
Hierarchical architecture of neo-sex chromosomes and accelerated adaptive evolution in tortricid moths
Abstract
Sex chromosomes can expand through fusion with autosomes, thereby acquiring unique evolutionary patterns. In butterflies and moths (Lepidoptera), these sex chromosome–autosome (SA) fusions occur relatively frequently, suggesting possible evolutionary advantages. Here, we investigated how SA fusion affects chromosome features and molecular evolution in leafroller moths (Lepidoptera: Tortricidae). Phylogenomic analysis showed that Tortricidae diverged ∼124 million years ago, accompanied by an SA fusion between the Merian elements M(20 + 17) and MZ. In contrast to partial autosomal fusions, the fused neo-Z Chromosome developed a hierarchical architecture, in which the three elements exhibit heterogeneous sequence features and evolutionary patterns. Specifically, the M17 part had a distinct base composition and chromatin domains. Unlike M20 and MZ, M17 was expressed at the same levels as autosomes in both sexes, compensating for the lost gene dosage in females. Concurrently, the SA fusion drove M17 as an evolutionary hotspot, accelerating the evolution of several genes related to ecological adaptation (e.g., ABCCs) and facilitating the divergence of closely related species, whereas the undercompensated M20 did not show such an effect. Thus, accelerated evolution under a novel pattern of dosage compensation may have favored the adaptive radiation of this group. This study demonstrates the association between a karyotype variant and adaptive evolution and explains the recurrent SA fusion in the Lepidoptera.
Large-scale chromosomal rearrangements such as fusions may promote ecological adaptation and speciation (Guerrero and Kirkpatrick 2014; Lucek et al. 2023). However, empirical evidence supporting this assumption remains scarce (Bracewell et al. 2017; Liu et al. 2022; Li et al. 2023). Butterflies and moths (Lepidoptera) exhibit a wide range of chromosome numbers, possibly owing to their holocentric chromosomes (Blackmon et al. 2017), which can tolerate large-scale rearrangements during meiosis. Therefore, Lepidoptera represents an ideal system for studying the evolution of chromosome rearrangements. In Lepidoptera, the sex Z Chromosome is frequently involved in chromosome fusions (Wright et al. 2024), giving rise to the so-called neo-sex chromosomes. These fusions increase the number of sex-linked genes, which can thus evolve faster than genes on other chromosomes—a phenomenon known as the Faster-X/Z effect (Coyne 2018). Theory predicts that sex chromosome–autosome (SA) fusions should be rare when the diploid autosome count is greater than 15 (Anderson et al. 2020). Thus, the relatively high proportion of SA fusions in Lepidoptera may be related to positive selection. Additionally, the rate of karyotypic variation is highly associated with species richness in Lepidoptera, suggesting a role for chromosome fusion in speciation (de Vos et al. 2020).
Chromosome fusions involving X/Z often synchronously generate sex-limited chromosomes, neo-Y/W, that degenerate through the suppression of recombination. Hemizygosity of genes linked to the neo-X/Z Chromosome then selects for dosage compensation (Natri et al. 2013; Sigeman et al. 2021). This process occurs slowly in mammals and birds and produces evolutionary strata with distinct divergence (Zhou et al. 2014). However, because of achiasmatic meiosis, lepidopteran females lack chromosomal recombination (Turner and Sheppard 1975; Marec 1996; Rasmussen et al. 1997), resulting in immediate suppression of recombination and rapid degeneration of the neo-W after SA fusion. In several lepidopteran species, sex chromosome dosage compensation (SCDC) has restored the gene dosage for the fusion-added part of the Z sex chromosome (added-Z) to autosomal levels (Gu et al. 2017, 2019), suggesting that the rapid degeneration of the neo-W is tolerable, although it is uncertain whether SCDC is common for added-Z. Overall, given the rapid degeneration of the neo-W, the exposure of recessive sites, and autosomal-level expression of hemizygous added-Z, we hypothesize that the intensity of selection for SA fusion is high in Lepidoptera.
Leafroller moths of the family Tortricidae represent one of the largest lepidopteran radiations (Mitter et al. 2017) with many economically important pest species (Brown 2022) and well-described karyotype evolution (Šíchová et al. 2013; Wright et al. 2024). In tortricids, one SA fusion occurred in the most recent common ancestor of Tortricinae and Olethreutinae, whereas two additional autosomal fusions occurred in the Olethreutinae (Šíchová et al. 2013; Béliveau et al. 2022). The SA fusion is hypothesized to be adaptive in tortricids owing to the involvement of detoxifying genes (Nguyen et al. 2013; Wan et al. 2019). In the olethreutine moth, Grapholita molesta, higher population differentiation was observed on the neo-Z Chromosome, which contains putative adaptive loci (Cao et al. 2022). Therefore, the SA fusion appears to have facilitated the divergence and adaptation of tortricid moths. SA fusion can influence adaptation and divergence in multiple ways. First, sex chromosomes evolve more rapidly (the Faster-X/Z effect) owing to unique evolutionary dynamics arising from their reduced effective population size and the exposure of recessive mutations to selection in the heterozygous sex (Charlesworth et al. 1987, 2018). Second, fusions could be selected if they tighten the linkage between adaptive alleles (Guerrero and Kirkpatrick 2014; Liu et al. 2022) or resolve sexual conflict via sex linkage (Kitano et al. 2009). Third, the evolution of SCDC on young sex chromosomes could facilitate the accumulation of alleles involved in speciation (Filatov 2018). Finally, fusions could alter the spatial arrangement of chromatin, consequently impacting the gene expression and recombination landscape (Gibcus and Dekker 2013; Vara et al. 2021).
Here, we report chromosome-level genomes of three economically important tortricid pests: G. molesta, Grapholita dimorpha, and Cydia pomonella. We sampled 24 genomes of related species, assigned their chromosomes to 32 ancestral linkage groups of lepidopterans (termed Merian elements) (Wright et al. 2024), and characterized the evolution of chromosome fusion in Tortricidae. The features of SA-fused chromosomes were explored in Tortricidae using transcriptomic and multiple genomic approaches. Then, we analyzed genomic divergence and adaptive evolution of the SA-fused chromosomes on using molecular evolution and population genetic analyses. Our results provide new insights into how karyotype evolution facilitates ecological adaptation and diversification of leafroller moths.
