were applied to the µColumns. After capture, beads were washed twice with pull-out wash buffer (100 mM Tris-HCl [pH 7.4], 10 mM EDTA, 1 M NaCl, and 0.1% Tween20). 4sU-RNA was eluted from beads using two rounds of 100 mM DTT and cleaned up with the RNeasy MinElute Cleanup kit (Qiagen, #74204) using 1.5× excess of ethanol for the RNA precipitation compared to the supplied protocol to ensure recovery of small RNA fragments. Concentration of 4sU-RNA was measured by Qubit (RNA high sensitivity assay; Thermo Fisher Scientific, #Q32852), and the size range was determined by high sensitivity RNA assay on a TapeStation (high sensitivity RNA ScreenTape; Agilent, #5067-5579).
Dot blot for quantification of 4sU incorporation
Dot blots was carried out as previously described with minor modifications (Gregersen et al. 2020). Five micrograms of total RNAs extracted with TRIzol (Thermo Fisher Scientific, #15596026) were used for biotinylation with 1 mg/mL EZ-Link HPDP-Biotin (Thermo Fisher Scientific, #21341) and incubated 3 h in the dark. RNA was purified from the biotin linker by addition of an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1 [vol/vol/vol]) to the biotinylated RNA. RNA from the aqueous phase was then precipitated with isopropanol and washed in 85% ethanol.
Purified RNA samples were blotted onto a Hybond-N membrane in a dot blot apparatus connected to a vacuum pump. The membrane was dried, blocked, and incubated as previously described (Gregersen et al. 2020). Biotin incorporation levels were accessed by developing membrane with 1:5 diluted ECL reagent, followed by chemiluminescence imaging. Finally, the membrane was stained with a methylene blue solution (0.5 M sodium acetate, 0.5% methylene blue) for 30 min following several washes in water.
Sequencing library preparation
For the US2O experiment, 4 ng of purified 4sU-labeled RNA was used for high-throughput sequencing library preparation with the NEBNext Ultra II directional RNA kit (NEB, #E7760) using Unique Dual Index UMI Adaptors (NEB, #E7416) following the protocol for degraded RNA (RIN 1–2) without additional RNA fragmentation. Libraries were amplified using 12 PCR cycles, concentration measured by Qubit (DNA high sensitivity assay), and size distribution assessed using TapeStation (high sensitivity D5000 ScreenTape; Agilent, #5067-5592). For the DLD-1 experiment, 10 ng of purified 4sU-labeled RNA was used for high-throughput sequencing library preparation with the NEBNext Ultra II directional RNA kit (NEB, #E7760) using Unique Dual Index UMI Adaptors (NEB, #E7416) following the protocol for degraded RNA (RIN 1–2) without additional RNA fragmentation. Libraries were amplified using nine PCR cycles, concentration measured by Qubit (DNA high sensitivity assay), and size distribution assessed using TapeStation (high sensitivity D5000 ScreenTape; Agilent, #5067-5592). The final library size distribution peaked at 390 nt, corresponding to an insert size of 260 nt. U2O2 libraries were sequenced on two P3 v100 flow cells (130 cycles) in paired-end mode on a NextSeq 2000 (Illumina) and DLD-1 samples sequenced on a single P3 v100 flow cell (130 cycles) in paired-end mode on a NextSeq 2000 (Illumina).
Sequencing data processing
Sequencing data were de-multiplexed using Illumina bcl2fastq and then quality checked with FastQC (Babraham Bioinformatics). For the single-ended libraries, we sequenced the UMI sequences and the DNA fragments in two separate cycles, which ended up with two reads for each sample (i.e., R1—DNA inserts, R2—UMIs). We extracted the UMI sequences from R2 and attached them to the headers of R1 using UMI-tools (Smith et al. 2017). The universal Illumina sequencing adapters were trimmed using Trim Galore! (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and cutadapt (Martin 2011) with default settings. Then, the reads were mapped to a merged reference of the human genome (GENCODE: GRCh38.primary_assembly.genome.fa) and yeast genome (Ensembl: Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa) using STAR (Dobin et al. 2013) with options: ‐‐runThreadN 10 ‐‐outSAMattributes NH HI AS NM MD XS ‐‐outSAMstrandField intronMotif ‐‐outSAMmultNmax 1 ‐‐outSAMunmapped None ‐‐outSAMtype BAM. Uniquely mapped reads were extracted from the aligned BAM files with SAMtools (Danecek et al. 2021). PCR duplicates were identified and removed with UMI-tools (Smith et al. 2017). The spliced reads that contained exon-exon junctions were identified and extracted from the others with SAMtools (Danecek et al. 2021). The remaining reads were used for downstream analysis of RNAPII elongation rates.
