
H3K27 and H3K9 methylation mask potential CTCF binding sites to maintain 3D genome integrity
Abstract
The three-dimensional (3D) genome structure is essential for gene regulation and various genomic functions. CTCF plays a key role in organizing topologically associated domains (TADs) and promoter-enhancer loops, contributing to proper cell differentiation and development. Although CTCF binds the genome with high sequence specificity, its binding sites are dynamically regulated during development, and aberrant CTCF binding is linked to diseases such as cancer and neurological disorders, and aging. However, the mechanisms controlling CTCF binding remain unclear. Here, we investigate the role of repressive chromatin modifications in CTCF binding using H3K9 methyltransferase-deficient immortalized mouse embryonic fibroblasts (iMEFs) and H3K27 methyltransferase EZH1/2 inhibitor. We find that H3K9 and H3K27 methylation regulate CTCF binding at distinct genomic regions, and their simultaneous loss induces drastic changes in CTCF binding. These changes are associated with alterations in 3D genome architecture and gene expression, suggesting that repressive chromatin modifications preserve proper chromatin organization by preventing aberrant CTCF binding. Additionally, whereas CTCF binding sites repressed by H3K9 methylation are bound by CTCF in early mouse embryos, those repressed by both H3K9 and H3K27 methylation remain inaccessible, with early embryo–specific H3K27 methylation forming at these sites. These findings implicate that H3K27 methylation plays a role for restricting CTCF binding in early embryos, ensuring proper genome organization during development.
CCCTC-binding factor (CTCF) is a conserved transcriptional regulator composed of 11 central zinc-finger domains (ZFs). It plays a crucial role in 3D genome organization and transcriptional regulation by mediating chromatin loop formation with cohesin, which acts as an insulator and contributes to the establishment of topologically associated domains (TADs) (Dixon et al. 2012; Nora et al. 2017). A large fraction of CTCF binding sites are shared across multiple cell types, but there is also a certain degree of cell type specificity, the regulation of which is influenced by transcription factors and epigenomic modifications, such as DNA methylation (Wang et al. 2012; Spracklin et al. 2023; Monteagudo-Sánchez et al. 2024). Recent studies have revealed that SETDB1 and EHMT1/2, which are histone H3 lysine 9 (H3K9) methyltransferases, prevent ectopic CTCF binding, particularly at transposable elements (Jiang et al. 2020; Gualdrini et al. 2022; Sun et al. 2024; Tam et al. 2024). Transposable elements constitute nearly 50% of the mouse genome, and some, such as SINE elements, contain CTCF-binding motifs that can occasionally function as insulators (Schmidt et al. 2012; Cournac et al. 2016; Ichiyanagi et al. 2021; Wang et al. 2024). H3K9 methylation is a key suppressor of transposable elements and may also prevent ectopic CTCF binding by blocking CTCF recognition at transposons genome-wide, thereby maintaining proper 3D genome organization. At least five H3K9 methyltransferases exist in mammals (Setdb1, Suv39h1, Suv39h2, Ehmt1, Ehmt2) (Allshire and Madhani 2018), and they regulate H3K9 methylation redundantly or independently, depending on genomic regions (Fukuda et al. 2021). For example, H3K9me2 in the inactive B compartment is redundantly regulated by all H3K9 methyltransferases (Fukuda et al. 2021), and H3K9me3 at LINE-1 elements is controlled by both SETDB1 and SUV39H1/2 (Bulut-Karslioglu et al. 2014; Liu et al. 2018; Robbez-Masson et al. 2018; Fukuda and Shinkai 2020).
Due to the redundancy of H3K9 methyltransferases, their roles in CTCF binding regulation have not been fully elucidated. Furthermore, the loss of H3K9 methylation leads to epigenomic reorganization, including the redistribution of H3K27me3 mediated by the Polycomb complex (Peters et al. 2003; Walter et al. 2016; Fukuda et al. 2023). For example, H3K27me3 can spread into regions previously marked by H3K9 methylation, forming new Polycomb domains and contributing to heterochromatin maintenance following the loss of H3K9 methylation (Fukuda et al. 2023). Such epigenomic reorganization is observed not only in developmental processes such as germ cell development and early embryogenesis but also in aging and cancer (Yang et al. 2023). The Polycomb complex is known to regulate chromatin architecture through the formation of Polycomb-associated domains (PADs) and chromatin compartments, such as the A/B compartments identified by principal component analysis (PCA) of Hi-C data, where compartment A corresponds to transcriptionally active regions and compartment B to inactive regions. (Du et al. 2020; Fukuda et al. 2023). However, its role in CTCF binding regulation and the functional implications of epigenomic reorganization in CTCF regulation remain largely unknown.
In this study, we utilize a series of H3K9 methyltransferase knockout cells, as well as a complete H3K9 methyltransferase-deficient cell line that we have established (Fukuda et al. 2023). Additionally, we employ H3K27 methyltransferase EZH1/2 inhibitor to create a system in which both H3K9 and H3K27 methylation are simultaneously depleted. Using this system, we performed CTCF ChIP-seq, RNA-seq, and Hi-C analyses to elucidate the redundant and independent roles of these repressive modifications in CTCF binding, transcription, and 3D genome regulation.