Results
Genome assembly and annotation
Long reads (85×–174×, Oxford Nanopore Technologies [ONT]) and Hi-C technologies were employed to obtain the genome assemblies of G. molesta, G. dimorpha, and C. pomonella, with respective sizes of 473 Mb, 486 Mb, and 604 Mb (Supplemental Fig. S1). Each genome contained 27 autosomes and a Z sex Chromosome. Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis showed a 97.7%–98.1% assembly completeness. The assemblies for G. molesta and C. pomonella were improved compared with previous versions (Wan et al. 2019; Cao et al. 2022). Gene annotation for the three assembled genomes yielded 14,234–14,513 genes. We found that 48.00%–59.35% of these assemblies consisted of repetitive sequences. We also annotated 24 genomes of related species based on protein homology and obtained 93.1–98.8% BUSCO completeness except for Leguminivora glycinivorella (85.1%) (Supplemental Table S1).
Evolution of chromosomal fusion in Tortricidae
Synteny comparisons between tortricid genomes and outgroup species showed high conservation for most chromosomes (Fig. 1A; for all chromosomes, see Supplemental Fig. S2), including the short chromosomes that are often involved in fusion events in Lepidoptera (Wright et al. 2024). Based on synteny, we assigned all chromosomes to the ancestral linkage groups of Lepidoptera (Merian elements; Autosomes: M1–M31; Ancestral Z Chromosome: MZ) (Wright et al. 2024). Consistent with previous reports (Wright et al. 2024), we detected a SA fusion comprising two ancient Merian elements (M17 + M20) and the Z Chromosome, forming the neo-Z Chromosome, F(20 + 17,Z) and two autosomal fusions specific to the Olethreutinae, F(5,13) and F(18,26) (Fig. 1A). We also identified several sporadic fusion events for single species and intrachromosomal inversions using synteny plots (Supplemental Fig. S2). We were unable to locate the fusion breakpoints by searching for the telomere sequence “(TTAGG) × 3” in any fused chromosomes of the studied tortricid species. By comparing gene positions, we found that the breakpoint was roughly central to the fused chromosomes of F(20 + 17,Z). In G. molesta, F(20 + 17,Z) comprised 18.9 Mb of M(20 + 17), and 17.1 Mb of MZ.
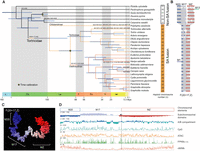
Evolution and features of the neo-Z Chromosome in Tortricidae. (A) Phylogenetic tree for 27 species of Tortricidae and outgroups. All branches had 100% bootstrap support. The node bars show 95% posterior densities for divergence times. Fusion events are marked on the branches. (B) Chromosome synteny of elements involved in SA fusion. (C) Three-dimensional architecture of F(20 + 17,Z) in Grapholita molesta. The white color denotes the fusion position. (D) Multiple features along F(20 + 17,Z) in G. molesta, including chromosome composition, subchromosomal domains, A/B compartments, the frequency of DNA methylation (CpG), the density of chromatin accessibility regions (ATAC-seq), gene expression levels (mean of FPKM, 0–100 are shown), and recombination rates (cM/Mb). The dotted line indicates the position of the SA fusion. The arrow denotes that the domain occupied by MZ has expanded to M17 (∼450 kb).
Using 6046 concatenated single-copy orthologs (SCOs), we constructed a phylogenetic tree for the Tortricidae. Molecular dating was used to set the upper limits for the timing of fusion events. The SA fusion occurred at the most recent common ancestor of the Tortricidae, ∼124 million years ago (Mya), and two autosomal fusions occurred ∼72 Mya in Olethreutinae (Fig. 1A).
Hierarchical features of neo-Z
To understand the consequences of SA fusion, we analyzed multiple chromosomal features of F(20 + 17,Z), including the guanine–cytosine (GC) content, the density of repeat transposable element (TE), and the exon density of four tortricid species (Polylopha cassiicola, Choristoneura fumiferana, C. pomonella, and G. molesta) and one outgroup species, Sesia bembeciformis along the phylogeny (Fig 1A). We found compartmentalized organization on F(20 + 17,Z), with a decrease in GC content and a local increase in the TE density at fusion breakpoints and clustering of genes in the low repeat region of M17 relative to chromosomes without SA fusion (Supplemental Fig. S3A). In contrast, these features appeared to be more evenly distributed along the other elements involved in autosome fusion (Supplemental Fig. S3B,C). SA fusion thus appears to have affected the sequence composition of F(20 + 17,Z).
We also investigated the effect of fusion on the three-dimensional (3D) chromosomal architecture. We constructed a 3D model of F(20 + 17,Z) using Hi-C data and found that F(20 + 17,Z) was complex in structure. The topology of M17 appeared independent of M20 and MZ and was less condensed (Fig. 1C). A distinctive topology formed the boundary between M(20 + 17) and MZ (Fig. 1B), corresponding to the TE-rich region (Supplemental Fig. S3A). This topology was further supported by the C-score (Fig. 1C, subchromosomal domains; Zheng and Zheng 2018). However, no such heterogeneity was observed in the two autosomal fusions (Supplemental Fig. S3B,C). Notably, the MZ domain extended to the tail of M17 (Fig. 1D, arrow). However, we did not find any reshaping of the two topologically associated domains at the end of M17 (Supplemental Fig. S4A). We also did not find any clear effect of the SA fusion on the A/B compartment distribution, neither at the broad chromosome level across multiple species nor in homologous coding regions (Supplemental Fig. S4B,C). Therefore, the 3D architecture of the three involved elements was less affected by SA fusion and remained independent after the fusion event.
The epigenetic signature also showed uneven changes along F(20 + 17,Z) (Fig. 1C). The frequency of CpG methylation (CpG; Mann–Whitney [M-W] U test, P = 0.029), the chromatin accessibility (ATAC, M-W U test, P < 0.001), and transcriptional expression (fragments per kilobase per million mapped [FPKM]; M-W U test, P < 0.001) were higher on M17 than on M20 and MZ. Moreover, we observed a clear discontinuity of chromosomal features between elements on F(20 + 17,Z). Overall, M17 occupied a unique topological domain and maintained a high degree of chromatin activity.
