
A deconvolution framework that uses single-cell sequencing plus a small benchmark data set for accurate analysis of cell type ratios in complex tissue samples
- Shuai Guo1,13,
- Xiaoqian Liu1,13,15,
- Xuesen Cheng2,13,
- Yujie Jiang1,3,
- Shuangxi Ji1,
- Qingnan Liang2,
- Andrew Koval1,3,
- Yumei Li2,
- Leah A. Owen4,5,6,16,
- Ivana K. Kim7,
- Ana Aparicio8,
- Sanghoon Lee9,
- Anil K. Sood9,
- Scott Kopetz10,
- John Paul Shen10,
- John N. Weinstein1,11,
- Margaret M. DeAngelis4,5,6,12,
- Rui Chen2,14 and
- Wenyi Wang1,14
- 1Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 2Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 3Department of Statistics, Rice University, Houston, Texas 77005, USA;
- 4Department of Ophthalmology, Jacobs School of Medicine and Biomedical Engineering, SUNY University at Buffalo, Buffalo, New York 14209, USA;
- 5Department of Population Health Sciences, University of Utah School of Medicine, Salt Lake City, Utah 84108, USA;
- 6Department of Ophthalmology and Visual Sciences, University of Utah School of Medicine, Salt Lake City, Utah 84132, USA;
- 7USA Retina Service, Harvard Medical School, Massachusetts Eye and Ear, Boston, Massachusetts 02114, USA;
- 8Department of Genitourinary Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77230, USA;
- 9Department of Gynecologic Oncology and Reproductive Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas 77230, USA;
- 10Department of Gastrointestinal Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 11Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 12VA Western New York Healthcare System, Buffalo, New York 14215, USA
Abstract
Bulk deconvolution with single-cell/nucleus RNA-seq data is critical for understanding heterogeneity in complex biological samples, yet the technological discrepancy across sequencing platforms limits deconvolution accuracy. To address this, we utilize an experimental design to match inter-platform biological signals, hence revealing the technological discrepancy, and then develop a deconvolution framework called DeMixSC using this well-matched, that is, benchmark, data. Built upon a novel weighted nonnegative least-squares framework, DeMixSC identifies and adjusts genes with high technological discrepancy and aligns the benchmark data with large patient cohorts of matched-tissue-type for large-scale deconvolution. Our results using two benchmark data sets of healthy retinas and ovarian cancer tissues suggest much-improved deconvolution accuracy. Leveraging tissue-specific benchmark data sets, we applied DeMixSC to a large cohort of 453 age-related macular degeneration patients and a cohort of 30 ovarian cancer patients with various responses to neoadjuvant chemotherapy. Only DeMixSC successfully unveiled biologically meaningful differences across patient groups, demonstrating its broad applicability in diverse real-world clinical scenarios. Our findings reveal the impact of technological discrepancy on deconvolution performance and underscore the importance of a well-matched data set to resolve this challenge. The developed DeMixSC framework is generally applicable for accurately deconvolving large cohorts of disease tissues, including cancers, when a well-matched benchmark data set is available.
Although recent advances in single-cell/nucleus RNA sequencing (sc/snRNA-seq) offer valuable insights into cell types and states in healthy (Haniffa et al. 2021) and diseased tissues (Gohil et al. 2021; Zeng et al. 2023), highly expensive and complex sample preparation procedures have restricted its widespread adoption in clinical settings (Li and Wang 2021). Bulk RNA-seq, on the other hand, retains its essential role, especially in large disease-based cohort studies, for which its cost-efficiency, streamlined sample processing, and high-throughput analytic capabilities establish it as the method of choice for both preliminary screenings and exhaustive population-level analyses (Ratnapriya et al. 2019; Stark et al. 2019; Cao et al. 2022). Nevertheless, bulk RNA-seq comes with a significant drawback: It captures averaged gene expression across heterogeneous cell types, thus confounding downstream analysis (Li and Wang 2021). To mitigate this drawback, deconvolution methods have been developed to delineate the cell type–specific signals from bulk RNA-seq data. Traditional bulk-based deconvolution methods (Anghel et al. 2015; Wang et al. 2018) employ bulk RNA-seq data from normal tissues or cell lines as the reference. They were typically constrained by low-resolution estimates, limited to identifying only two or three cellular components within the bulk samples. The progress of sc/snRNA-seq techniques opens the door to the emergence of single-cell-based deconvolution methods (Newman et al. 2019; Tsoucas et al. 2019; Wang et al. 2019; Aliee and Theis 2021; Dong et al. 2021; Erdmann-Pham et al. 2021; Chu et al. 2022; Fan et al. 2022; Cobos et al. 2023), which tap into the granularity of even a modest set of single-cell data to provide far superior resolution in estimating cell type proportions in complex tissues, thereby offering a cost-effective alternative.
Single-cell-based deconvolution methods are not without their disadvantages, however. Though affording remarkable resolution, they encounter a substantial challenge in achieving precision and accuracy. The limitations arise from inconsistencies in gene expression profiles between bulk and sc/snRNA-seq data. Those inconsistencies are attributable to technique variations in sample acquisition, preparation, and sequencing (Aird et al. 2011; Wery et al. 2013; Denisenko et al. 2020; Stoler and Nekrutenko 2021; Hippen et al. 2023). Such inconsistencies, which we refer to as “technological discrepancies,” have caused prior deconvolution studies to produce suboptimal estimates of cell type proportions, particularly when unpaired sc/snRNA-seq data serve as the reference for deconvolving publicly available large bulk cohorts (Sturm et al. 2019; Jin and Liu 2021; Fan et al. 2022). Most existing benchmarking designs (Sturm et al. 2019; Cobos et al. 2020; Jin and Liu 2021) often employ data sets such as simulated pseudobulk data, cell line mixtures, or publicly available data, none of which are tailored to reveal the negative effect of technological discrepancy. Researchers have become aware of these discrepancies (Dietrich et al. 2022; Sutton et al. 2022; Cobos et al. 2023; Hippen et al. 2023). A recent study (Hippen et al. 2023) generated matched bulk and scRNA-seq data from seven high-grade serous ovarian cancer (HGSC) samples as a benchmark and discussed the impact of technological discrepancy on deconvolution analysis. However, current attempts to address these issues have achieved only limited success (Newman et al. 2019; Dong et al. 2021; Cobos et al. 2023). CIBERSORTx (Newman et al. 2019) implements a batch effect correction step but offers limited improvements in deconvolving complex bulk tissues. The ensemble approach of SCDC (Dong et al. 2021) uses matched bulk and scRNA-seq data from two normal tissue samples (e.g., mouse breast) but lacks generalizability to patient cohorts. The most recent SQUID (Cobos et al. 2023) builds on top of DWLS (Tsoucas et al. 2019) with a Bisque-based linear transformation step (Jew et al. 2020) to align matched bulk and scRNA-seq data; it can distort gene expression profiles, risking overcorrection. Therefore, there is still need for methods to effectively mitigate such discrepancy by taking full advantage of the well-matched benchmark data set.
In this paper, we offer a new solution to improve deconvolution performance. To accomplish this, we generate a specialized benchmark data set of 24 healthy retinal samples, ensuring technological discrepancy as the main confounding factor. Using this data set, we demonstrate that technological discrepancy significantly affects the expression profiles of bulk and single-nucleus data and thus reduces the accuracy of existing single-cell-based deconvolution methods. Against this backdrop, we introduce a novel deconvolution method called DeMixSC, which employs a benchmark data set and an improved weighted nonnegative least-squares (wNNLS) framework (Ruppert and Wand 1994) to identify and adjust for genes consistently affected by technological discrepancy. DeMixSC is generalizable to any tissue type, given a small representative benchmark data set, to effectively deconvolve a large tissue-type-matched bulk cohort. We validated the improved deconvolution performance of DeMixSC by comparing it on our benchmark data set with eight existing deconvolution methods. When applied to 453 peripheral retinal samples from patients with age-related macular degeneration (AMD) (Ratnapriya et al. 2019), DeMixSC achieved more realistic cell type estimates that reflect subtle changes in cell type proportions among AMD grades, suggesting its reliability and generalizability in real-world settings. DeMixSC further exhibited superior deconvolution performance on an HGSC cohort (Lee et al. 2020) by employing the available HGSC benchmark data set (Hippen et al. 2023). We expect DeMixSC to fill the gap in resolving the technological discrepancy in bulk deconvolution and serves as an accurate and adaptable tool for estimating cell type proportions.