Results
H3K9 and H3K27 methylation deficiency reshapes CTCF binding profiles
To investigate the function of H3K9 and H3K27 methyltransferases on CTCF binding profiles in mammalian cells, we performed CTCF chromatin immunoprecipitation sequencing (ChIP-seq) in wild-type (WT) immortalized mouse embryonic fibroblasts (iMEFs), Setdb1 KO iMEFs (Kato et al. 2018), Setdb1/Suv39h1/Suv39h2 triple knockout (TKO) iMEFs (Fukuda et al. 2021), Setdb1/Suv39h1/Suv39h2/Ehmt1/Ehmt2 quintuple knockout (5KO) iMEFs (Fukuda et al. 2023), and WT, Setdb1 KO, TKO, and 5KO iMEFs treated with the EZH1/2 dual inhibitor DS3201 (hereafter referred to as DS) (Yamagishi et al. 2019). We previously demonstrated that a 7-day treatment with 1 µM DS leads to an almost complete loss of H3K27me3 in iMEFs (Fukuda et al. 2023); therefore, we applied this condition for CTCF ChIP-seq analysis in this study. CTCF ChIP-seq was performed with two biological replicates for each sample, and two clones (#14 and #55) of the 5KO iMEFs were used. Cluster analysis of the CTCF ChIP-seq data showed a high correlation between the replicates (Fig. 1A). To investigate the features of CTCF binding regulated by H3K9 and H3K27 methyltransferases, we identified differentially enriched peaks (DE peaks) in each sample compared to WT using DiffBind (FDR < 0.05) (Ross-Innes et al. 2012). CTCF peaks that showed increased binding upon KO and/or DS treatment (referred to as “Increased DE peaks”)—including both newly emerged peaks and pre-existing peaks with significantly elevated enrichment—and those with decreased binding (“Decreased DE peaks”)—including both peaks with reduced enrichment and those completely lost. In samples lacking H3K9 methyltransferases (Setdb1 KO, TKO, 5KO), the number of “Increased” DE peaks in each KO cell was greater than those of “Decreased” DE peaks (Fig. 1B). This suggests that H3K9 methyltransferases primarily function to prevent CTCF binding, consistent with previous analyses in Setdb1-deficient cells (Gualdrini et al. 2022; Sun et al. 2024; Tam et al. 2024) and Ehmt2-deficient cells (Jiang et al. 2020). On the other hand, in all cell types, DS treatment produced a larger increase in the number of “Decreased” DE peaks than in those of “Increased” DE peaks signals (Fig. 1B). Treatment of each H3K9 methyltransferase-deficient cell line with DS resulted in more extensive changes in CTCF binding than the sum of CTCF changes observed in the deficient cells and those induced by DS treatment in WT cells, and this trend is particularly pronounced in 5KO cells (Fig. 1B). In 5KO+DS cells, 16,521 “Increased” and 13,665 “Decreased” DE peaks were identified. Thus, both number of “Increased” and “Decreased” DE peaks in 5KO+DS cells correspond to ∼35% of the average number of peaks observed in WT (average number of WT peaks: 42,451). This suggests that the loss of both H3K9me and H3K27me3 leads to highly dynamic changes in CTCF binding.
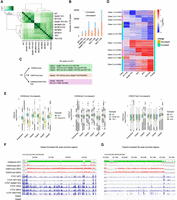
Dysregulation of CTCF binding profiles by H3K9/K27 methylation deficiency. (A) Clustering analysis of CTCF ChIP-seq data. Each sample analyzed in two biological replicates, showing a high correlation between replicates. (B) The number of differentially enriched (DE) peaks. A bar graph represents the number of CTCF accumulation increased or decreased in each sample. (C) The classification method for DE peaks. (D) Heat map of CTCF enrichment among DE classes. (E) Epigenomic profiles at increased DE peaks. H3K9me3 enrichment in WT and Setdb1 KO iMEFs (left), H3K9me2 enrichment in WT, Setdb1 KO, and TKO iMEFs (middle), and H3K27me3 enrichment in WT, TKO, and 5KO iMEFs (right). Statistical tests were performed using t-tests, and multiple testing correction was applied using the Benjamini–Hochberg (BH) method. All statistical results are provided in Supplemental Table S1. (F) Representative Class 2 increased DE peaks enriched regions. (G) Representative Class 5 increased DE peaks enriched regions.
To elucidate the regulatory mechanisms of the DE peaks, we classified DE peaks into five categories: Class 1: DE peaks observed in Setdb1 KO but not in WT+DS (Setdb1-dependent change); Class 2: DE peaks observed in TKO but not in Setdb1 KO and WT+DS (Suv39h1/2-dependent change); Class 3: DE peaks observed in 5KO but not in Setdb1 KO, TKO, and WT+DS (Ehmt1/2-dependent change); Class 4: DE peaks observed in WT+DS but not in Setdb1 KO, TKO, and 5KO (Ezh1/2-dependent change); Class 5: DE peaks observed only in 5KO+DS (regulated by both H3K9 and K27 methyltransferases) (Fig. 1C,D; Supplemental Fig. S1A). All peaks used in the DiffBind analysis were defined as consensus peaks, including those newly identified in samples other than WT iMEFs, but excluding the differentially enriched peaks. Motif enrichment analysis of each class revealed an accumulation of CTCF-binding motifs across all classes (Supplemental Fig. S1B). Additionally, motifs such as THRB, NR2F2, BMAL1, and NPAS are particularly enriched in “Increased” DE peaks observed in H3K9 methyltransferase-deficient cells, suggesting that H3K9 methyltransferases inhibit CTCF binding to motifs colocalized with these transcription factors (Supplemental Fig. S1B). Genomic distribution is largely different among classes. “Increased” Class 2, Class 3, and Class 5 are more enriched in intergenic regions (prop.test, P < 2.2 × 10−16) (Supplemental Fig. S1C) and the B compartments (Supplemental Fig. S1D). This is consistent with the fact that EHMT1/2 (G9a/GLP) and SUV39H1/2 target gene-poor intergenic regions and the B compartments (Fukuda et al. 2021), and H3K27me3 spreads into gene-poor B compartments after the loss of H3K9 methylation (Fukuda et al. 2023). “Increased” Class 2, Class 3, and Class 5 exhibit a relatively high frequency of B-to-A conversion in TKO, 5KO, and 5KO+DS, respectively, suggesting a potential association between increased CTCF binding and compartment conversion (Supplemental Fig. S1D). Unlike “Increased” Class 2, Class 3, and Class 5, “Increased” Class 1 is primarily found in the A compartment, consistent with the role of Setdb1 in regulating H3K9 methylation mainly in the A compartment (Supplemental Fig. S1D; Fukuda et al. 2021). Contrary to “Increased” classes, “Decreased” classes are predominantly found in genic regions, except for Class 1 (Supplemental Fig. S1C). Additionally, the compartment distribution of “Decreased” classes is the opposite of “Increased” classes, with Class 1 being enriched in the B compartment, whereas Class 2, 3, and 5 are more frequently found in the A compartment (Supplemental Fig. S1D).