The distinctive pattern of SCDC is associated with selective pressure
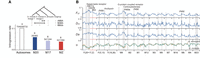
Unlike birds, the W Chromosome is extremely degenerated in butterflies and moths, sometimes even absent (Sahara et al. 2012). In G. molesta, this was confirmed by the female read coverage along F(20 + 17,Z), which was reduced by half compared with males (Supplemental Fig. S5). Therefore, the dosage of the M20 and M17 genes should be compensated in female tortricids. We analyzed the gene expression in G. molesta per individual elements of F(20 + 17,Z) and found that, in both sexes, MZ maintained half of the ancestral autosomal average as represented by the outgroup S. bembeciformis, which represents the ancestral pattern of SCDC in Lepidoptera (Fig. 2A). In M(20 + 17), genes from M17 were expressed (FPKM > 0.1) at the same levels as autosomes in both sexes (Fig. 2A; Supplemental Fig. S6A). However, genes from M20 exhibited similar levels of expression as those linked to MZ in females, whereas their expression levels were not reduced in males (Fig. 2A), indicating dysregulation between the sexes. The SCDC patterns were robust across various minimum FPKM cutoff criteria from 0.1 to five (Supplemental Fig. S6B) and were overall heterogeneous along F(20 + 17,Z) (Fig. 2B).

Patterns of sex chromosome dosage compensation (SCDC) of neo-Z in G. molesta. (A) Comparisons of gene expression among chromosomal elements by bootstrapping of FPKM. The purple and yellow lines, respectively, indicate the median of raw FPKM and the average of resampled FPKM medians. The vertical axis shows the distribution density in the range from zero to 0.5. (B) A model of SCDC in the neo-Z Chromosome. The arrows indicate the up- or downregulation of expression in chromosomal elements.
The high expression level of M17 was consistent with its high chromatin accessibility described above (Fig. 1C, ATAC). Moreover, M17 contained more active genes (452 of 506 active genes) than M20 (126 of 160 active genes) and MZ (450 of 553 active genes; chi-square tests, P < 0.005), indicating that the high accessibility of M17 depended on the activity but not the density of genes along F(20 + 17,Z). Therefore, the differences in expression between elements were caused by differences in regulation rather than by coincidental clustering of highly or poorly expressed genes when compartmentalizing Merian elements.
Expression levels can influence the efficiency of natural selection. We employed a multiple linear regression approach to examine this correlation in the Tortricidae. After accounting for the background nucleotide diversity for each gene, we found a significant correlation between functional genetic diversity (πN) and expression levels for genes both on the neo-Z (t-test, P = 0.028) (Supplemental Fig. S7) and on autosomes (t-test, P < 0.001). This finding was consistent with the more intense selection pressure on highly expressed genes, particularly those on M17.
Accelerated molecular evolution following SA fusion
The differences in the pattern of SCDC may influence the perceived extent of Faster-Z evolution (Mank et al. 2010). Given the novel features and SCDC pattern, we investigated whether M17 demonstrated a different rate of molecular evolution. We estimated the ratio of nonsynonymous to synonymous substitutions (dN/dS or ω) with the 6533 SCOs (M20: 83; M17: 250; MZ: 261; autosomes: 5939) across 20 genomes of Tortricidae (Fig. 1A) and found that ω was significantly higher on M17 and significantly lower on M20 (M-W U tests, P < 0.001) (Fig. 3A). This difference in M20 and M17 was not observed in an outgroup species without SA fusion (Fig. 3B).

Comparison of molecular evolution rates among chromosomal elements on neo-Z. (A,B) Violin plots of gene-wide ω on different chromosomal elements within the SA fusion (Tortricidae) and non-SA fusion (outgroups) clades. Significant differences from Mann–Whitney U tests (α = 0.01) are marked using uppercase letters. The horizontal lines show the medians. (C,D) Molecular evolution rate (ω) of different chromosomal elements. The term ω0,1, indicates sites under purifying selection (ω0) or neutral evolution (ω1) in both Tortricidae and the outgroups. The term ω2a,2b, indicates sites under purifying selection (ω2a) or neutral evolution (ω2b) in the outgroups but under positive selection in the Tortricidae.
We asked whether phylogenetic clades with the SA fusion (Tortricidae) had different rates of molecular evolution from those without SA fusion. Within a phylogenetic framework that includes six outgroup species without F(20 + 17,Z) (Fig. 1A), we conducted a branch-site model using CodeML in PAML (Yang 2007) to test for variation in the ω-values of codon sites under negative and positive selection between tortricids and the outgroups. For concatenated SCOs, the rates of purifying selection were similar among elements (ω ≈ 0.045) (Fig. 3C), but the values of ω for positively selected sites were much higher on the fused M17 and MZ (ω > 30, Fig. 3D) than on other chromosomal elements. Elevated ω can be caused by the relaxed purifying selection or the intensified positive selection. To further clarify the changes in selection pressure on M17, RELAX (Wertheim et al. 2015) was employed; the program introduces a parameter K that indicates overall relaxation (K < 1) or intensification (K > 1) of selection. The results showed that positive selection for M17 and ABCC genes (see below, Supplemental Table S4) was significantly intensified after the SA fusion (Supplemental Fig. S8), consistent with the comparison of ω among chromosomal elements (Fig. 3C,D).
We conducted a branch-site model analysis for each SCO and detected 1697 positively selected genes (PSGs; PLRT < 0.05) in tortricid moths (Supplemental Table S4). Among these, 219 PSGs with a false-discovery rate < 0.05 were enriched for 123 GO terms (Q < 0.05) (Supplemental Table S5), including several functions related to cell morphogenesis. Moreover, the 46 PSGs (PLRT < 0.05) on M17 were enriched for a major function (P < 0.05) (Supplemental Table S6) associated with the Wnt signaling pathway, a classical pathway related to morphology (Parr and McMahon 1994; Martin et al. 2012). We found several large-effect genes under strong positive selection, including ABCCs and HERC2 (Supplemental Table S5). These genes were well documented to be associated with pigmentation (Kayser et al. 2008) and ecological adaptation (Beran and Petschenka 2022). The results suggested that M17 may promote ecological and phenotypic divergence of tortricids.