Results
Using benchmark data to assess technological discrepancy
We designed and generated a specialized benchmark data set to assess the technological discrepancy between bulk and sc/sn sequencing platforms (Fig. 1A; Supplemental Fig. S1). This data set comprises 24 healthy retinal samples from donors’ eyes collected within 6-h postmortem (ages of death between 53 and 91) (Supplemental Table S1), for two batches of sequencing experiments. Both bulk and snRNA-seq profiling was performed on each sample from the same single-nucleus suspension aliquot using a template-switching method to generate full-length cDNA libraries (see Methods). Because single-cell protocols can be biased toward retaining certain cell types (Mereu et al. 2020), hence changing the cell type proportions, this special approach maximizes our chance that the matched sequencing data share approximately the same cell type proportions. We performed cell type annotation for snRNA-seq data with known markers (see Methods) (Supplemental Table S2). The resulting snRNA-seq data were summed to create matched pseudobulk RNA-seq data (see Methods). We hypothesized that major differences in gene expression profiles between the matched pseudobulk and real-bulk RNA-seq would be owing to technological factors, rather than biological signals.
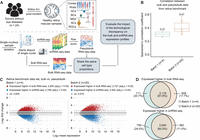
Assessing technological discrepancy between bulk and single-cell sequencing platforms using matched single-nucleus aliquots. (A) Workflow for generating a benchmark data set. We collect 24 healthy human retinal samples within 6 h postmortem. An illustration shows the layer and cell compositions of the human retina. Seven major cell types include photoreceptors (rod and cone cells), bipolar cells (BCs), retinal ganglion cells (RGCs), horizontal cells (HCs), amacrine cells (ACs), and Müller glia cells (MGs). Three minor cell types are not depicted in the illustration: astrocytes, microglia cells, and retinal pigment epithelial cells (RPEs). Samples are isolated into single-nucleus suspensions. The same aliquot of single nucleus is used for both bulk and snRNA-seq profiling. The matched pseudobulk mixtures are generated as conventionally done by summing UMI counts across cells from all cell types in each sample. This data generation pipeline guarantees the matched bulk and snRNA-seq data share the same cell type proportions, which enables us to evaluate the impact of technological discrepancy (i.e., the shot-gun sequencing procedure) on the bulk and snRNA-seq expression profiles. (B,C) The influence of technological discrepancy at the sample and gene level, respectively. (B) Spearman's correlation coefficient across genes between the matched real-bulk and pseudobulk RNA-seq data for one sample at a time for both batches. The correlations were calculated using quantile-normalized expression data (relative abundances). (C) MA-plots displaying the mean expression levels of all genes between matched real-bulk and pseudobulk data. Differentially expressed (DE) genes are identified using the paired t-test with Benjamini–Hochberg (BH) adjustment. Red represents genes expressed higher in the real bulk, and blue represents genes expressed higher in the pseudobulk. The horizontal dotted lines denote a twofold change between matched real-bulk and pseudobulk data. (adj.p) Adjusted P-values. (D) Venn diagrams showing genes consistently expressed higher in the bulk (top, overlap of red dots in panel C) or the snRNA-seq generated pseudobulk (bottom, overlap of blue dots in panel C) between the two batches, which were generated using different tissue samples and a different time.
We observed much larger batch differences between real-bulk and pseudobulk data than the small differences in cell type distributions across samples in snRNA-seq or differences between the two experimental batches (Supplemental Fig. S2A–E). Total read counts from bulk RNA-seq data were significantly lower than total UMI counts from matched pseudobulk data (Supplemental Fig. S2F). Assuming that the difference in read depth does not impact the relative expression of each gene, we expected gene expression correlation to be a better metric for identifying technological discrepancy. We observed a low-to-moderate correlation of gene expression, consistent across samples, between the paired bulk data sets (mean Spearman's correlation coefficient = 0.31 for batch-1 and 0.41 for batch-2) (Fig. 1B). Further differential expression (DE) analysis between the paired bulk and pseudobulk samples identified more than 5000 DE genes in each experimental batch (adjusted P-values < 0.05) (Fig. 1C), with >60% of those genes overlapping across the experiments (Fig. 1D; Supplemental Fig. S3). We next converted the retina bulk data to transcripts per million (TPM) to account for gene length effects, considering the incomplete gene coverage from the 10x Genomics single-cell platforms, and observed even lower correlations with paired pseudobulk data (mean Spearman's correlation coefficient = 0.18 for batch-1 and 0.25 for batch-2), indicating that TPM normalization did not ameliorate these discrepancies. We further analyzed a benchmark data set from seven primary HGSC samples (Hippen et al. 2023) with matched single-cell and three types of bulk data: dissociation with poly(A) enrichment (Disso&poly(A)+), dissociation with rRNA depletion (Disso&rRNA−), and tissue chunk with rRNA depletion (Chunk&rRNA−; see Methods) (Supplemental Fig. S4A). The HGSC benchmark data set exhibited significant technological discrepancy (more than 5000 DE genes) between bulk and pseudobulk RNA-seq data, with consistent DE patterns across seven samples (Supplemental Fig. S4B,C).
Our observations suggest a consistent technological effect across experiments. In broader contexts, factors such as library preparation, RNA capture efficiency, reverse transcription protocol, and sequencing depth could serve as potential sources of technological discrepancy (Tung et al. 2017; Denisenko et al. 2020; Stoler and Nekrutenko 2021). We, therefore, expect that the reference matrices derived from sc/snRNA-seq data will not fully represent cell type–specific expression profiles in bulk samples (Cobos et al. 2023; Hippen et al. 2023). Given such discrepancies, the performance of existing deconvolution methods is compromised, as their key assumption about the representative reference is violated.
Overview of DeMixSC
Here, we present our novel deconvolution framework, DeMixSC, and illustrate how it addresses the observed consistent technological
discrepancy in order to enhance the estimation accuracy of cell type proportions. The DeMixSC framework, as depicted in Figure 2, is built upon the commonly used wNNLS approach (Ruppert and Wand 1994; Tsoucas et al. 2019; Wang et al. 2019) with several essential improvements (see Methods) (Supplemental Note). Concretely, for a subject j, DeMixSC estimates its cell type proportions, denoted by 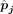 , by minimizing a composite of two weighted squared error terms,
, by minimizing a composite of two weighted squared error terms,
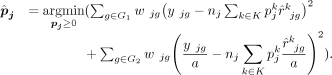

Overview of DeMixSC. The DeMixSC framework for deconvolution analysis of bulk RNA-seq data using sc/sn RNA-seq data as a reference.
(A) The framework starts with a benchmark data set of matched bulk and sc/snRNA-seq data with the same cell type proportions.
Pseudobulk mixtures are generated from the sc/sn data. DeMixSC identifies genes in G1 and G2 with the matched real-bulk and pseudobulk data. The non-DE genes are considered stably captured by both sequencing platforms
(blue), whereas the DE genes are more impacted by the technological discrepancy (orange). (B) DeMixSC then employs a normalization procedure to perform the alignment between two bulk RNA-seq data sets (e.g., with ComBat).
(C) DeMixSC estimates cell type proportions under a weighted nonnegative least square (wNNLS) framework with two improvements:
(1) partitioning and adjusting genes with high technological discrepancy and (2) a new weight function. The final estimates
are obtained when the algorithm either converges or reaches the prespecified maximum number of iterations. Here, G1 is genes with low technological discrepancy, G2 is genes with high technological discrepancy, a is a user-defined positive constant that serves as an adjustment factor,  is the reference matrix derived from the sc/snRNA-seq data, y is the observed expression in bulk RNA-seq data,
is the reference matrix derived from the sc/snRNA-seq data, y is the observed expression in bulk RNA-seq data, 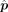 is the vector of estimated cell type proportions, and
is the vector of estimated cell type proportions, and 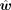 is the estimated gene weights.
is the estimated gene weights.