Next, we investigated the relationship between increase in CTCF binding and chromatin modifications. “Increased” Class 1, which is regulated by Setdb1, exhibited a significant decrease in both H3K9me2 and H3K9me3 in Setdb1 KO (H3K9me2: adj. P = 1.3 × 10−176, H3K9me3: adj. P ≈ 0) (Fig. 1E; Supplemental Table S1). In contrast, in the “Increased” Class2, H3K9me3 is maintained in Setdb1 KO (indeed more enriched, adj. P = 2.7 × 10−24). In TKO, however, H3K9me3 is lost (Supplemental Fig. S1E). Furthermore, H3K9me2 in this class is markedly reduced in the TKO (adj. P = 8.5 × 10−260) (Fig. 1E; Supplemental Table S1). These results suggest that both H3K9me2 and H3K9me3 in this class are primarily maintained by Suv39h1/2. In “Increased” Class 3, where CTCF binding is elevated in 5KO, H3K9me3 is markedly reduced in Setdb1 KO (adj. P = 2.3 × 10−70) (Fig. 1E; Supplemental Table S1), whereas H3K9me2 remains highly retained even in the TKO (Fig. 1E; Supplemental Table S1). Because H3K9me2 is completely lost in 5KO (Fukuda et al. 2023), it is likely that H3K9me2 mediated by EHMT1/2 plays a role in suppressing CTCF binding in this class. Therefore, the differences among “Increased” Class 1, Class 2, and Class 3 can be explained by the distinct genomic regions where each H3K9 methyltransferase regulates H3K9 methylation. For H3K27me3, “Increased” Class 4 does not exhibit strong accumulation of H3K27me3 even in WT, suggesting that H3K27 methyltransferases may not directly regulate CTCF binding in this class (Fig. 1E; Supplemental Table S1). Additionally, in “Increased” Class 2 and Class 5, H3K9 methyltransferase deficiency led to an increase in H3K27me3 in TKO and 5KO iMEFs (adj. P < 1.3 × 10−56) (Fig. 1E; Supplemental Table S1). Although both Class 2 and Class 5 show increased H3K27me3, CTCF binding is elevated in Class 2 despite the presence of H3K27me3, whereas in Class 5, CTCF binding increases only after H3K27me3 is removed (Fig. 1E–G; Supplemental Table S1). CTCF binding in Class 2 and Class 5 regions does not increase in WT+DS (Fig. 1D; Supplemental Fig. S1A), suggesting that H3K27me3 is dispensable for regulation of “Increased” Class 2 regions and the increase in CTCF binding for Class 5 regions may be not an indirect effect of DS treatment. These findings also suggest that, in addition to H3K27me3, other factors may cooperate to repress CTCF bindings in Class 5. Regarding the “Decreased” class, an increase in H3K9me3 is observed in Class 1 upon Setdb1 KO (adj. P = 0), and an increase in H3K9me2 is observed in Class 2 upon TKO (adj. P = 9.2 × 10−14) (Supplemental Fig. S1F,G). This suggests that the reorganization of H3K9 methylation states due to the loss of each H3K9 methyltransferase inhibits CTCF binding.
Because certain fractions of CTCF binding are sensitive to DNA methylation (Wang et al. 2012; Kim et al. 2015), we examined the relationship between “Increased” CTCF binding sites and DNA methylation by reanalyzing the previously reported global DNA methylation status of WT and 5KO iMEFs (Supplemental Fig. S1H; Fukuda et al. 2023). The results show that significant reduction of DNA methylation is induced in 5KO at “Increased” sites other than Class 4. It is reported that the contribution of DNA demethylation to the newly emerged CTCF binding in Setdb1 KO mESCs is mostly dispensable (Tam et al. 2024). However, the certain fractions of “Increased” sites observed in the H3K9 methyltransferases KO iMEFs may be dependent on DNA demethylation.
H3K9 methyltransferases inhibit CTCF binding at repetitive sequences, which can be further compensated by H3K27 methyltransferases
All H3K9 methyltransferases are known to repress transposons by catalyzing H3K9 methylation (Matsui et al. 2010; Maksakova et al. 2013; Bulut-Karslioglu et al. 2014), and it was reported that SETDB1 and EHMT2 deposits H3K9 methylation on transposons, such as SINE B3, to prevent CTCF binding (Jiang et al. 2020; Gualdrini et al. 2022; Sun et al. 2024; Tam et al. 2024). In addition, H3K27me3 is known to accumulate on transposons in conditions where H3K9 methylation is low, such as in female primordial germ cells (Huang et al. 2021) or H3K9 methyltransferase-deficient cells (Fukuda et al. 2023). These findings may suggest that SUV39H1/2 and H3K27 methyltransferases also inhibit CTCF binding at transposons. To explore this, we examined the types of transposons enriched in each class. Consistent with previous reports that SETDB1 inhibits CTCF binding at SINE B3 (Gualdrini et al. 2022; Sun et al. 2024; Tam et al. 2024), SINE B3 is strongly enriched in “Increased” Class 1 (Fig. 2A; Supplemental Table S2). Furthermore, SINE B3 is also significantly enriched in “Increased” Class 2, which is regulated by Suv39h1/2, and in “Increased” Class 3, which is regulated by Ehmt1/2. This suggests that CTCF binding at SINE B3 is suppressed by all H3K9 methyltransferases (Fig. 2A; Supplemental Table S2). However, only about 9% of all SINE B3 copies overlap with CTCF binding sites identified in this study, suggesting that most SINE B3 elements are not bound by CTCF (Fig. 2B). An analysis of the epigenomic profile of SINE B3 reveals that, compared to the consensus peak, H3K9me3 levels are higher in WT for “Increased” Class 1, 2, and 3 (Fig. 2C). Upon Setdb1 KO, H3K9me3 levels at SINE B3 decrease in “Increased” Class 1, whereas “Increased” Class 2 remains within regions of high H3K9me3 (Fig. 2C; Supplemental Fig. S2A). H3K9me2 is depleted at SINE B3 in WT but is elevated exclusively in “Increased” Class 3 under TKO conditions (Fig. 2C; Supplemental Fig. S2A). A subset of SINE B3 copies in “Increased” Class 2 shows high levels of H3K27me3 enrichment in TKO, suggesting that H3K27me3 on these SINE B3 copies does not exert an inhibitory effect on CTCF binding (Supplemental Fig. S2B). These findings indicate that H3K9 methylation plays a central role in inhibiting CTCF binding to SINE B3 and that distinct H3K9 methyltransferases act on different copies of SINE B3. Phylogenetic analysis of SINE B3 showed that each class is not assigned to distinct clusters, suggesting that the regulatory differences among SINE B3 copies are not due to differences in SINE B3 subfamilies (Fig. 2D). Given that compartment patterns differ between classes (Supplemental Fig. S1D), we examined the compartment distribution of SINE B3 and found significant differences: SINE B3 in “Increased” Class 1 is enriched in the A compartment, Class 2 in the B compartment, and Class 3 in an intermediate level (Fig. 2E,F). These findings suggest that SINE B3 undergoes differential CTCF suppression by distinct H3K9 methyltransferases depending on the chromatin compartment in which it resides.