Evolution of neo-Z in recently diverged species
Because of the high-molecular evolution rate of M17, we compared evolutionary dynamics between fused F(20 + 17,Z) and autosomes in two recently diverged species, G. molesta and G. dimorpha. First, we employed an ABBA–BABA approach to testing for relative resistance to interspecific gene flow among chromosomal elements. The introgression rate (defined as the proportion of non-BBAA patterns) of the three elements on F(20 + 17,Z) was significantly lower than that of autosomes (chi-squared tests, all P < 0.01) (Fig. 4A), indicating that a stronger barrier to interspecific gene flow may be established on M(20 + 17) of F(20 + 17,Z), where MZ has the lowest introgression rate, although such a pattern may also be expected even in the absence of gene flow between lineages (Presgraves 2018).
Evolution of neo-Z in two recently diverged species, G. molesta and G. dimorpha. (A) The introgression rates defined as (ABBA + BABA)/(ABBA + BABA + BBAA) among different chromosomal elements. The insert shows the ABBA-BABA pattern with (ABBA + BABA) or without (BBAA) introgression. The significance levels and the raw data of (ABBA + BABA)/(ABBA + BABA + BBAA) are shown at the top of the column. (B) Genomic divergence in patterns of FST, DXY, Da, and π. The 14 longest chromosomes are shown. The gray bars indicate the genomic islands of divergence that overlap with genes.
Second, genome resequencing of G. molesta (n = 54) and G. dimorpha (n = 17) indicated higher average differentiation on F(20 + 17,Z) than on autosomes (Fig. 4B) according to the values of FST, DXY, and Da (the accumulation of nucleotide differences after divergence), and lower intraspecific nucleotide diversity (M-W U tests, P < 0.001), suggesting divergent selection on the fused F(20 + 17,Z). Notably, several highly differentiated regions (referred to as genomic islands) were identified by the values of FST and Da (Fig. 4B, gray bar; Supplemental Fig. S9). In these islands, we identified multiple genes or gene families related to host adaptation (for the longest 14 chromosomes, see Fig. 4B; Supplemental Table S7) such as the ABCC4 transporter on M17 that was also detected in previous analysis (Supplemental Table S4) and the sweet-taste receptor that may be related to host preference (Agnihotri et al. 2016). We also identified genes with discrete functions in other regions, such as the Wnt-related gene that is frequently associated with morphological differences, including the wing type in Lepidoptera (Parr and McMahon 1994; Martin et al. 2012), and Cytochrome P450 genes on autosomes.
Repatterning of recombination and signals of selection on neo-Z
One major hypothesis supporting the adaptive role of chromosomal fusions is their ability to facilitate the linkage between adaptive alleles by creating regions of low recombination (Charlesworth and Charlesworth 1979; Yeaman 2013; Liu et al. 2022). To test this hypothesis, we inferred the recombination landscape of F(20 + 17,Z) based on SNP data in G. molesta or the GC content in other related species. The GC content was presumed to be positively correlated with the recombination rate (Galtier 2004; Lesecque et al. 2013).
In G. molesta, the estimated genome-wide recombination rate (r) of autosomes ranges from 2.07 to 3.57 cM/Mb, whereas that of F(20 + 17,Z) was 0.88 cM/Mb (Fig. 5A). The recombination landscape showed a “U” shape for most long chromosomes, including the fused chromosomes (Fig. 5A). For F(20 + 17,Z), such a “U” shape was conserved after the SA fusion, as suggested by the GC content of multiple species in the Tortricinae (Supplemental Fig. S3A). For the outgroup species, S. bembeciformis, the U-shaped recombination landscape was also observed for most chromosomal elements, including M(20 + 17), MZ, M5, M13, M18, and M26 (Fig. 5A). Therefore, the recombination rates of related chromosomes were reduced after fusion, especially for the regions around the fusion breakpoints. This reduction is attributed not only to the loss of independence of involved elements during meiosis but also to the placement of certain sequences (e.g., M17) in the middle region of the chromosomes, as these regions exhibited lower recombination potential. We also showed that r was negatively correlated with interspecific FST and Da in F(20 + 17,Z) and autosomes (Fig. 5B,C), suggesting the possible role of recombination repatterning by fusion in shaping genomic divergence.
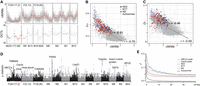
Landscapes of recombination and selection in G. molesta. (A, top) The recombination rate (in units of cM/Mb) of the seven longest chromosomes in G. molesta; (bottom) the intron GC content of the upper-corresponding chromosome elements in outgroup Sesia bembeciformis, reflecting the recombination landscape prior to the fusion. The orange lines indicate the 10th-degree polynomial regression curve. Horizontal dashed lines indicate the average recombination rate for each chromosome. The horizontal axis of GC content in S. bembeciformis was scaled to suit the chromosome length of G. molesta. (B,C) Correlations of the recombination rate with interspecific FST and Da in 100 kb sliding windows. (D) Signals of selective sweeps in G. molesta. The purple horizontal line shows the cutoff for 0.5‰ μ-statistics. (E) Distributions of linkage for different regions of chromosomal elements and the two gene clusters that are marked in red in D.
If fusions facilitate adaptation by strengthening the linkage between adaptive alleles, we would expect an increased signature of selection on the fused chromosomes. In G. molesta, we found that M17 was one of the four elements with a significantly higher number of selected loci, accounting for variation in the length of elements (Supplemental Table S7). Interestingly, the selection signals for M17 showed a tendency to cluster at the fusion point (Fig. 5D). By examining the sites with the top 0.5‰ of the μ-statistics, we identified genes with discrete functions (Supplemental Table S8). Notably, we again identified the ABCC (n = 3) and Ces3 (n = 2) gene clusters on M17, which showed tighter linkage compared with other regions (Fig. 5E). These results suggest that the SA fusion may have provided an evolutionary opportunity for these ecological performance genes and facilitate the adaptation of tortricid species. However, the accelerated evolution of M17 may also benefit from its unusual gene content, although no signals were observed for the other 19 ABCC-paralogs on the autosomes.