Here, yjg is the observed expression value of gene g from subject j in the bulk RNA-seq data, nj is the number of all cells, 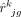 is the estimated cell type–specific expression value of cell type k in the reference matrix derived from sc/snRNA-seq data, and wjg is an associated weight. The gene sets G1 and G2 comprise genes with minimal and substantial impact by technological discrepancy, respectively. The first innovation of DeMixSC
is a gene partitioning approach that identifies and adjusts the expression levels of genes that exhibit consistently high
technological discrepancy (G2). We do so with a small representative benchmark data set such as our special matched RNA-seq data from 24 retinal samples
(Fig. 2A). DeMixSC uses a DE analysis between bulk and matched pseudobulk RNA-seq data to segregate genes with low inter-platform
discrepancy (G1) from those highly affected by technological discrepancy (G2; see Methods). It then employs a partitioned loss function and adjusts genes from G2 by rescaling their expressions by a positive constant adjustment factor a to mitigate the influence of technological discrepancy (see Methods) (Supplemental Note).
is the estimated cell type–specific expression value of cell type k in the reference matrix derived from sc/snRNA-seq data, and wjg is an associated weight. The gene sets G1 and G2 comprise genes with minimal and substantial impact by technological discrepancy, respectively. The first innovation of DeMixSC
is a gene partitioning approach that identifies and adjusts the expression levels of genes that exhibit consistently high
technological discrepancy (G2). We do so with a small representative benchmark data set such as our special matched RNA-seq data from 24 retinal samples
(Fig. 2A). DeMixSC uses a DE analysis between bulk and matched pseudobulk RNA-seq data to segregate genes with low inter-platform
discrepancy (G1) from those highly affected by technological discrepancy (G2; see Methods). It then employs a partitioned loss function and adjusts genes from G2 by rescaling their expressions by a positive constant adjustment factor a to mitigate the influence of technological discrepancy (see Methods) (Supplemental Note).
The second innovation of DeMixSC comes from our proposed weight function 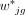 , which is given by
, which is given by
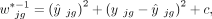 where
where 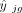 denotes the fitted expression value of gene g in subject j. This weight function comprises three terms: the squared fitted expression, the squared residual, and a baseline constant,
which is distinct from previously proposed weights (Tsoucas et al. 2019; Wang et al. 2019; Dong et al. 2021; Fan et al. 2022; Cobos et al. 2023). The fitted term addresses genes with high expression levels; the squared residual accounts for the remaining variance after
fitting; and the baseline constant c adds a reasonable upper bound on the weight (see Methods) (Supplemental Note). These two innovations enable DeMixSC to more effectively address the technological discrepancy compared with nondifferential
weighting approaches, for example, test statistics (see Methods).
denotes the fitted expression value of gene g in subject j. This weight function comprises three terms: the squared fitted expression, the squared residual, and a baseline constant,
which is distinct from previously proposed weights (Tsoucas et al. 2019; Wang et al. 2019; Dong et al. 2021; Fan et al. 2022; Cobos et al. 2023). The fitted term addresses genes with high expression levels; the squared residual accounts for the remaining variance after
fitting; and the baseline constant c adds a reasonable upper bound on the weight (see Methods) (Supplemental Note). These two innovations enable DeMixSC to more effectively address the technological discrepancy compared with nondifferential
weighting approaches, for example, test statistics (see Methods).
DeMixSC runs as a three-tier model in application. First, DeMixSC uses a specifically designed benchmark data set to identify and adjust genes with high inter-platform discrepancy (Fig. 2A). Second, to deconvolve a large unmatched bulk RNA-seq data set, DeMixSC aligns the large bulk cohort with the bulk RNA-seq data in the small benchmark data set (Fig. 2B; Zhang et al. 2020) to generalize the technological discrepancy detected. Last, DeMixSC runs the refined wNNLS framework iteratively for deconvolution (Fig. 2C), allowing for dynamic updates as the model fit improves and progressively enhancing estimation accuracy. A diagram (Supplemental Fig. S5) complementary to Figure 2 visualizes the complete workflow of DeMixSC with more technical details. Our main prerequisite is a matched tissue type between the small benchmark data set and the large, targeted cohort.
DeMixSC includes a quantile normalization step and a batch effect correction step, both of which operate under specific assumptions. Quantile normalization assumes symmetric DE between the conditions and similar gene expression distributions across the samples. Batch effect correction requires the bulk benchmark data to share a similar tissue microenvironment with the large cohort. We note that DeMixSC is compatible with most batch effect correction methods.
Comparing the estimation accuracy of DeMixSC with that of other existing deconvolution methods
Using our retina benchmark data, we compared the performance of DeMixSC with that of eight existing deconvolution methods (Newman et al. 2019; Tsoucas et al. 2019; Wang et al. 2019; Aliee and Theis 2021; Dong et al. 2021; Erdmann-Pham et al. 2021; Chu et al. 2022; Cobos et al. 2023): AutoGeneS, BayesPrism, CIBERSORTx, DWLS, MuSiC, RNAseive, SCDC, and SQUID (see Methods) (Fig. 3A). The retinal tissue samples in our benchmark data set comprised 10 distinct cell types. We focused our evaluation of different deconvolution methods on seven major cell types (amacrine cells [ACs], bipolar cells [BCs], cone cells, horizontal cells [HCs], Müller glial cells [MGs], retina ganglion cells [RGCs], rod cells) (Fig. 1A), which on average accounted for 98% of the total cell population (Liang et al. 2023).
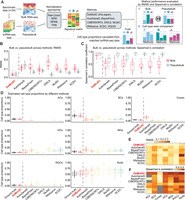
Comparing the estimation accuracy of DeMixSC to existing deconvolution methods. (A) Workflow for the deconvolution benchmarking design. We use benchmark data from retinal samples. The cell count proportions for each cell type are used as ground truth for the corresponding tissue samples. We assess the deconvolution performance of DeMixSC and seven existing methods for both bulk and pseudobulk mixtures. In addition to the raw counts, we also test RPM, RPKM, and TPM. The deconvolution performance is assessed by RMSE and Spearman's correlation coefficient. Note the results by SQUID are discussed in the text only. (B,C) Boxplots showing the deconvolution performance of eight deconvolution methods for the bulk and pseudobulk data. RMSE and Spearman's correlation coefficient values are calculated across seven major cell types for each sample, with gray denoting pseudobulk and red denoting real bulk. Smaller RMSEs or larger Spearman's correlations indicate a higher accuracy in proportion estimation. (D) Boxplots showing the distributions of deconvolution estimates at the cell type level for all 24 retinal samples. Each color corresponds to a given deconvolution method, with black denoting the ground truth, and each panel corresponds to a given cell type. (E,F), An overview of deconvolution performance at the cell type level across the eight methods using RMSE and Spearman's correlation coefficient, respectively. Lighter colors correspond to lower RMSE or Spearman's correlation coefficient values. Gray indicates NA.