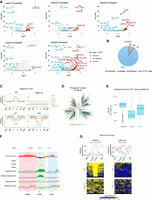
Prevention of additional CTCF binding at repetitive elements by H3K9/K27 methylation. (A) Enrichment of repeat type in increased DE peaks. The volcano plots represent the enrichment of each repeat type in DE peaks, with the x-axis showing log2(FC) and the y-axis showing −log10(adjusted P-value). Repeat types that are significantly enriched or depleted in each class compared to consensus peaks (adj. P-value < 0.01) are highlighted in red and blue, respectively. All raw data used in A were described in Supplemental Table S2. (B) Fraction of SINE B3 transposons overlapping with CTCF peaks. “Consensus,” “Increased,” “Decreased,” and “Non-CTCF peak” represent the fractions of SINE B3 copies that overlap with the “Consensus peaks,” “Increased” DE peaks, “Decreased” DE peaks, and those that do not overlap with any of the CTCF peaks identified in this study, respectively. (C) Enrichment of H3K9 methylation around SINE B3 transposons overlapping with “Increased” DE peaks. (D) Phylogenetic analysis of SINE B3 transposons. SINE B3 transposons overlapping with “Increased” DE peaks are color-coded according to their respective classes. (E) The distribution of Hi-C PC1 values in genomic regions where SINE B3 transposons overlap with “Increased” DE peaks. (F) Representative genomic regions where SINE B3 transposons overlap with “Increased” DE peaks. SINE B3 elements overlapping with Class 1 are predominantly found in strong A compartments, those overlapping with Class 2 are mainly in B compartments, and those in Class 3 are frequently located in intermediate regions between Class 1 and Class 2. (G) Epigenome profiles around IAPLTR2_Mm elements overlapping with “Increased” Class 5. In WT iMEFs, H3K9me3, regulated by SETDB1, is enriched. However, when H3K9 methylation is lost, H3K27me3 becomes enriched instead.
Unlike the “Increased” Class 1/2/3, the “Increased” Class 4—which is regulated by H3K27 methyltransferases—shows no enrichment of any transposons (Fig. 2A; Supplemental Table S2). However, various transposons including L1 and ERV are enriched in “Increased” Class 5, which is controlled by both H3K9 and H3K27 methyltransferases (Fig. 2A; Supplemental Table S2). These findings suggest that H3K9 and H3K27 methylation redundantly suppress potential CTCF binding sites within transposons. These transposons do not accumulate in Class 4, suggesting that, in the presence of H3K9 methylation, H3K27me3 does not function to inhibit CTCF binding on these transposons (Fig. 2A). H3K27me3 is redistributed to transposons enriched in “Increased” Class 5 (IAPLTR2_Mm, L1Fd_2) under 5KO conditions (Fig. 2G; Supplemental Fig. S2C). This suggests that the redistribution of H3K27me3 to transposons following the loss of H3K9 methylation plays a role in preventing CTCF binding at these transposons.
Unlike the “Increased” Class, the “Decreased” Class shows only a weak accumulation of a limited number of transposons, such as the IAPEY family in Class 1 (Supplemental Fig. S2D; Supplemental Table S2). GC-rich elements and simple repeats with high GC content are strongly enriched, particularly in “Decreased” Class 3, 4, and 5 (Supplemental Fig. S2D; Supplemental Table S2). Because GC-rich regions are often found in promoter regions, this enrichment aligns with the fact that these classes are frequently located in promoters (Supplemental Fig. S1C). These results indicate that H3K9 and H3K27 methyltransferases function both to promote and inhibit CTCF binding on repetitive elements, although the details of the difference are currently unknown.
Association between changes in CTCF binding profiles and alterations in gene expression and insulation
CTCF functions as an insulator, regulating three-dimensional genome structure and gene expression (Rao et al. 2014). To investigate how the changes in CTCF binding we identified affect gene expression and genome architecture, we reanalyzed our previously conducted RNA-seq and Hi-C data (Fukuda et al. 2023). To analyze the association between CTCF DE peaks and gene expression changes, we identified differentially expressed genes (DE genes) in each sample and calculated the enrichment of DE genes around DE peaks (Fig. 3A; Supplemental Fig. S3A). Specifically, genes upregulated in 5KO and 5KO+DS are highly enriched around “Increased” Class 3 and Class 5, respectively. In contrast, genes downregulated in Setdb1 KO, TKO, 5KO, and 5KO+DS are strongly enriched around “Decreased” Class 1, Class 2, Class 3, and Class 5, respectively (Fig. 3B; Supplemental Fig. S3B; Supplemental Table S2). Therefore, changes in CTCF binding caused by the loss or inhibition of H3K9/K27 methyltransferases may alter the expression of nearby genes.
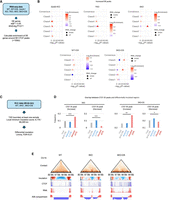
Association of DE peaks with changes in gene expression and 3D genome structure. (A) A process for analyzing the association between DE peaks and changes in gene expression. (B) A dot plot showing the enrichment of differentially expressed genes around DE peaks. The x-axis represents −log10(adj. P-value). The enrichment results for upregulated and downregulated genes are represented by circles and triangles, respectively. The degree of enrichment (log2[fold change]) is color-coded, with red indicating enrichment and blue indicating depletion triangles and circles, respectively. The degree of enrichment (log2[Enrichment]) is color-coded, with red indicating enrichment and blue indicating depletion. The details of the statistical calculations are described in the “Statistical analysis” section of the Methods. All raw data used in B are described in Supplemental Table S3. (C) A process for analyzing the association between DE peaks and changes in insulation. (D) Frequency of overlap between CTCF DE peaks and differentially insulated regions. (E) Representative regions of differential insulation overlapping with “Increased” DE peaks. The figure displays, from top to bottom, the contact heat map, insulation map, CTCF ChIP-seq signal, RNA-seq signal, and compartment information. In this region, an increase in CTCF binding, the emergence of new insulation sites, an increase in gene expression, and a B-to-A compartment change are observed in both 5KO and 5KO+DS iMEFs.