Discussion
The adaptive differentiation of many lepidopteran species has been hypothesized to be related to frequent SA fusion events (Carabajal Paladino et al. 2019; de Vos et al. 2020). Here, our analysis of F(20 + 17,Z) further strengthens this association in the Tortricidae. Our genomic analyses at the family scale, between sibling species, and within tortricid species found that the autosome involved in SA fusion has high rates of molecular evolution. To elucidate the underlying mechanisms, we investigated how SA fusion affects the nuclear architecture, gene expression, and recombination landscape. Comparisons between the neo-Z and corresponding unfused chromosomes of outgroups demonstrated that the changes in multiple features mediated by SA fusion have impacted the evolutionary dynamics of related synteny blocks as well as the recent adaptation and divergence in the Tortricidae.
On the neo-Z, three distinct subchromatin domains corresponding to the three ancestral chromosomal elements suggest a limited effect of fusion on the 3D architecture. This contrasts sharply with the chromatin remodeling observed in mice (Vara et al. 2021), muntjac deer (Yin et al. 2021), and yeast (Di Stefano et al. 2020). This specific chromosome structuring could be significant, as multiple-level chromatin organization has been shown to influence gene expression, recombination, and mutation in various species (Li et al. 2021; Gaskill and Harrison 2022). For example, we observed a decrease in the recombination rates at the boundaries of M17 (Fig. 1C), indicating that structure of the M17 domain contributes to its low recombination rate. However, such structuring was not observed in the fused autosomes. The difference may reflect a distinction between autosomes and sex chromosomes. The hierarchical 3D architecture also seems to have affected the distribution of TEs, including the accumulation of TEs at both ends of M(20 + 17) in relatively younger species such as G. molesta and C. pomonella. Because of the close association of TEs with heterochromatin (Pimpinelli et al. 1995), changes in TEs distribution may in turn exacerbate heterochromatinization and the dysregulation of gene expression and may reduce selection in certain chromosome regions such as M20.
Moreover, the hierarchical 3D architecture may be related to the complex pattern of SCDC. We observed a TE-rich heterochromatin block at the boundaries of M17 and MZ. This architecture is also frequently observed in the SA fusion of other species (Viegas-Péquignot et al. 1982; Dutrillaux and Dutrillaux 2023) such as the monarch butterfly Danaus plexippus (Gu et al. 2019). The impact of such heterochromatin block is largely unexplored, but it may affect gene expression and evolution. In humans, a similar heterochromatin block sequesters the ancestral X segment from the fused autosome, thereby preventing the transcriptional regulatory pattern from the ancestral X from spreading to the fused autosome (Ratomponirina et al. 1986; Dutrillaux and Dutrillaux 2023). Coincidentally, the neo-Z Chromosomes [F(14,Z)] in D. plexippus (Gu et al. 2019; Wright et al. 2024) and the present F(20 + 17,Z) in G. molesta possess a hierarchical pattern of SCDC. Furthermore, we showed that the hierarchical architecture after fusion was associated with the GC and TE composition (Supplemental Fig. S3). Therefore, conservation of the 3D architecture may contribute to SCDC and serve as the genomic basis for the differentiation of molecular evolution among chromosomal elements.
Consistent with the prediction (Cicconardi et al. 2021), we revealed reduced recombination owing to SA fusion. However, in the sex chromosome system of Lepidoptera, the Z Chromosome likely experiences a relatively higher recombination rate than the autosomes, because in meiosis, two-thirds of the Z Chromosomes but only half of the autosomes recombine as a result of the female achiasmatic meiosis (Székvölgyi et al. 2015). This contradictory observation concerning the recombination rate may require alternative explanations. For example, a higher reproductive cost of males (Bateman 1948) may reduce the effective population size and recombination opportunities for the Z Chromosome, and the linkage selection of haploid Z Chromosomes in females may reduce genetic diversity and the extent of homozygosity, resulting in local inefficiency of meiotic crossover. Although the suppression of recombination may reduce the efficiency of selection, it can still facilitate adaptation by reinforcing the association of adaptive sites, as suggested by the present study, as well as studies in other butterflies and fishes (Wellband et al. 2019; Cicconardi et al. 2021; Liu et al. 2022). The severely reduced recombination rate may also result in a reduced GC content and an increased repeat content around the fusion breakpoint. This is of interest, as the accumulation and activity of TEs have been shown to impact the temporal and spatial expression of genes and promote speciation (Feiner 2016; Singh et al. 2020). Moreover, reduced recombination can also result in an increased efficiency of background selection and resistance to interspecific gene flow. Thus, the repatterning of the recombination landscape following SA fusion may affect adaptive divergence and indirectly contribute to reproductive isolation, in contrast to the effect of hybrid sterility owing to karyotypic differences (Yoshida et al. 2023).
Sex chromosomes are known to evolve faster in many taxa. However, we did not observe a more rapid evolution in M20 or MZ, possibly owing to their lower expression levels, or even dysregulation between sexes for M20. Conversely, for M17 with active expression, the Faster-Z effect was clearly present. Given that this phenomenon was also observed in D. plexippus, including the complex pattern of SCDC (Gu et al. 2019) and the accelerated rate of evolution (Mongue et al. 2022) of the Merian elements M14 on the Z Chromosome, our study points to a general feature of karyotype evolution of Lepidoptera: SA fusion–SCDC–Accelerated evolution. In the presented study, one accelerated gene, ABCC4, may be involved in host adaptation (Wei et al. 2021; Beran and Petschenka 2022) and thus favor the coprosperity of the Tortricidae with their host plant (Fagua et al. 2017; Brown 2022). Therefore, SA fusion may have far-reaching consequences in the diversification of the Tortricidae.
The role of SA fusion in accelerated evolution may be specific to Lepidoptera because there is no recombination in female meiosis. As a result, the neo-W can rapidly degenerate and favor efficient fixation of favorable variants on the added-Z (e.g., M17) (Charlesworth et al. 1987; Lasne et al. 2017). In contrast, younger vertebrate lineages may need a longer time to degenerate all neo-Y/W. Furthermore, the first challenge for some chromosome fusion in monocentric species is the inactivation of one centromere; a factor that delays the selection of genes compared with the holocentric Lepidoptera. Our results provide an explanation for the recurrent SA fusion events in Lepidoptera.