Overall, DeMixSC achieved the lowest root mean squared error (RMSE) and the highest Spearman's correlation coefficient in deconvolving bulk RNA-seq data, with mean values of 0.03 and 0.86 (Fig. 3B,C). Moreover, DeMixSC produced comparable RMSEs and Spearman's correlation coefficients for deconvolving bulk and pseudobulk RNA-seq data (mean RMSE: bulk 0.03, pseudobulk 0.03; mean Spearman's correlation: bulk 0.86, pseudobulk 0.92). These results suggest that DeMixSC adjusts well to undesired technological discrepancies. In contrast, existing methods performed reasonably well for pseudobulk but much worse for bulk data. In that sense, technological discrepancies that compromise deconvolution accuracy remained unaddressed by other existing approaches. Specifically, AutoGeneS showed a higher RMSE for pseudobulk data, likely owing to its inability to distinguish between rod and cone cells, which share largely similar expression profiles (Fig. 3D). DWLS excelled in deconvolving pseudobulk samples but fell short for bulk RNA-seq data, possibly because of overfitting. Using the tree-based deconvolution in MuSiC or the ensemble option in SCDC did not improve their accuracy (Supplemental Fig. S6). CIBERSORTx presented slightly better performances than others in both bulk and pseudobulk data, likely because of its batch effect correction step. Looking further at the cell type level, we observed systematic biases across other methods. Most methods underestimate the proportions of ACs, BCs, and cones while overestimating HCs and rods (Fig. 3D; Supplemental Fig. S7). DeMixSC accurately estimated the proportions of all seven major cell types and improved the deconvolution results for ACs, BCs, cones, HCs, and MGs (mean RMSE: 0.01, 0.04, 0.03, 0.02, 0.03, respectively) (Fig. 3D,E). DeMixSC also performed better in correlations of the estimated versus the true cell proportions, particularly for the top three prevalent cell types (rods, MGs, and BCs, Spearman's correlation coefficients of 0.78, 0.73, and 0.58, respectively) (Fig. 3F).
In addition, we tested the robustness of these methods under varied data formats (Dillies et al. 2013), including reads per million mapped reads (RPM); reads per kilobase of transcript, per million mapped reads (RPKM); and TPM (see Methods) and found DeMixSC to be robust to data normalization procedures (Supplemental Fig. S8). In line with previous benchmarking studies (Cobos et al. 2020), we found using raw counts as input is sufficient to obtain good results. Finally, SQUID delivered the least desirable results in this benchmarking study (mean RMSE and Spearman's correlation in bulk data: 0.25 and 0.31). The issue with SQUID possibly lies in its data transformation step (Jew et al. 2020), which has the potential to misrepresent gene expression profiles. In summary, our DeMixSC framework has achieved the most accurate deconvolution among the compared methods by successfully addressing the key issues with the technological discrepancy between sequencing platforms. Regarding the required sample size for the benchmark data set, we found that DeMixSC exhibited satisfying deconvolution performance with a sample size of four, and its performance becomes stable when the sample size is more than seven in the retina data (Supplemental Fig. S9).
Applying DeMixSC to human peripheral retina bulk RNA-seq data
AMD is characterized by deterioration of retina and choroid that leads to substantial decreased visual acuity, with loss of cone and rod cells as a major manifestation. It is the leading cause of blindness among the elderly population globally (Fleckenstein et al. 2021). However, the molecular and cellular events that underlie AMD remain poorly understood, impeding the development of effective treatments (Khanani et al. 2022). Understanding the molecular and cellular dynamics is essential for targeting the progression of AMD. We aim to examine cell type proportion changes during AMD progression using bulk RNA-seq data from 453 human peripheral retina samples (see Methods) (Ratnapriya et al. 2019). Among these, 105 have been scored in the Minnesota grading system as grade 1 (MGS1), 175 as MGS2, 112 as MGS3, and 61 as MGS4. An MGS1 rating indicates a non-AMD healthy retina, and an MGS4 rating indicates AMD. MGS2 and MGS3 represent intermediate stages (Olsen and Feng 2004).
We ran DeMixSC to first align the AMD cohort with the bulk data from our specialized benchmark data set of retina samples and then to estimate cell type proportions in the AMD cohort (see Methods) (Fig. 4A). For the reference matrix in wNNLS, we constructed a consensus reference by integrating expression profiles from seven single-nucleus samples (see Methods) to achieve reliable deconvolution. DeMixSC produced overall robust deconvolution estimates among the consensus and each individual single-nucleus references at both the cell type and sample levels, only with low-to-moderate correlations observed in some conditions owing to variations in the ranking of low-abundance cell types across samples (Fig. 4B,C; see Methods). DeMixSC achieved cell type proportions that are closer to experimental measures for non-AMD samples (Liang et al. 2019), with mean RMSE of 0.04 and mean Spearman's correlation coefficient of 0.75 (see Methods; Supplemental Table S3). DeMixSC revealed changes in cell type proportions between non-AMD and AMD samples (Fig. 4D). We observed statistically significant decreases in photoreceptors, including rod cells (P-value = 0.047) and cone cells (P-value = 0.035), and HCs (P-value = 0.005). Besides, DeMixSC identified increases in glial cells, specifically astrocytes (P-value = 0.006) and MGs (P-value = 0.002). The increase of BCs is of marginal significance (P-value = 0.068). These changes in cell type proportions showed consistent patterns across the progression of AMD severity from MGS1 to MGS4 (Supplemental Fig. S10), reflecting the progressive nature of the disease. For comparison, we deconvolved the same cohort with MuSiC2 (Fan et al. 2022), CIBERSORTx (Newman et al. 2019), and SQUID (Cobos et al. 2023), in which MuSiC2 was chosen for its added ability to leverage conditionally stable genes from healthy references in analyzing diseased tissue. Among the four methods compared, DeMixSC exhibited the least bias for three out of seven major cell types (ACs, BCs, and rod cells) (Supplemental Table S3; Supplemental Fig. S11). CIBERSORTx is the least biased for cones and MGs; MuSiC2 showed the least bias for HCs and RGCs; and SQUID demonstrated the most biased estimates across all cell types. Additionally, although DeMixSC, CIBERSORTx, and MuSiC2 all detected a decrease in rod cells in AMD, only DeMixSC identified a statistically significant reduction in cone cells, consistent with AMD pathology affecting both photoreceptors (Curcio et al. 1996). We further evaluated DeMixSC framework on the AMD cohort under different benchmark alignment conditions (see Methods) (Supplemental Table S3; Supplemental Fig. S12). Limma (Ritchie et al. 2015) also effectively corrected batch effects and yielded comparable deconvolution performance (mean RMSE: 0.05; mean Spearman's correlation: 0.68), whereas both no batch correction and VSN (Huber et al. 2002) showed worse results (mean RMSE: 0.12 and 0.20; mean Spearman's correlation: 0.57 and −0.61), highlighting the importance of effective batch correction.
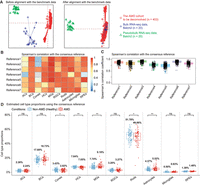
Using DeMixSC to deconvolve a large cohort of human peripheral retinal samples. (A) PCA plots of both the retina cohort data and the benchmark data. Red denotes the bulk data to be deconvolved; blue denotes the benchmark bulk data; and green denotes the benchmark pseudobulk data. (B,C) Panels demonstrating the robustness of DeMixSC to different reference matrices at both the cell type and sample levels. Higher correlation coefficients indicate better performance. (D) Distributions of DeMixSC estimated cell type proportions of Ratnapriya et al. (2019) data using consensus references. Each panel corresponds to a given cell type. The P-values for Student's t-tests comparing the estimated cell type proportions between non-AMD (healthy) and AMD groups are denoted as follows: (ns) not significant, P-value > 0.05; (*) P-value ≤ 0.05; (**) P-value ≤ 0.01; and (***) P-value ≤ 0.001.
Currently it is believed that adult retinal photoreceptors (cone and rod cells) cannot regenerate after injury (Menon et al. 2019; Fleckenstein et al. 2021; Khanani et al. 2022). We hypothesize that photoreceptor loss reduced the total cell count, hence inflating the rest of the cell proportions in AMD. Indeed, we found that losing 12.53% of total photoreceptors resulted in the observed subtle drop for rod cells (2.72%) and cone cells (0.61%; see Methods). The increased proportion of BCs primarily resulted from photoreceptor loss, whereas MGs and Astrocytes showed actual increases beyond this effect (see Methods). These results align well with the current understanding that photoreceptor degeneration is accompanied by reactive gliosis (Pfeiffer et al. 2020; Tomita et al. 2021), characterized by glial cell activation and proliferation. In summary, our findings demonstrated DeMixSC's ability to capture subtle yet biologically relevant changes in retinal cell composition in AMD.