The association of both upregulated and downregulated genes with CTCF binding changes in 5KO and 5KO+DS prompted us to investigate insulation changes in these cells. Using our previously reported Hi-C data from WT, 5KO, and 5KO+DS (Fukuda et al. 2023), we extracted insulated regions at 40-kb resolution and identified regions where insulation changed in 5KO and 5KO+DS (Fig. 3C). We detected 472 and 523 regions with increased insulation and 1477 and 1073 regions with decreased insulation in 5KO and 5KO+DS, respectively (Supplemental Fig. S3C). In both 5KO and 5KO+DS, regions with elevated insulation significantly overlap with “Increased” CTCF peaks, whereas regions with reduced insulation significantly overlap with “Decreased” CTCF peaks (Fig. 3D). A total of 20 regions in 5KO and 74 regions in 5KO+DS among the increased DE peaks exhibited enhanced insulation and were located within 100 kb of differentially expressed genes. Several representative regions are shown in Figure 3E and Supplemental Figure S3D. These results suggest that H3K9 methylation and H3K27 methylation function independently or cooperatively to maintain the homeostasis of CTCF binding, 3D genome structure, and gene expression.
Prevention of CTCF binding by H3K9 methylation at early embryo–specific CTCF binding sites
The epigenome and 3D genome structure undergo dynamic changes during development. Recently, an analysis of CTCF binding dynamics in mouse early embryos reported that cleavage stage-specific CTCF binding sites are derived from the SINE B2 and B3 families (Wang et al. 2024). These cleavage stage-specific CTCF binding sites acquire H3K9me3 during development, leading to the loss of CTCF binding (Wang et al. 2024). This report prompted us to compare CTCF DE peaks, which we identified, with the CTCF binding and epigenome profiles during early development using publicly available data (Liu et al. 2016; Wang et al. 2018, 2024). The CTCF binding levels at “Increased” DE peaks dynamically fluctuate during early development (Supplemental Fig. S4A). The CTCF enrichment in “Increased” classes, except for Class 5, which is regulated by both H3K9 and K27 methyltransferases, are low in MII oocytes, peak at the two-cell stage, and then gradually decrease (Supplemental Fig. S4A). In contrast, H3K9me3 follows the opposite pattern, being lowest at the two-cell stage and increasing thereafter (Supplemental Fig. S4B). The CTCF binding levels of “Increased” Class 5 remains low throughout development (Supplemental Fig. S4A). Because the global levels of CTCF and H3K9me3 fluctuate significantly during development, we set CTCF Stable Peaks as internal control regions, where CTCF binding remains unchanged across all iMEF conditions analyzed. The CTCF/H3K9me3 levels in each developmental stage were then normalized using the median value of the Stable Peaks. Additionally, because CTCF-mediated chromatin loops emerge from the eight-cell stage onward (Wang et al. 2024), we focused our analysis on the eight-cell stage and Inner Cell Mass (ICM). H3K9me3 levels at “Increased” Class 1/2/3—CTCF binding regions repressed by H3K9 methyltransferases—are lower at the eight-cell stage than in the ICM, whereas CTCF binding is higher at the eight-cell stage. This trend is most pronounced in Class 1, which is regulated by Setdb1 (Fig. 4A,B). This trend is not observed in “Increased” Class 4 and 5 (Fig. 4A,B). These findings suggest that CTCF sites inhibited by H3K9 methylation, but not by H3K27me3, in iMEFs are in a low H3K9me3 state at the eight-cell stage, creating a permissive chromatin environment for CTCF binding. To investigate whether “Increased” Class1 peaks are associated in early embryo–specific 3D genome structures, we compared the insulation status between the eight-cell stage and ICM using previously reported Hi-C data (Du et al. 2017; Wang et al. 2024). The results show that “Increased” Class 1 specifically functions as an insulator at the eight-cell stage (Fig. 4C,D). Consistent with low CTCF binding throughout early development in “Increased” Class 5, Class 5 does not show insulation function at the eight-cell stage (Supplemental Fig. S4C). When examining the H3K27me3 profile at the eight-cell stage, Class 5 exhibits particularly high levels of H3K27me3, embedding within eight-cell stage-specific H3K27me3 domains (Fig. 4E,F). To investigate this statistically, we defined eight-cell stage-specific H3K27me3 domains as 100-kb bins in which all three replicates of eight-cell H3K27me3 ChIP-seq showed positive log2(ChIP/Input) signals, whereas both replicates of iMEF H3K27me3 ChIP-seq showed negative log2(ChIP/Input) signals. Based on this criterion, 6955 out of 26,700 bins were identified as eight-cell-specific H3K27me3 domains. We then compared the overlap frequency of consensus peaks and “Increased” Class 5 peaks with these regions using a proportion test. The results showed that “Increased” Class 5 peaks were significantly more enriched in the eight-cell-specific H3K27me3 regions (P < 2.2 × 10−16, 9.0% in consensus peak and 26.3% in “Increased” Class 5 peaks). These findings suggest that H3K27me3 may prevent additional CTCF binding during early embryo development, when H3K9 methylation becomes unstable.