In summary, our findings have revealed the potential of SA fusion for reshaping chromosome features and driving rapid divergence of fused chromosomes among taxa. Our data, together with the observations of novel features on other autosomal elements involved in SA fusion (Gu et al. 2019; Mongue et al. 2022), lead us to speculate that the evolutionary advantage of SA fusion may be a common phenomenon in Lepidoptera. The variable chromosome numbers in Lepidoptera provide valuable opportunities to study the evolutionary mechanism of SA fusion (Wright et al. 2024). However, further studies on additional species are required to gain deeper insight into the relationship between SA fusion and adaptive radiation of tortricid moths as well as other lepidopteran taxa.
Methods
Genome assembly and annotation
Larvae of G. dimorpha and C. pomonella were collected from pomes and used to prepare the ONT and Hi-C libraries. The library construction for G. dimorpha and C. pomonella used single larvae. DNA extraction, library construction, and sequencing were performed by GrandOmics. We downloaded the ONT and Hi-C data of G. molesta (obtained from the NCBI BioProject database [https://www.ncbi.nlm.nih.gov/bioproject/] under accession number PRJNA627114) for the previous genome assembly (Cao et al. 2022). The raw ONT reads were assembled into contigs using NextDenovo v2.5.0 (Hu et al. 2024). Redundant fragments were removed using Purge_dups v1.2.6 (Guan et al. 2020). The Hi-C reads were then mapped to the clean contigs using BWA (Li and Durbin 2009). The contigs were assembled into chromosome-level scaffolds using YaHS 1.2a.1 (Zhou et al. 2023) guided by the Hi-C mapping information. The chromosome-level scaffolds were manually corrected using Juicertools v1.22.01 (Durand et al. 2016). Finally, the assembled chromosomes were polished using NextPolish v1.4.1 (Hu et al. 2020), with two rounds using the ONT reads and two rounds using the Illumina reads.
Repeat sequences were detected using RepeatMasker v4.1.2 (Tarailo-Graovac and Chen 2009) with the options “-no_is -norna -xsmall -q.” This analysis was conducted against the Repbase (Bao et al. 2015) and Dfam (Storer et al. 2021) databases, as well as a species-specific repeat library identified by RepeatModeler2 (Flynn et al. 2020). PASA (Haas et al. 2003) was used for transcript-based gene annotation. Briefly, the transcript reads (Supplemental Table S1) were mapped to the genome using HISAT2 (Pertea et al. 2016) with the option “‐‐dta” and assembled using StringTie v2.2.1 (Pertea et al. 2016). Ab initio–based annotation was performed using Helixer v0.3.2 (Stiehler et al. 2021) utilizing the pretrained model “invertebrate_v0.3_m_0200.” Protein homology annotation was performed by mapping the amino acid sequences of Plutella xylostella to the target genomes using Miniprot v0.12 (Li 2023) with the options “‐‐gff-only –gff.” Gene annotations predicted by the three methods were combined using EVidenceModeler v2.1.0 (Haas et al. 2008) using equal weights. For other species, we annotated their genomes using a homology-based method as described above. Although this method cannot annotate species-specific genes, the annotated common genes provide sufficient data for subsequent searches for interspecific orthologs. Functional annotations were performed using the egg-NOG-mapper online tools (Cantalapiedra et al. 2021). We used BUSCO v5.4.7 (Manni et al. 2021), against the Lepidoptera_odb10 data set, to evaluate the completeness of the assemblies and annotations.
Phylogenetic analysis
We collected 27 chromosome-level genomes related to Tortricidae from the NCBI (Supplemental Table S2). OrthoFinder v2.5.4 (Emms and Kelly 2019) was used to identify their shared SCOs. We generated multiple sequence alignments for nucleotides at the codon level for each gene using ParaAT v2.0 (Zhang et al. 2012). All 6046 genes were then concatenated for phylogenetic tree construction. The tree was constructed using FastTree (Price et al. 2010). We utilized MCMCTree in the PAML package v4.10.0 (Yang 2007) for phylogenetic dating, incorporating two secondary calibrated time points. The first calibrated time range for the most recent common ancestor of Olethreutinae and Tortricinae (58.4–87.74 Mya) was obtained from a published phylogenic tree (Fagua et al. 2017), whereas the second calibrated time range for the common ancestor of S. bembeciformis and Zeuzera pyrina (101.5–122.3 Mya) was obtained from TimeTree (Kumar et al. 2017). For the Markov chain Monte Carlo (MCMC) runs, samples were drawn every 50 steps over a total of 10,000,000 steps following a 100,000-step burn-in. The convergence of the MCMC output was checked using Tracer v1.7.2 (Rambaut et al. 2018).
Chromosome synteny
We utilized the MSCANX pipeline in JCVI utility libraries (Tang et al. 2024) for protein-based comparative synteny analysis. The pipeline was used to extract, clean, align, screen (‐‐minspan=30), and visualize the ortholog transcript sequences for the 27 genomes. The fusion breakpoints were determined approximately by the gene positions between the fused and unfused homologous chromosomes. We assigned the names of the ancestral linkage groups in Lepidoptera (Merian elements, M1–M31, and MZ) based on their homology (Wright et al. 2024). The tandem fusion events were denoted as “F().” For example, the fusion of M5 and M13 would be denoted as F(5,13).
Chromosomal features
To analyze basic chromosomal features, we used genomes of six moth species (G. molesta, C. pomonella, C. fumiferana, P. cassiicola, S. bembeciformis, and Z. pyrina). The exon density, repeat sequence density, and GC content were calculated in 100 kb nonoverlapping sliding windows using BEDTools (Quinlan 2014). To explore the features of DNA methylation, we detected signals of CpG methylation using MultiNanopolish (Hu et al. 2021) based on the raw signals of ONT sequencing and obtained methylation frequencies using the built-in calculate_methylation_frequency.py. The methylation frequency was remapped into 50 kb genomic sliding windows using BEDTools. To explore chromatin accessibility, an adult male and an adult female of G. molesta were used for ATAC sequencing (Frasergen). The short reads were mapped to the genome assembly using BWA (Li and Durbin 2009). The accessible regions were identified using MACS3 (https://github.com/macs3-project/MACS) with the pooling of the two samples and the option “callpeak -q 0.01.” We calculated the peak density in 50 kb sliding windows. We compared accessibility at the gene level to account for the effect of gene density on the ATAC peak density. Specifically, if the coding region of a gene and its upstream 1 kb did not overlap with at least one ATAC peak, it was considered “closed.” The chi-squared test was used to assess the differences in the number of closed genes between chromosomal elements.