Applying DeMixSC to HGSC
HGSC is the most common and lethal subtype of epithelial ovarian cancer, yet it remains poorly understood (Veneziani et al. 2023). A major challenge in comprehending and treating HGSC lies in its extensive tumor heterogeneity, characterized by the presence of diverse cell populations. This heterogeneity contributes to differential responses to therapy and various clinical outcomes. Accurate deconvolution analysis of HGSC bulk cohorts with well-documented clinical follow-ups is crucial for dissecting cellular interactions underlying disease progression and treatment response.
We compared the deconvolution performance of DeMixSC to seven existing methods using a HGSC benchmark data set (see Methods) (Supplemental Fig. S4A; Hippen et al. 2023). DeMixSC notably outperformed other methods when deconvolving Disso&poly(A)+ samples, achieving the lowest RMSE (mean: 0.09) and the highest Spearman's correlation coefficient (mean: 0.72) (Fig. 5A). At the cell type level, DeMixSC accurately estimated the proportions of all 13 cell types in the Disso&poly(A)+ samples, with marked improvements in the deconvolution of epithelial cells, endothelial cells, and T cells (Supplemental Fig. S13A). In comparison, the next best-performing method, CIBERSORTx, had a mean RMSE of 0.13 and a mean Spearman's correlation of 0.49 for the Disso&poly(A)+ data type. Additionally, we evaluated DeMixSC on two other data types (Disso&rRNA− and Chunk&rRNA−) from the HGSC data set, which were made with a lower level of technical matchness with the scRNA-seq data. DeMixSC did not outperform other methods on these two data types (mean RMSE: 0.14 and 0.14; mean Spearman's correlation: 0.27 and 0.30; for Disso&rRNA− and Chunk&rRNA−, respectively) (Fig. 5A; Supplemental Fig. S13B,C), suggesting that the benchmark data set needs to be specifically designed for optimal performance.
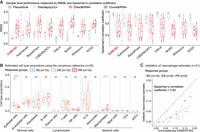
Using DeMixSC to deconvolve HGSC samples. (A) Boxplots showing the deconvolution performance of eight deconvolution methods for the pseudobulk and three types of bulk data in the HGSC benchmark data set. RMSE values and Spearman's correlation coefficients are calculated across 13 cell types for each sample. Smaller RMSEs or larger Spearman's correlations indicate higher accuracy in proportion estimation. (B) Distributions of DeMixSC estimated cell type proportions of Lee et al. (2020) data using consensus references. Each panel corresponds to a given cell type. (NK cells) natural killer cells, (ILC) innate lymphoid cells, (DC) dendritic cells macrophages, and (pDC) plasmacytoid dendritic cells. The P-values for Student's t-tests comparing the estimated cell type proportions across R0, ER, and PR groups are denoted as follows: (ns) not significant, P-value > 0.05; (*) P-value ≤ 0.05; (**) P-value ≤ 0.01; and (***) P-value ≤ 0.001. (C) Scatter plot comparing DeMixSC estimates of macrophages with immunofluorescent measures (CD68/CD163) in 21 HGSC samples. The black dashed line represents the diagonal, and the gray solid line indicates the linear fit across the data points.
To demonstrate the generalizability of DeMixSC, we utilized the Disso&poly(A)+ data as the benchmark data set to deconvolve an unmatched HGSC cohort (Lee et al. 2020) with detailed clinical annotations (see Methods). This cohort contains 30 primary treatment-naïve tumor samples, categorized into three groups based on their responses to treatment: those who underwent complete gross resection (R0, n = 10), those who received neoadjuvant chemotherapy with an excellent response (ER; n = 10), and those with a poor response (PR; n = 10). As in the AMD deconvolution analysis, we applied DeMixSC, MuSiC, CIBERSORTx, and SQUID for deconvolution (see Methods). DeMixSC achieved the most biologically realistic estimations of cell type proportions among all compared methods (Fig. 5B; Supplemental Fig. S14). DeMixSC was the only method that successfully captured proportion differences in epithelial cells (R0 vs. ER, P-value = 0.013; R0 vs. PR, P-value = 0.007) and macrophages (R0 vs. ER, P-value = 0.085; R0 vs. PR, P-value = 0.044) across three distinct response groups. Notably, DeMixSC revealed a decrease in epithelial cells from patients with no need for chemotherapy (R0) to only showing partial response to chemo-treatment (PR) (Supplemental Fig. S14), aligning with previous clinical observations (Caudle et al. 2010). Additionally, DeMixSC identified a consistent trend of increased proportion of macrophages from R0 to PR groups, suggesting that higher pretreatment macrophage infiltrations may be associated with decreased treatment response. The deconvolution-based estimates were further validated by immunofluorescence staining (Spearman's correlation coefficient = 0.82, see Methods) (Fig. 5C; Lee et al. 2020). In contrast, the other three methods produced lower Spearman's correlations with the staining results (MuSiC: 0.60; SQUID: 0; and CIBERSORTx: 0.56) and failed to discern biological differences across response groups, although the CIBERSORTx results showed similar trends (Supplemental Fig. S14).
Discussion
This study addresses the technological discrepancy between bulk and sc/sn RNA-seq data in order to improve the deconvolution accuracy of bulk RNA-seq data. We constructed a specialized benchmark data set of healthy retina samples and demonstrated the impact of technological discrepancy on existing single-cell-based deconvolution methods (Newman et al. 2019; Tsoucas et al. 2019; Wang et al. 2019; Aliee and Theis 2021; Dong et al. 2021; Erdmann-Pham et al. 2021; Chu et al. 2022; Fan et al. 2022; Cobos et al. 2023). Using this benchmark data set, we introduce the DeMixSC deconvolution method that makes innovative improvements to the wNNLS framework to address the consistently observed technological discrepancy at the gene level. The distinct advantage of DeMixSC lies in its superior deconvolution accuracy and broad generalizability. As demonstrated in the benchmarking study, DeMixSC achieves more accurate estimates of cell type proportions than other existing deconvolution methods. In our application to complex retina samples from patients with AMD, DeMixSC was able to accurately delineate seven to 10 cell types and identify subtle yet critical changes in cell type proportions. Furthermore, DeMixSC succeeded in deconvolving ovarian cancer data by utilizing a publicly available HGSC benchmark data set, in which it achieved considerably more accurate deconvolution performance and discovered proportional differences associated with different treatment responses. Our studies support the capability and generalizability of DeMixSC in deconvolving large heterogeneous bulk cohorts, only requiring a small set of tissue-type-matched benchmark data. DeMixSC is computationally efficient, completing the analysis of 453 AMD samples within 5 min, and exhibits robust convergence against different starting values (see Methods) (Supplemental Fig. S15).
Generation of the benchmark data set in DeMixSC is crucial for accurate and reliable estimation of cell type proportions. Our study employed a specifically tailored cDNA library preparation procedure to generate the benchmark data set of retinal samples. A critical step in the data generation process is to ensure the “matchness” of paired bulk and snRNA-seq data. In our procedure, the cDNA library for bulk RNA-seq was generated using the Smart-seq v4 ultralow input RNA kit procedure, a protocol similar to that used in snRNA-seq. The improved performance of DeMixSC in large cohort bulk data demonstrates the benchmark data generation is a worthwhile one-time investment. One available benchmark data set can be utilized for unlimited times to deconvolve any large cohort of the same tissue type. Second, the required sample size for the benchmark data set is small. Eight samples were sufficient to ensure accurate deconvolution in the retina benchmark data set. Single-cell data for the tissue of interest are already being generated and are needed to apply existing single-cell-based deconvolution methods. Saving the remainder-dissociated cell/nucleus suspension for a minimum of eight bulk RNA-seq experiments, an additional step that typically costs less than $2000, can provide valuable benchmark data for enhanced deconvolution accuracy. In addition, given its importance to the success of DeMixSC, we expect that the specialized benchmark data can improve other deconvolution methods, such as the deep learning-based Scaden (Menden et al. 2020) and the guided topic modeling-based GTM-decon (Swapna et al. 2023), by providing insights into cross-platform technical discrepancies.