Single base pair–resolution read coverage counting
We counted the single base pair-resolution read coverage from the mapped reads. Prior to counting, the mapped reads in BAM format were split into genes transcribed from the forward and reverse strands. We then counted the read coverage for each gene from its transcription start site to its transcription end site with the bamCoverage function from R package bamsignals (DOI:10.18129/B9.bioc.bamsignals) with custom arguments: bampath = my.bam, gr = gene.gr, filteredFlag = 1024, paired.end = “ignore”, mapqual = 20, verbose = FALSE. Here, only nonoverlapping protein-coding and lncRNA genes that ranged from 30 to 300 kilobase pairs in lengths were included into the gene.gr file to avoid miscounting of the read coverage among genes. The coverage values were then normalized to the size factors calculated from yeast spike-ins using DESeq2 (Love et al. 2014). If the spike-in is not available, then it could be normalized to the sample library size.
Wave front identification
To identify the position where the wave front ends in coverage profiles of the DRB/TTchem-seq2 data, we first smoothed the coverage profiles using the gene body. Then, we summed up all the smoothed coverage signals
along the gene body which resulted in cumulative sum curves. Because the coverage profiles are positive and continuous between
the TSS and the wave end, the cumulative sum curves are above the diagonal (Fig. 2D). We thus identified the “elbow” of this curve by finding the position that gives the smallest cosine values between this
point and the start point and end point. Based on the law of cosines, we used the following formula for the calculation:
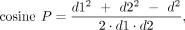 in which P represents any point on the cumulative curve, d is the Euclidian distance between the start and end points on the curve, d1 is the Euclidian distance between P and the start point, and d2 is the Euclidian distance between P and the end point. Finding the smallest cosine P identifies the “elbow” position of the cumulative curve which is the wave front position of the coverage profile. To avoid
the bias due to the accumulation of reads around the promoter-proximal pausing site, we defined the start point to be 1000
bp downstream from the TSS.
in which P represents any point on the cumulative curve, d is the Euclidian distance between the start and end points on the curve, d1 is the Euclidian distance between P and the start point, and d2 is the Euclidian distance between P and the end point. Finding the smallest cosine P identifies the “elbow” position of the cumulative curve which is the wave front position of the coverage profile. To avoid
the bias due to the accumulation of reads around the promoter-proximal pausing site, we defined the start point to be 1000
bp downstream from the TSS.
Gene-specific RNAPII elongation rate calculation
We calculated RNAPII elongation rates by taking the distance traveled by RNAPII as a function of time. This was done by fitting a line between the wave fronts and the elongation times after RNAPII had been released from the pausing sites for each gene. To improve accuracy, only genes that present at least three preceding wave fronts were kept for the fitting. We did not use the origin point (0, 0) for the fitting, which resulted in constant RNAPII elongation rates estimation in the gene bodies. Elongation rates were represented by the slope of the linear regression line. To keep genes with a high goodness of fit, we filtered out genes that showed adjusted R2 values <0.95.
Comparison of gene numbers between DRB/TTchem-seq and DRB/TTchem-seq2 data
To fairly compare the estimated number of genes between DRB/TTchem-seq and DRB/TTchem-seq2, we downsampled reads of the DRB/TTchem-seq data set from U2OS cells (Pappas et al. 2023) and the newly generated DRB/TTchem-seq2 data set from U2OS cells, both to 28 million per sample (after mapping, uniquely mapped reads extraction, and PCR deduplication). Following the RNAPII elongation rate calculation procedure as outlined in Figure 2A, we calculated single-gene elongation rates for both data sets and compared the final number of individual genes that ended up with RNAPII elongation rates estimation.
Data simulation
To check if the wave fronts in the DRB/TTchem-seq2 data are correctly identified, we simulated data in silico to represent the nascent transcription waves post-DRB release. Because the main objective was to generate coverage profiles to mimic the DRB/TTchem-seq2 data, we simply assumed that RNAPII is released from the promoter pausing site at a constant rate and RNAPII is elongating at a constant speed along gene bodies. We defined the following terms: RNAPII release frequency r [/cell/min]; RNAPII elongation rate v [kb/min]; DRB release time T [min]; gene length L [kb].