Hi-C analysis
We assessed the effect of SA fusion on chromatin 3D features using Hi-C data (Supplemental Table S2). The two parts of the paired-end Hi-C reads were individually mapped to the genome assembly using BWA with the options “mem -A1 -B4 -E50 -L0.” The Hi-C contact matrix was constructed, normalized, and plotted in bin sizes of 20 kb and 50 kb using HiCExplorer v3.7.3 (Wolff et al. 2020). The 20 kb contact matrix of F(20 + 17,Z) of G. molesta was fed into a 3DMax model in GenomeFlow v2.0 (Trieu et al. 2019), with 2000 interactions and a conversion factor of 0.84 to construct a 3D model of the chromatin. CscoreTool (Zheng and Zheng 2018) was used to calculate the C-score to identify the A/B compartments using 50 kb bins. To test whether fusion caused A/B compartment switching, we calculated and compared the average C-scores of SCO regions between species of Tortricidae (G. molesta, G. dimorpha, C. pomonella, and C. fumiferana) and non-SA fused outgroups (S. bembeciformis and Z. pyrina). The hicFindTADs function with a 20 kb contact matrix was used to identify the topologically associating domain around the fusion point of F(20 + 17,Z) in G. molesta and S. bembeciformis.
RNA sequencing analysis
Three male and three female adults of G. molesta and an adult male S. bembeciformis (Supplemental Table S2) were used to compare the expression levels among different chromosomal elements. To accurately assess the pattern of SCDC, we removed the gonads and antennae of G. molesta to reduce sex-biased genes. RNA-seq libraries were prepared using a VAHTSTM mRNA-seq V2 library prep kit (Vazyme) and sequenced on the Illumina NovaSeq 6000 platform. RNA reads were aligned to the genome assembly using HISAT2 (Kim et al. 2019). FPKM of predicted genes for each individual was obtained using TPMCalculator (Vera Alvarez et al. 2019). To examine the difference in expression among chromosomal elements, we performed 1000 resampling analyses using R v4.1.2 (R Core Team 2024) with a sample size of 100 and calculated the median for each resampled set.
The correlations between gene expression and selection pressure in G. molesta were measured using a multiple linear regression model: πn ∼ log(FPKMmean) + πs + chromosome_type in R v4.1.2. Here, πn and πs represent the nucleotide diversity for nonsynonymous and synonymous sites, respectively. FPKMmean represents the mean of FPKM across samples. The factor “chromosome_type” was either autosome or F(20 + 17,Z). We excluded 1138 genes with no polymorphism or no expression in the analysis.
Molecular evolutionary analysis
To investigate differences in the evolutionary rates of chromosome elements, we calculated the ratio of the number of nonsynonymous substitutions to synonymous substitutions (dN/dS or ω) among tortricid species (Supplemental Table S1). L. glycinivorella was excluded because of the low completeness of the genome assembly. OrthoFinder 2.5.4 was run again for the remaining 26 species to find shared SCOs. KaKs_Calculator 3.0 (Zhang 2022) with the “MYN” method was used to calculate ω-values for each SCO. The differences in ω between chromosomal elements (M20, M17, MZ, and autosomes) were analyzed by M-W U test.
We applied the branch-site model using Codeml in the PAML package v4.10.0 under the phylogenetic framework to test whether sites had significant differences in ω between the SA fused group (the tortricids) and the unfused group (the outgroups). We performed chromosomal element-level comparisons by concatenating codon sequences of SCOs into supergenes for different elements (80 for M20, 242 for M17, 261 for MZ, and 5463 for other autosomes). RELAX (Wertheim et al. 2015) was used to clarify the changes in selection pressure for concatenated SCOs on M17 through SA fusion. RELAX estimates the variable ω across sites in three categories: purifying, neutral, or positive selection. We used RELAX to estimate K-values and employed a likelihood ratio test (LRT) to compare tortricids with the outgroups. K > 1 and PLRT < 0.05 were considered to indicate significant intensified selection, whereas the results showing K < 1 and PLRT < 0.05 were considered to indicate significant relaxed selection.
We also applied the branch-site model for each SCO to identify PSGs. LRTs were used to calculate P-values. We used RELAX to elucidate the pattern of selection acting on ABCC genes after the SA fusion. The homologs of PSGs in G. molesta were used for a Gene Ontology (GO) enrichment analysis against all genes annotated in the G. molesta genome. The TBtools program (Chen et al. 2020) was used to perform the GO enrichment analysis. We applied a Bonferroni correction for LRTs and choice PSGs with a false-discovery rate < 0.05 for the GO enrichment.
DNA sequencing and variant genotyping
G. molesta and its sibling species, G. dimorpha, were chosen to investigate the evolution of fused chromosomes in recently diverged species. A G. funebrana individual was used as an outgroup to test for interspecific gene flow. All samples were collected from damaged tree shoots and fruits from orchards and immediately frozen in liquid nitrogen and stored at −60°C. For G. molesta and G. dimorpha, we obtained whole-genome resequencing data for 72 individuals, in which 40 sequences were newly generated (Supplemental Tables S2, S3). DNA was extracted from whole larvae using the magnetic beads method. Sequencing libraries were generated using Illumina DNA library preparation kits. The resulting libraries were sequenced using a 150 bp paired-end model on the Illumina NovaSeq 6000 platform, aiming for 20–40× coverage per sample. The Illumina reads were cleaned using fastp (Chen et al. 2018) and aligned to the G. molesta genome using BWA (Li and Durbin 2009). The mapped reads were sorted and marked for duplicates using SAMtools (Danecek et al. 2021). We called whole-site variants for each species separately using FreeBayes v1.3.6 (Garrison and Marth 2012) with the options “‐‐report-monomorphic ‐‐limit-coverage 40 ‐‐min-coverage 5 ‐‐min-alternate-fraction 0.2 -k -j -n 4 -0 -=.” We called variants in a haploid model for the female's neo-Z Chromosome and converted the result to homozygous diploid format if the downstream analysis was incompatible with the haploid data. Sites with missing individual variants > 0.5 were excluded from downstream analyses.