The advance represented by DeMixSC is noteworthy, but there is potential room for improvements in future work. The key to DeMixSC rests on effectively identifying and down-weighting genes with high technological discrepancy. A potential challenge arises in gene identification when applying DeMixSC to tissue types (e.g., tumors) with high cellular plasticity. In that scenario, a stratified categorization of genes into three distinct groups can be beneficial: technologically stable genes, biologically stable genes (e.g., global tumor signature genes) (Cao et al. 2022), and the remaining unstable genes. Moreover, DeMixSC can be expected to gain from machine learning models to simultaneously identify and adjust genes. Additionally, alternative methods to ComBat (Zhang et al. 2020) for aligning the large cohort with the benchmark data set can be considered when dealing with tumor samples, which often are highly heterogeneous with complex batch structures. DeMixSC also holds the potential to address the challenge of missing cell types in single-cell reference samples by analyzing the residual information from the deconvolution process (Ivich et al. 2024).
Considering such potential adaptations, we anticipate that DeMixSC will prove useful in cancer research. By using a concise benchmark data set derived from matched tissue specimens, DeMixSC can be leveraged to accurately deconvolve large bulk cohorts acquired through either surgical or biopsy samples. DeMixSC's enhanced deconvolution accuracy can improve the reliability of downstream cell type–specific DE analysis with any methods that rely on estimated cell type proportions (Luca et al. 2021; Wang et al. 2021). This capability can be expected to accelerate the discovery of cell subtypes and cell type–specific markers among diverse patient groups with a variety of different types of cancer.
Methods
Ethics approval and consent to participate
Institutional approval for patient consent to donate their eyes was obtained from the University of Utah, and the study adhered to the principles of the Declaration of Helsinki. All retinal tissues were deidentified in accordance with HIPAA privacy rules.
Human retina sample collection
These samples were obtained from 24 individuals between age of 73 to 91 who had passed away because of respiratory or heart failure or from a myocardial infarction (Supplemental Table S1). Human donor eyes were obtained through the Utah Lions Eye Bank. For this study, we included samples collected within 6 h postmortem. Dissections of donor eyes were performed immediately following a published protocol (Owen et al. 2019). Macular retinal tissue was collected using a 6 mm disposable biopsy punch (Integra 33–37), flash-frozen, and stored at −80°C. Only one eye was used per donor, and donors with any history of retinal degeneration, diabetes, macular degeneration, or drusen were excluded from the study. Additionally, each donor underwent an ophthalmology check to ensure that the eye was in a healthy condition.
Generation of benchmark data from 24 human retinal samples
Single-nucleus mRNA sequencing
Nuclei were isolated with prechilled fresh-made RNase-free lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, 0.02% NP-40). The frozen tissue was resuspended and triturated to break the tissue structure in lysis buffer and homogenized with a Wheaton dounce tissue grinder. Isolated nuclei were filtered with 40 µm flow cell strainer and stained with 4′,6-diamidino-2-phenylindole (DAPI; 10 µg/mL) before fluorescence-activated cell sorting (FACS) on an FACSAria III cell sorter (BD Biosciences) in the Cytometry and Cell Sorting Core at Baylor College of Medicine (BCM). All snRNA-seq was performed at the Single Cell Genomics Core (SCGC) at BCM. Single-nucleus cDNA library preparation and sequencing were performed following the manufacturer's protocols (https://www.10xgenomics.com). A single-nucleus suspension was loaded on a chromium controller to obtain a single-cell GEMS (gel beads-in-emulsions) for the reaction. The snRNA-seq library was prepared with chromium single-cell 3′ reagent kit v3 (10x Genomics). The product was then sequenced on an Illumina NovaSeq 6000 (https://www.illumina.com).
Bulk mRNA sequencing of retina single-nucleus suspension
To ensure the “matchness” of paired bulk and snRNA-seq data, the mRNA library for bulk RNA-seq followed the same pipeline as for snRNA-seq. Specifically, matched samples with snRNA-seq were used for RNA isolation by applying TRIzol (Invitrogen) to the separated single-nucleus resuspension. cDNA was prepared from ∼1 ng of total RNA by using the SMART-Seq v4 ultralow input RNA kit according to the manufacturer's directions (Takara). The libraries were made using Nextera XT library prep (Illumina). Full-length RNA-seq was performed on NovaSeq 6000 sequencers according to the manufacturer's directions (Illumina).
Benchmark design for matched single-cell/nucleus and bulk RNA-seq data
The workflow for the benchmark design was summarized in Supplemental Figure S1. Two steps were essential for the “matchness” of the paired bulk and sc/snRNA-seq in the benchmark data set. First, tissue chunks needed to be dissociated into cell or nucleus suspensions, and the paired bulk and sc/snRNA-seq profiling was carried out using the same aliquot (Supplemental Fig. S1A). This process guaranteed the two sequencing data sets share approximately equal cell type proportions. Second, it was necessary to employ the same cDNA library preparation protocol for both sequencing data (Supplemental Fig. S1B). In our study, both bulk and single-nucleus cDNA libraries were generated using the poly(A) enrichment method. These two critical steps together ensured that any technological discrepancies stem solely from the sequencing platforms.
Preprocessing of snRNA-seq and bulk RNA-seq data
Retina snRNA-seq unique molecular identifier (UMI) count matrices were obtained using CellRanger (version 3.1.0) (Zheng et al. 2017) following the official guide to estimate absolute counts and were then processed using the Seurat package (version 3.6.0) (Hao et al. 2021). Specifically, for each snRNA-seq data set, we first removed genes expressed in <5% of cells and then filtered out cells with either fewer than 500 total UMIs or 200 expressed genes, or >50% total UMI counts derived from mitochondrial genes. The total numbers of transcripts of each cell were then normalized to 10,000, followed by a natural log transformation. Highly variable genes were detected and used for principal component analysis (PCA). Cells were then clustered using the Seurat package at a resolution of 0.5.
For bulk RNA-seq data, the quality of raw sequencing data was first evaluated by FastQC (version 0.11.9) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and low-quality reads and adapters were then trimmed by Trimmomatic (version 0.4.0) (Bolger et al. 2014). Next, reads that passed quality control were aligned to GRCh38 using the two-pass mode of STAR (version 2.7.7b) (Dobin et al. 2013), and read counts were obtained by the featureCounts (Liao et al. 2014) function from the Subread package (version 1.22.2) following the standard pipeline.
Cell type annotation for snRNA-seq data
Seven major cell types, including cone cells, rod cells, HCs, BCs, ACs, RGCs, and MGs, were annotated using known marker genes (Supplemental Fig. S2A,B; Supplemental Table S2; Liang et al. 2019; Menon et al. 2019). For the deconvolution analysis of bulk AMD retinal samples (Ratnapriya et al. 2019), we included additional three minor cell types, including astrocytes, microglia cells, and retina pigmental epithelium (RPE).
Generation of ground-truth proportion and pseudobulk mixtures
With each annotated snRNA-seq data, the true proportion of each cell type was calculated as the number of cells in the cell type divided by the total number of cells. Pseudobulk mixtures corresponding to each bulk were calculated by adding up the UMI counts from all the annotated cells per gene from the matched snRNA-seq data.
Statistical analysis
We used paired Student's t-tests to identify the differentially expressed (DE) genes between matched bulk and pseudobulk RNA-seq data. The P-values for DE analysis were adjusted for multiple testing by the Benjamini–Hochberg (BH) method (Benjamini and Hochberg 1995). We used Student's t-tests to compare the estimated cell type proportions between non-AMD and AMD conditions from different deconvolution methods. We used Wilcoxon rank-sum tests to compare the sequencing read depth between bulk and pseudobulk data.