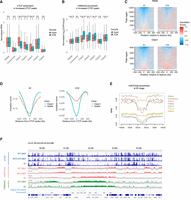
Association of DE peaks with 3D genome structures in mouse early embryos. (A) CTCF accumulation in “Increased” DE peaks in early mouse embryos. The y-axis represents CTCF accumulation, corrected by the median value of Stable CTCF peaks in each developmental stage. CTCF accumulation in DE peaks is tested between the eight-cell stage and ICM using a t-test. (B) H3K9me3 accumulation in “Increased” DE peaks in early mouse embryos. The y-axis represents H3K9me3 accumulation (log2[ChIP/Input]), corrected by the median value of Stable CTCF peaks in each developmental stage. H3K9me3 accumulation in DE peaks is tested between the eight-cell stage and ICM using a t-test. (C,D) A tornado plot showing the insulation status in Stable and “Increased” Class1 DE peaks in eight cells and ICM. Panel C displays the results for each region in a heat map, and panel D shows the average values of regions. The x-axis represents the distance from CTCF peaks. (E) Enrichment of H3K27me3 in the eight-cell stage around “Increased” DE peaks. Class 5 is embedded in regions with high H3K27me3 enrichment. (F) Representative regions of “Increased” Class5 DE peaks embedded in H3K27me3 enriched regions in the eight-cell stage.
Discussion
In this study, we utilized various H3K9 methyltransferase-deficient cells and H3K27 methyltransferase EZh1/2 inhibitor to elucidate the specificity and redundancy of each enzyme in the regulation of CTCF. We demonstrated that both types of methylation play a role in orchestrating 3D genome architecture and transcription by inhibiting CTCF binding. Loss of both H3K9 and H3K27 methylation resulted in large changes in the CTCF binding profile, indicating that these modifications function redundantly to maintain the robustness of the CTCF binding profile and 3D genome organization. Furthermore, classification of CTCF binding changes under different cellular conditions enabled us to identify the characteristics of genomic regions where each enzyme exhibits specificity or functions cooperatively. Setdb1 suppresses CTCF binding predominantly in the A compartment, Suv39h1/2 act mainly in the B compartment, and Ehmt1/2 contribute to repression in both (Supplemental Fig. S1D). Notably, CTCF sites corepressed by both H3K9 and H3K27 methylation are strongly enriched in the B compartment (Supplemental Fig. S1D). These observations suggest that distinct H3K9 methyltransferases exert compartment-specific control over CTCF accessibility and that dual-layered repression by H3K9 and H3K27 methylation is a characteristic feature of the B compartment. This raises an important question: Why is such a stringent repression of CTCF necessary specifically in the B compartment? One possibility is that the B compartment harbors repeat-rich, gene-poor regions that require robust epigenetic silencing to maintain genomic stability. CTCF binding in these regions could disrupt compact heterochromatin structure, induce inappropriate chromatin looping, or interfere with transcriptional repression of transposable elements. Such chromatin destabilization may predispose these regions to larger-scale genomic rearrangements, including segmental duplications, insertions, or deletions, particularly in repeat-rich and structurally fragile domains of the B compartment. To assess this possibility, it would be informative to perform long-read whole-genome sequencing (e.g., Pacific Biosciences [PacBio] HiFi or Oxford Nanopore Technologies [ONT]) in cells lacking both H3K9 and H3K27 methylation, in order to detect subtle or large structural variants that may arise from the loss of repressive constraints. Combined with high-resolution chromatin interaction mapping and transposon activity profiling, this approach could help determine whether the derepression of CTCF in heterochromatic regions contributes to genomic instability and structural evolution.
CTCF primarily peaks in the A compartment and is excluded from the B compartment (Supplemental Fig. S1D). Our analysis of cells deficient in both H3K9 and H3K27 methylation demonstrated that these modifications inhibit CTCF binding within the B compartment and prevent the formation of sub-TAD structures in the B compartments.
Transposons, which occupy about half of the mammalian genomes, can serve as major sources of binding sites for various transcription factors, but they are often covered by repressive chromatin modifications. It has been previously known that H3K9 methylation regulates CTCF binding at SINE B2/B3 transposons (Sun et al. 2024; Tam et al. 2024). In this study, we also observed an increase in CTCF binding on SINE B2/B3 elements upon the loss of H3K9 methylation (Fig. 2A). Furthermore, analyses of various H3K9 methyltransferase-deficient cell lines revealed that the suppression of CTCF binding on SINE B2/B3 depends on the chromatin compartment where the SINE elements are inserted and involves different H3K9 methyltransferases (Fig. 2E). Although H3K9 methylation deficiency is known to activate transposon expression (Fukuda and Shinkai 2020), our previous RNA-seq analysis did not detect increased expression of SINE B2/B3 (Fukuda et al. 2025). Furthermore, published reports also claimed that reduction of H3K9me2/3 on SINE B2/B3 impacts on CTCF binding without alteration of their transcription (Gualdrini et al. 2022; Tam et al. 2024). All studies used poly(A) selected RNA for RNA-seq analysis, which might have failed to adequately capture SINE B2/B3 transcripts. Recently developed techniques such as melRNA-seq (Mori and Ichiyanagi 2021), which are optimized for detecting SINE expression, may help clarify how the gain of CTCF binding affects SINE transcription. In addition, H3K9 methylation loss is known to result in reduced DNA methylation (Jiang et al. 2020; Fukuda et al. 2023). Whether changes in DNA methylation contribute to the altered CTCF binding at SINE B2/B3 is an intriguing question. However, the low-depth WGBS used in this study was insufficient to assess methylation status at individual copies. Due to multimapping issues, WGBS remains limited in its ability to resolve the DNA methylation landscape of individual transposon copies. This limitation may be overcome by using direct DNA methylation detection through nanopore sequencing, which remains a future direction of our research.
Our analysis of CTCF binding in cells deficient in both H3K9 methylation and H3K27me3 revealed additional potential CTCF binding sites that are normally repressed by these two repressive histone modifications. Furthermore, we found that not only the previously focused SINE B2/B3 transposons but also a wide variety of transposons can potentially function as CTCF binding sites (Fig. 2A). Recently, we reported that, in the absence of both H3K9 methylation and H3K27me3, uH2A functions to maintain compartmentalization (Fukuda et al. 2025). This raises the possibility that uH2A may also mask additional potential CTCF binding sites.