For the RNAPII molecules that have been released from the pausing site, the elongation times t (t > 0) for them are
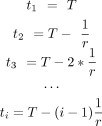 For a specific nascent RNA that is being elongated, the length of it, l (l ≤ L), is defined by
For a specific nascent RNA that is being elongated, the length of it, l (l ≤ L), is defined by
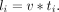 Summing up all the transcribed RNAs, we got the total nascent transcripts during the whole DRB release time T
Summing up all the transcribed RNAs, we got the total nascent transcripts during the whole DRB release time T
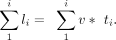 The above formulas describe all the nascent transcripts made by RNAPII post-DRB release. However, we only added 4sU in the
last 5 min of the DRB/TTchem-seq2 experiment, meaning we only partially labeled the nascent RNAs. To simulate DRB/TTchem-seq2 data, the length of the labeled RNA transcript is defined by
The above formulas describe all the nascent transcripts made by RNAPII post-DRB release. However, we only added 4sU in the
last 5 min of the DRB/TTchem-seq2 experiment, meaning we only partially labeled the nascent RNAs. To simulate DRB/TTchem-seq2 data, the length of the labeled RNA transcript is defined by
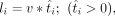 in which
in which 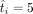 when
when 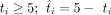 when ti < 5.
when ti < 5.
Genomic features
Genomic features were extracted from the transcriptome annotation file in GTF format (GRCh38.gencode.v43.gtf). To simplify the analysis, we took the union of all the annotated transcripts for each gene. Gene lengths were calculated by taking the distances between TSS and TES for each gene. For exons features, we kept the union of the nonoverlapping exons for each gene. Then, we subtracted the exons from the gene track to get the independent introns. GC contents were obtained by first retrieving the DNA sequence for each feature and then dividing the total number of G and C bases by the total number of A, T, G, and C. Exon density was calculated as the ratio of the length of exons to the length of the gene.
Published DRB/TTchem-seq data analysis
Published DRB/TTchem-seq data were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE215990, GSE165368, and GSE148844. The raw reads were processed as DRB/TTchem-seq2 libraries using the method described above and outlined in Figure 2A. Because there are no control samples in these data sets, we simply counted the read coverage for each gene from TSS to TES and then identified the fronts of the transcription waves using our computational method. Wave fronts that do not proceed with time were filtered out. We then fit a linear regression line between the distance between TSS and wave fronts and the DRB release time. The RNAPII elongation rate fitting with adjusted p value ≥ 0.95 were kept as good estimates. The filtered RNAPII elongation rates were used for the comparisons between biological conditions.
Correlation analysis between elongation rates obtained in U2OS cells and ENCODE ChIP-seq data
The ChIP-seq data of histone modifications, elongation factors, and RNAPII shown in Figure 5 and Supplemental Figures S9 and S10 were from ENCODE (https://www.encodeproject.org/). All the data were downloaded in the format of normalized bigWig files in which the bigWig signals were represented by fold change between target and input. To explore the correlation between elongation rates and ChIP-seq signals, we categorized the 3009 genes with characterized elongation rates in U2OS cells into three equal-sized groups (n = 1003 per group) based on their elongation rates. Specifically, elongation rates of the “low” group ranged from 0.14 to 3.93 kb/min; “medium” group ranged from 3.93 to 4.28 kb/min; and “high” group ranged from 4.28 to7.02 kb/min. We then plotted the ChIP-seq coverage profiles of RNAPII-associated factors and histone modifications for these three groups of genes by taking the mean fold change values across genes in each group. The plots were generated with the ggplot2 in R (R Core Team 2022). Details of the ChIP-seq data used in this study are listed in Supplemental Table S2. The heat maps were generated using ComplexHeatmap (Gu 2022) in R (R Core Team 2022).
Data access
Genome-wide data supporting the findings of this study have been deposited in the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE269247. The code used for processing DRB/TTchem-seq2 data and estimating RNAPII elongation rates is available both as Supplemental Code and at GitHub (https://github.com/haiyueliu/elongation_rate).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Anders Krogh and Jesper Svejstrup as well as lab members for helpful discussions and comments on the manuscript. We also thank the lab of Jiri Bartek from providing U2OS cells and Brian Strahl's lab for the courtesy of providing the DLD-1 derived cell line. Sequencing was performed at the ICMM/reNEW/CPR genomic platforms at University of Copenhagen. Work in the lab of L.H.G. is funded by a Hallas-Møller emerging investigator grant from the Novo Nordisk Foundation (NNF20OC0059959), a Sapere Aude grant from Independent Research Fund Denmark (0165-00092B), and a Horizon Europe European Research Council starting grant (TranscriptStress, 101161245). Work in the Center for Gene Expression is supported by Center of Excellence grant (DNRF166) from the Danish National Research Foundation.
Author contributions: L.H.G. and H.L. conceived the study. H.L. performed all bioinformatic analyses. L.H.G. performed experiments. H.L. and L.H.G. wrote the manuscript. L.H.G. provided funding and managed the project.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280852.125.
-
Freely available online through the Genome Research Open Access option.
- Received April 29, 2025.
- Accepted September 25, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