Interspecies genomic divergence
For G. molesta and G. dimorpha, we retained their shared sites and merged the data sets using BCFtools (Danecek et al. 2021). The indels or sites located at repeat regions were filtered out, resulting in a data set with 112,864,330 variants. We conducted ABBA-BABA analysis using Dsuite (Malinsky et al. 2021) to assess the relative resistance to gene flow among chromosomal elements of neo-Z Chromosome and autosomes. Four groups with 36 sequenced samples were included in this analysis. The groups comprised a G. funebrana sample (SXTY) as the outgroup, a G. dimorpha population (HBYC), and two G. molesta populations (SCCD and HBYC) (Supplemental Table S3). G. dimorpha and G. molesta are sympatric at HBYC to facilitate the discovery of introgression. The two G. molesta populations represented distinct lineages located in the Sichuan Basin and other regions of China (Cao et al. 2022). In this analysis, the BBAA pattern corresponded to a standard species tree between G. molesta and G. dimorpha, whereas ABBA or BABA indicated gene flow between G. dimorpha and a G. molesta lineages (Fig. 4B). We used chi-squared tests to compare the resistance to gene flow between different chromosomal elements by testing the number of ABBA + BABA occurrences relative to ABBA + BABA + BBAA.
Interspecific FST, DXY, and nucleotide diversity (πG.m and πG.d) were calculated using popgenWindows.py (https://github.com/simonhmartin/genomics_general). Because DXY is sensitive to ancestral diversity, potentially affecting fair comparisons between sex chromosomes and autosomes, we calculated the net nucleotide differences after the divergence (denoted as δ or Da) (Nei and Li 1979) as Da = DXY − (πG.m + πG.d)/2. We used a LOESS smoothing algorithm to identify putative peaks of divergence regions based on the values of FST and Da in 100 kb genomic windows. Briefly, only regions with values greater than the sum of LOESS smoothing values and one standard deviation were considered as peaks. Only peaks identified based on both FST and Da were retained. After identifying the peaks, we manually identified the genes at the top of the peak top, guided by FST values in 20 kb genomic windows. Peaks that did not overlap with any genes were discarded.
Recombination rates
For the whole-site variants of G. molesta, only biallelic single-nucleotide polymorphisms (SNPs) with minor allele frequencies > 0.05 and missing rates < 0.2 were retained, resulting in a data set with 25,561,036 SNPs. ReLERNN v1.0.0 (Adrion et al. 2020) was used to infer the genome-wide recombination rate in G. molesta. To generate simulated data for the training set, we inferred the historical effective population size using SMC++ (Terhorst et al. 2017). This inference was based on the biallelic variation of autosomes and spanned 4 × 106 generations, with a mutation rate of 2.9 × 10−9 per site per generation (Keightley et al. 2015), and a generation time of 0.25 years (Amat et al. 2021). The simulation used an upper bound for “rho/θ” of 35, an assumed mutation rate of 2.9 × 10−9, a generation time of 0.25 years, and the demographic history inferred by SMC++ (Supplemental Fig. S10). The neo-Z Chromosome was evaluated individually from 24 male samples. The output in crossover rate per base (c/bp) was converted to the units of centimeters per million-base (cM/Mb) by multiplying by 108. The result was then mapped to 100 kb sliding windows using BEDTools. Additionally, the intron GC content was calculated to reflect the ancestral recombination landscape, and the results were visualized using the Integrative Genomics Viewer (IGV) (Thorvaldsdóttir et al. 2013) with the median function. The correlations of recombination rates with FST and Da (in 100 kb sliding windows) were estimated using Spearman's rank correlation.
Selective sweeps
Putative selective sweeps were identified using RAiSD v2.9 (Alachiotis and Pavlidis 2018), using the μ-statistic that integrates information from linkage disequilibrium, site frequency spectrum, and site vectors. The μ-statistics were calculated in windows with 50 SNPs. We used the chi-squared test and a linear regression model to determine whether the number of outliers on chromosomal elements was significantly higher than expected given the sequence length. PopLDdecay v3.42 (Zhang et al. 2019) was used to examine the decay of linkage disequilibrium for chromosomal elements and gene region detected above.
Data access
The genome assemblies for G. molesta, G. dimorpha, and C. pomonella generated in this study have been submitted to the NCBI Genomes database (https://www.ncbi.nlm.nih.gov/home/genomes/) under accession numbers GCA_022674325.2, GCA_038095585.1, and GCA_038396475.1, respectively. The raw data of genome assembly, whole-genome resequencing, ATAC-seq, and transcriptome sequencing generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA1030308, PRJNA1032306, PRJNA1031399, and PRJNA1064642, respectively. The custom scripts are available at the GitHub repository (https://github.com/yangfangyuan0102/tortricid_neo-z) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Zhen Ye for help in phylogenomic analyses. We thank Qi Zhou, Yanhua Qu, Deyan Ge, and Jilong Cheng for helpful comments on the manuscript. This work was supported by the National Natural Science Foundation of China (32070464 and 32272543), the Key Laboratory of Environment Friendly Management on Fruit and Vegetable Pests in North China (Co-construction by MOA of the PRC and Province), the Beijing Key Laboratory of Environmental Friendly Management on Pests of North China Fruits (BZ0432), and the Program of Beijing Academy of Agriculture and Forestry Sciences (JKZX202208). P.N. was supported by the Grant of the Czech Science Foundation (23-06455S).
Author contributions: S.-J.W. conceived the study. L.-J.C. conducted resources preparation and sequencing. Z.-Z.M. and J.-C.C. contributed to the materials and experiments. W.S. contributed to the genome assembly and annotation. F.Y. conducted the data analysis. F.Y., L.-J.C., and P.N. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279569.124.
- Received May 10, 2024.
- Accepted November 26, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