The state of H3K9 methylation dynamically changes during early development, and accordingly, genomic regions that usually do not bind CTCF, including transposons, can become specifically bound by CTCF in early embryos (Wang et al. 2024). We revealed that CTCF binding sites that are repressed by H3K9 methyltransferases (“Increased” Class1/2/3) in iMEFs show decreased H3K9 methylation and increased CTCF binding in early embryos (Fig. 4A,B). Thus, H3K9 methyltransferases prevent the formation of an early embryo-like 3D genome architecture in somatic cells. On the other hand, CTCF binding sites that are suppressed by both H3K9 methylation and H3K27me3 in iMEFs (classified as “Increased” Class 5) are associated with H3K27me3-enriched domains in early embryos (Fig. 4E,F) and exhibit persistently low CTCF binding throughout early development (Supplemental Fig. S4A). “Increased” Class 5 peaks are predominantly located in the B compartment (Supplemental Fig. S1D), where H3K9 methylation is typically abundant. However, upon the loss of H3K9 methylation, these sites gain H3K27me3 and form H3K27me3 domains (Fig. 1E,F). Such domain formation of H3K27me3 is also observed in aging cells (Yang et al. 2023), raising the possibility that H3K27 methylation may help preserve 3D genome architecture in such condition. In mouse early embryos, “Increased” Class 5 exhibits a unique epigenomic state characterized by the colocalization of H3K9me3 and H3K27me3, along with suppression of CTCF binding (Fig. 4A,B,E,F). Same as in “Increased” Class5, “Increased” Class 2, which is regulated by Suv39h1/2, also resides in the B compartment (Supplemental Fig. S1D); however, its chromatin features in early embryos differ substantially from those of Class 5. Specifically, Class 2 shows embryo-specific CTCF binding and a marked reduction of H3K9me3 at the eight-cell stage, whereas Class 5 retains high levels of H3K9me3 and remains refractory to CTCF binding (Fig. 4A,B). In iMEFs, both classes display similar epigenomic dynamics (Fig. 1E). What drives the differences between Class 2 and Class 5 remains unknown. It will be important to investigate whether they differ in sequence features, 3D genome organization, epigenomic status, or transcriptional activity. In any case, the fact that Class 5 regions are robustly protected from CTCF binding suggests that CTCF occupancy at these sites may be detrimental to the organism. Future studies are needed to elucidate the developmental and physiological significance of this suppression.
The molecular mechanism by which H3K27me3 inhibits CTCF binding remains unclear. Whereas DNA methylation is known to suppress CTCF binding, DNA methylation and H3K27me3 are anticorrelated, and DNA methylation levels decrease in regions where H3K27me3 infiltrates in 5KO cells (Fukuda et al. 2023). “Increased” Class 5 peaks already showed strong reduction of DNA methylation in 5KO iMEFs compared to WT iMEFs (Supplemental Fig. S1G). Therefore, it is unlikely that DNA methylation is responsible for inhibiting CTCF binding in H3K27me3-rich regions. Although “Increased” Class 2 also shows an increase in H3K27me3 levels in TKO and 5KO (Fig. 1E), similar to Class 5, it differs in that CTCF binding is increased regardless of H3K27me3 enrichment. This observation raises the possibility that, in Class 2, there may be a mechanism that counteracts the repressive effect of H3K27me3 on CTCF binding. Alternatively, it may suggest that H3K27me3 alone is not sufficient to block CTCF binding and that additional factors—such as specific chromatin modifications, chromatin architecture, or corepressors—are required for effective suppression. Understanding the mechanistic basis for this differential response will be essential for uncovering how chromatin states influence CTCF binding in a context-dependent manner. Further studies are needed to elucidate the mechanism by which H3K27me3 suppresses CTCF binding.
In conclusion, this study revealed previously unidentified potential CTCF binding sites and proposed a new regulatory layer that maintains the CTCF binding profile. Our findings provide new insights into the mechanisms controlling 3D genome architecture and gene expression regulation, shedding light on how these mechanisms contribute to cellular development and tissue-specific regulation.
Methods
Cell culture
iMEFs were maintained in Dulbecco's Modified Eagle's Medium (Nacalai Tesque, 08458-16) containing 10% fetal bovine serum (Biosera, FB1061), MEM Non-Essential Medium, and 2-Mercaptoethanol (Nacalai Tesque, 21417-52). To inhibit EZH1/2 catalytic activity, iMEFs were cultured for 7 days with 1 μM DS3201.
CTCF ChIP-seq
iMEFs (5 × 105) were crosslinked in 1 mL PBS supplemented with 0.1 mL Crosslinking Buffer (100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 50 mM HEPES [pH 8.0], 11% PFA) at 25°C for 20 min. The reaction was quenched by adding 0.125 M glycine. After washing with PBS, the pellet was resuspended in 300 µL SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 1% SDS, protease inhibitor). Chromatin was sonicated using a Picoruptor (Diagenode) with a cycle of 30 sec on/30 sec off for a total of five cycles. The lysate was then centrifuged at 15,000 rpm for 10 min at 4°C. The supernatant was diluted with 1.2 mL Chromatin Dilution Buffer (10% Tris-HCl [pH 7.5], 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1× cOmplete EDTA-free protease inhibitor [Roche, 11836170001], 1 mM PMSF). From the diluted chromatin solution, 100 µL was set aside for input control, and the remaining portion was incubated with 8 µg of anti-CTCF antibody (Active Motif, #61311) and 10 μL of Protein A/G agarose beads (Santa Cruz, sc-2003). The mixture was rotated at 4°C overnight. After rotating the solution, the beads were sequentially washed with low salt buffer (150 mM NaCl, Tris-HCl [pH 8.0], 2 mM EDTA, 1% Triton X-100, 0.1% SDS), high salt buffer (500 mM NaCl, Tris-HCl [pH 8.0], 2 mM EDTA, 1% Triton X-100, 0.1% SDS), LiCl buffer (25 mM LiCl, 10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1% NP-40, 1% DOC), and TE, and were suspended in Salting method buffer (20 mM Tris [pH 8.0], 10 mM EDTA, 400 mM NaCl). Input and ChIP samples were de-crosslinked by RNase A, SDS, and Proteinase K treatment, and purified with a QIAquick PCR purification Kit (Qiagen).