DeMixSC deconvolution framework
DeMixSC is a reference-based model built upon the wNNLS deconvolution framework with several improvements. Our model explicitly
requires a benchmark data set for training. To begin with, we revisit the core equation of existing deconvolution methods
(Ruppert and Wand 1994; Tsoucas et al. 2019; Wang et al. 2019; Aliee and Theis 2021; Dong et al. 2021; Fan et al. 2022; Cobos et al. 2023), which is
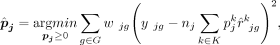 (1)
where yjg is the observed expression (relative abundance) of gene g from subject j in the bulk RNA-seq data,
(1)
where yjg is the observed expression (relative abundance) of gene g from subject j in the bulk RNA-seq data, 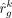 is the estimated expression value of gene g in cell type k in the reference matrix derived from sc/snRNA-seq data, wjg is the weight of each gene g for subject j, nj is the total number of cells in subject j, and
is the estimated expression value of gene g in cell type k in the reference matrix derived from sc/snRNA-seq data, wjg is the weight of each gene g for subject j, nj is the total number of cells in subject j, and 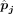 is the estimated vector of cell type proportions. The main drawback of Model 1 is that it does not address technological
discrepancies observed in our benchmark data. To explain this, we split the squared term in Equation 1 into two components, and rewrite the model as
is the estimated vector of cell type proportions. The main drawback of Model 1 is that it does not address technological
discrepancies observed in our benchmark data. To explain this, we split the squared term in Equation 1 into two components, and rewrite the model as
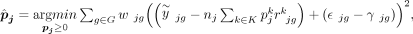 (2)
where
(2)
where 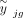 is the true expression value of gene g in the bulk data with
is the true expression value of gene g in the bulk data with 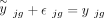 ,
, 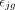 is the measurement noise for gene g in subject j at the bulk level, γjg is the accumulated measurement noise at the single-cell level, and
is the measurement noise for gene g in subject j at the bulk level, γjg is the accumulated measurement noise at the single-cell level, and 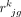 is the true cell type–specific reference matrix (see Supplemental Note). The component
is the true cell type–specific reference matrix (see Supplemental Note). The component 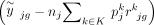 consists of the true bulk-level expression
consists of the true bulk-level expression 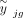 and the true expression value derived based on the true cell type–specific mean expression n
and the true expression value derived based on the true cell type–specific mean expression n 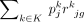 . This component reflects the true estimation error that we aim to minimize. The component
. This component reflects the true estimation error that we aim to minimize. The component  defines the difference in noises introduced by the bulk (
defines the difference in noises introduced by the bulk (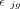 ) and the sc/snRNA-seq (γjg) data, which represents the measurable technological discrepancy between sequencing platforms. Genes with highly inconsistent
expressions, or equivalently inconsistent noises, across different platforms suffer from high technological discrepancy (see
Supplemental Note). Thus, when the technological discrepancy overtakes the true signal, instead of minimizing estimation errors, this model
is geared toward minimizing the technological discrepancy and is no longer fitting the expression profiles of individual bulk
samples.
) and the sc/snRNA-seq (γjg) data, which represents the measurable technological discrepancy between sequencing platforms. Genes with highly inconsistent
expressions, or equivalently inconsistent noises, across different platforms suffer from high technological discrepancy (see
Supplemental Note). Thus, when the technological discrepancy overtakes the true signal, instead of minimizing estimation errors, this model
is geared toward minimizing the technological discrepancy and is no longer fitting the expression profiles of individual bulk
samples.
To address the issue with the technological discrepancy in Model 1, we introduce DeMixSC, which estimates cell type proportions
by minimizing a partitioned loss function, as shown below:
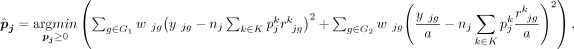 (3)
where G1 is a set of genes hardly affected by technological discrepancy, and G2 contains genes highly affected by technological discrepancy. DeMixSC employs a DE analysis to identify genes affected by
technological discrepancy (G2). This process begins with a paired t-test on matched bulk and pseudobulk data. Genes with BH-adjusted P-values less than 0.05 are selected and ranked from high to low based on mean expression across both data types. The top-ranking
genes are most impacted by technological discrepancy and are designated to G2. To mitigate this technological discrepancy, DeMixSC introduces a positive adjustment factor (a) to rescale the gene expression and thereby reduce the contribution of squared residuals from genes in G2. Rather than excluding them, DeMixSC preserves the high discrepancy genes in G2 in the wNNLS model, acknowledging their potential biological significance and contribution to mixed expression levels. The
size of G2 and the value of a are user-definable parameters in DeMixSC, allowing for flexibility in different analytical contexts. We have tested the model's
performance with various G2 gene selections and values of a (Supplemental Fig. S16), and we set the size of G2 to be 5000 and a to be 1000 by default.
(3)
where G1 is a set of genes hardly affected by technological discrepancy, and G2 contains genes highly affected by technological discrepancy. DeMixSC employs a DE analysis to identify genes affected by
technological discrepancy (G2). This process begins with a paired t-test on matched bulk and pseudobulk data. Genes with BH-adjusted P-values less than 0.05 are selected and ranked from high to low based on mean expression across both data types. The top-ranking
genes are most impacted by technological discrepancy and are designated to G2. To mitigate this technological discrepancy, DeMixSC introduces a positive adjustment factor (a) to rescale the gene expression and thereby reduce the contribution of squared residuals from genes in G2. Rather than excluding them, DeMixSC preserves the high discrepancy genes in G2 in the wNNLS model, acknowledging their potential biological significance and contribution to mixed expression levels. The
size of G2 and the value of a are user-definable parameters in DeMixSC, allowing for flexibility in different analytical contexts. We have tested the model's
performance with various G2 gene selections and values of a (Supplemental Fig. S16), and we set the size of G2 to be 5000 and a to be 1000 by default.
In addition, we introduce a new weight function (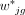 ) to reduce the influence of highly expressed genes and assign lower rankings for genes with large variances:
) to reduce the influence of highly expressed genes and assign lower rankings for genes with large variances:
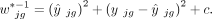 (4)
The current literature uses either the squared fitted value
(4)
The current literature uses either the squared fitted value 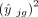 (Tsoucas et al. 2019; Cobos et al. 2023) or the variance
(Tsoucas et al. 2019; Cobos et al. 2023) or the variance 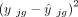 (Wang et al. 2019; Dong et al. 2021; Fan et al. 2022) in the weight, but never both. The constant term c is introduced for controlling the range of the weight. A range of positive values can be appropriate for the constant c (Supplemental Fig. S17). We treat c as a tuning parameter in the DeMixSC software and set it to 2 by default for users’ convenience. Using the summation of these
three terms in Equation 4 as our new weight function improves model fit, accounts for variability, and enhances the numerical stability of the DeMixSC
framework. Detailed mathematical derivation is provided in the Supplemental Note. Implementation of the DeMixSC framework is available as an R package (R Core Team 2023) at GitHub (https://github.com/wwylab/DeMixSC).
(Wang et al. 2019; Dong et al. 2021; Fan et al. 2022) in the weight, but never both. The constant term c is introduced for controlling the range of the weight. A range of positive values can be appropriate for the constant c (Supplemental Fig. S17). We treat c as a tuning parameter in the DeMixSC software and set it to 2 by default for users’ convenience. Using the summation of these
three terms in Equation 4 as our new weight function improves model fit, accounts for variability, and enhances the numerical stability of the DeMixSC
framework. Detailed mathematical derivation is provided in the Supplemental Note. Implementation of the DeMixSC framework is available as an R package (R Core Team 2023) at GitHub (https://github.com/wwylab/DeMixSC).