Preparation of ChIP-seq library
The ChIP DNA was fragmented by Picoruptor (Diagenode) for 10 cycles of 30 sec on, 30 sec off. Then, the ChIP library was constructed with a KAPA Hyper Prep Kit (KAPA BIOSYSTEMS) and SeqCap Adapter Kit A (Roche) according to the manufacturer's instructions. The concentration of the ChIP-seq library was quantified by a KAPA Library quantification Kit (KAPA BIOSYSTEMS). ChIP sequencing was performed on a HiSeq X platform (Illumina). We performed two biological replicates for ChIP-seq.
Hi-C data analysis
Hi-C data processing was done by using Docker for the 4DN Hi-C pipeline (v43, https://github.com/4dn-dcic/docker-4dn-hic). The pipeline includes alignment (using the mouse genome, mm10) and filtering steps. After filtering valid Hi-C alignments, HIC format Hi-C matrix files were generated by Juicer Tools (Durand et al. 2016) using the reads with MAPQ > 10. The A/B compartment (compartment score) profiles (in 250-kb bins) in each chromosome (without sex chromosome) were calculated from HIC format Hi-C matrix files (intrachromosomal KR normalized Hi-C maps) by Juicer Tools (Durand et al. 2016) as previously described (Miura et al. 2018). We averaged Hi-C PC1 values in each 250-kb bin from two biological replicates for the downstream analysis. We used GENOVA tools (van der Weide et al. 2021) for producing aggregate TAD plots, insulation analysis, and pyramid plots.
The insulation score was calculated for each 40-kb bin using GENOVA tools. In GENOVA, the insulation score is calculated using a sliding window that computes the average contact signal for each bin. This value is then normalized by the genome-wide average signal to produce the final insulation score. For each 40-kb bin, the local minimum of the insulation score was identified within a ±200-kb window (upstream 200 kb and downstream 200 kb). Bins with local minima were assigned as TAD boundaries. Out of 66,095 bins, 8178 were identified as TAD boundaries in at least one sample. Next, the insulation scores of all identified TAD boundaries were compared between WT and other conditions. Regions with significant changes in insulation (differential insulation) were identified using Limma (FDR < 0.05).
ChIP-seq analysis
CTCF ChIP-seq analysis was performed using Nextflow with the nf-core/chipseq pipeline (version 2.0.0) (Ewels et al. 2020). The analysis was conducted using the mouse mm10 genome as the reference. Differential peaks were identified using DiffBind (FDR < 0.05) (Ross-Innes et al. 2012) based on narrow peaks called by MACS2 (Zhang et al. 2008) in the Nextflow pipeline. The commonality of differential peaks across samples was analyzed using BEDTools (Quinlan and Hall 2010), and peaks were classified into five categories accordingly.
RNA-seq analysis
Adaptor sequences and low-quality bases in reads were trimmed using Trim Galore! (version 0.3.7) (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). To calculate RPM values in each 250-kb bin, trimmed reads were aligned to the mm10 genome build using Bowtie (version 0.12.7) (Langmead et al. 2009) with –m 1. Duplicated reads were removed using SAMtools version 0.1.18 (Li et al. 2009). The number of mapped reads in each 250-kb bin was counted by featureCounts (Liao et al. 2014), then, RPM values in each 250-kb bin were calculated. To identify differentially expressed genes or repeats, the trimmed reads were mapped to the mouse GRCm38 genome assembly using TopHat (v2.1.1) with –g 1 (Trapnell et al. 2009). After read mapping, the number of reads mapped in genes or repeats was counted by TEtranscripts (v1.4.11) with default parameters) (Jin et al. 2015). We performed two biological replicates for RNA-seq and identified DE genes by DESeq2 (adj. P-value < 0.05, FC > 2) (Love et al. 2014).
Visualization of NGS data
The Integrative Genomics Viewer (IGV) (Robinson et al. 2011) was used to visualize next-generation sequencing (NGS) data. For the Hi-C contact matrix and correlation matrix, we used Juicer Tools (Durand et al. 2016).
Statistical analysis
All statistical analyses were performed using R (R Core Team 2022). All methods for statistical analysis and P-values in this study are described in each figure legend and included in each figure, respectively. In Figure 1E and Supplemental Figure S1E, statistical comparisons between WT and the other samples were performed using a t-test, followed by Benjamini–Hochberg correction for multiple testing. In Figure 2A and Supplemental Figure S2D, the fold changes and P-values were calculated by comparing the number of transposable elements (TEs) overlapping with consensus peaks versus those overlapping with each DE peak class. In Figure 3B and Supplemental Figure S3B, the P-values were calculated using the prop.test function in R, based on four key numbers: (1) the total number of genes analyzed; (2) the number of genes overlapping with differentially bound peaks (DE peaks); (3) the total number of differentially expressed genes; and (4) the number of DE genes that also overlap with DE peaks. In Figure 3D, the frequency of overlapping CTCF DE peaks was compared between all insulated regions and differentially insulated regions, and statistical significance was assessed using a proportion test (prop.test). In Figure 4A, statistical comparisons between developmental stages were performed using a t-test.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE GSE297986. All accession numbers for the NGS data used in this study are provided in Supplemental Table S4.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
Y.S. was supported by a RIKEN internal research fund (Pioneering project “Genome building from TADs”) and by the Japan Society for the Promotion of Science (JSPS) (Grant-in-Aid for Transformative Research Areas [A], 25H01302; Grant-in-Aid for Scientific Research [A], JP22H00413; and Grant-in-Aid for Scientific Research on Innovative Areas [Research in a proposed research area], JP18H05530). K.F. was supported by the JSPS (Grant-in-Aid for Transformative Research Areas [A], 25H01302; and Grant-in-Aid for Early-Career Scientists, 22K15044). We thank the staff of the Support Unit for Bio-Material Analysis (BMA) at the RIKEN Center for Brain Science (CBS) Research Resources Division (RRD) for NGS library construction, DNA sequencing, and flow cytometry. We also thank our colleagues at Shinkai laboratory for their support and valuable comments.
Author contributions: K.F. and Y.S. designed and conceived the study. K.F. and Y.S. supervised the study and interpreted the data. C.S. performed molecular and cellular experiments and generated the ChIP-seq, RNA-seq, and Hi-C-seq libraries. K.F. performed informatics analysis of generated NGS data. K.F. and Y.S. wrote the manuscript and prepared figures. All authors read, discussed, and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280732.125.
- Received April 2, 2025.
- Accepted July 28, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