Evaluation of gene partitioning and weight function
To validate the efficacy of our proposed gene partitioning and weight function, we tested an alternative method with the retina
bulk benchmark data. We used the technological discrepancy (i.e., the test statistics from the paired t-test) as inverse weights and did not rescale the gene expressions using adjustment factor (i.e., no gene partition). The
tested inverse weights were calculated as the t-statistics: 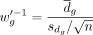 , where
, where 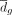 is mean of the differences between paired bulk and pseudobulk samples (i.e.,
is mean of the differences between paired bulk and pseudobulk samples (i.e., 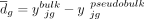 ) of gene g,
) of gene g, 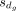 is the standard deviation of the differences, and n is the number of sample pairs. This approach yielded decreased deconvolution accuracy, with mean RMSE increasing from 0.03
to 0.13 and mean Spearman's correlation decreasing from 0.86 to 0.73.
is the standard deviation of the differences, and n is the number of sample pairs. This approach yielded decreased deconvolution accuracy, with mean RMSE increasing from 0.03
to 0.13 and mean Spearman's correlation decreasing from 0.86 to 0.73.
Evaluation of batch correction methods
We tested DeMixSC framework on the AMD retina cohort with four batch correction approaches: no correction, ComBat (implemented in DeMixSC) (Zhang et al. 2020), Limma (Ritchie et al. 2015), and VSN (Huber et al. 2002). Each method was applied following its standard procedure.
Data normalization of bulk mixtures
We applied the following data normalizations to the bulk raw count matrices (Dillies et al. 2013): (1) RPM, (2) RPKM, and (3) TPM. Both RPKM and TPM include an additional step that uses the gene length to obtain normalized counts per million.
Convergence property of the DeMixSC algorithm
To evaluate how robust DeMixSC is against different initial values, we randomly selected a sample from the AMD retina cohort
as a case study. To create different initial values, we set three different scale factors 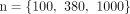 . For each scale factor, we chose 10 extreme starting values for the proportions
. For each scale factor, we chose 10 extreme starting values for the proportions 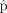 , with the proportion of one out of 10 cell types being one and the rest being zero. Finally, we used the 30 pairs of
, with the proportion of one out of 10 cell types being one and the rest being zero. Finally, we used the 30 pairs of 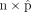 to initialize the wNNLS framework and then compared the estimates of DeMixSC.
to initialize the wNNLS framework and then compared the estimates of DeMixSC.
Computational deconvolution with existing methods
Eight deconvolution methods that use the same scRNA-seq data as the reference were tested in our benchmarking study (Newman et al. 2019; Tsoucas et al. 2019; Wang et al. 2019; Aliee and Theis 2021; Dong et al. 2021; Erdmann-Pham et al. 2021; Chu et al. 2022; Cobos et al. 2023). We first used the default settings of each method as described in the GitHub repository or the websites (AutoGeneS: https://github.com/theislab/AutoGeneS; BayesPrism: https://github.com/Danko-Lab/BayesPrism; CIBERSORTx: https://cibersortx.stanford.edu/; DWLS: https://github.com/dtsoucas/DWLS; MuSiC: https://github.com/xuranw/MuSiC; RNAseive: https://github.com/songlab-cal/rna-sieve; SCDC: https://github.com/meichendong/SCDC; and SQUID: https://github.com/favilaco/deconv_matching_bulk_scnRNA). For CIBERSORTx, we followed the recommended built-in batch correction method for the deconvolution analysis of bulk samples (batch mode = S). Additionally, we evaluated the performance of the tree-guided deconvolution of MuSiC (Wang et al. 2019) and the ensemble option of SCDC (Dong et al. 2021). For tree-guided MuSiC, we first performed hierarchical clustering on the single-cell reference data set; based on the hierarchical clustering results, we grouped cone and rod cells to form a mega cell cluster (Supplemental Fig. 6A), and each of the remaining cell types also formed a cluster. Cell type–specific marker genes of cones and rods were obtained using FindAllMarkers function from Seurat (Hao et al. 2021) package under the bimod likelihood ratio test. We ran MuSiC deconvolution first at the cell cluster level and then again within the rod and cone clusters. For the SCDC ensemble option, we ran deconvolution on SCDC with three different sc references; then, we ran the SCDC_ENSEMBLE function to obtain the ensemble deconvolution results. For deconvolving the AMD cohort, we used MuSiC2 (Fan et al. 2022) following the tutorial provided with default settings (https://github.com/Jiaxin-Fan/MuSiC2).
Evaluation metrics for the deconvolution performance
-
We evaluated the performance of each method using (1) RMSE at both sample (RMSEj) and cell type (RMSEk) levels:
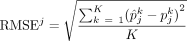 and
and 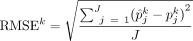 , where
, where 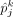 denotes the estimated cell proportion by the investigated method for cell type k and sample j, and
denotes the estimated cell proportion by the investigated method for cell type k and sample j, and 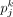 is the corresponding ground truth. We use J to denote the total number of samples and K to denote the total number of cell types. A smaller RMSE value indicates a better deconvolution performance.
is the corresponding ground truth. We use J to denote the total number of samples and K to denote the total number of cell types. A smaller RMSE value indicates a better deconvolution performance.
-
We evaluated the Spearman's correlation coefficient (ρ) at both sample (ρj) and cell type (ρk) levels. A higher Spearman's correlation coefficient indicates a better deconvolution performance.
Deconvolution analysis on the human healthy retina benchmark data set
The retina benchmark data set comprised two batches, batch-1 (n = 4) and batch-2 (n = 20). To account for potential batch effects and ensure optimal deconvolution performance for each method, the analysis was performed separately for each batch. All deconvolution methods were performed using the same single-nucleus reference derived from the retinal benchmark data set.
Deconvolution analysis on the human diseased retina cohort (AMD)
Data acquisition and quality control
The expression matrix of the AMD cohort comprised 523 samples and was obtained from Ratnapriya et al. (2019)'s study under the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) accession number GSE115828. We conducted quality control following the pipeline described in their original study. Samples were filtered out owing to ambiguous clinical features (n = 26), poor sequencing results (n = 24), inconsistent genotyping results (n = 14), and divergent ancestry (n = 6). A total of 70 samples were removed, with a total of 453 samples remaining to be used to perform the deconvolution analysis. We further evaluated DeMixSC's performance on genetically diverse populations by including six samples with ancestry diverging from European-American, which were previously excluded based on QC criteria (Ratnapriya et al. 2019; Fan et al. 2022). DeMixSC achieved comparable results (mean RMSE: 0.04; mean Spearman's correlation: 0.75) to our original analysis, demonstrating DeMixSC's potential applicability across diverse genetic backgrounds.
Computational deconvolution with consensus reference
We performed deconvolution using four methods: DeMixSC, MuSiC2, CIBERSORTx, and SQUID. The three existing methods were selected
for the following reasons: (1) MuSiC2 leveraged conditionally stable genes from healthy references to analyze diseased tissue;
(2) CIBERSORTx was ranked second in our benchmarking study; and (3) SQUID also deconvolved unmatched large bulk cohorts using
benchmark data. To run each method, we generated a consensus reference by integrating seven samples from batch-2 (samples
5, 10, 12, 18, 19, 21, and 23) (Supplemental Table S1). We selected these samples as they adequately represented these three minor cell types: astrocytes, microglia cells, and
RPE. For each sample, we randomly selected up to 500 cells per cell type, using all available cells for types with fewer than
500 cells. Relative abundance 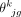 and cell size
and cell size 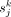 for each cell type k were calculated for each sample j. A consensus reference matrix r was subsequently derived by multiplying the averaged relative abundance and the averaged cell size across the selected samples.
Mathematically, the consensus reference is defined as
for each cell type k were calculated for each sample j. A consensus reference matrix r was subsequently derived by multiplying the averaged relative abundance and the averaged cell size across the selected samples.
Mathematically, the consensus reference is defined as 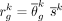 , where
, where 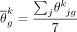 and
and 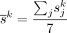 are the averaged abundance and averaged cell size over the seven samples, respectively.
are the averaged abundance and averaged cell size over the seven samples, respectively.
Validating deconvolution performance using reference proportions from healthy human peripheral retina
To validate methods performance in deconvolving the AMD cohort, we compared the estimated cell type proportions of non-AMD samples (n = 105) with the previously reported proportions in healthy human peripheral retina tissues (Liang et al. 2019). The non-AMD samples were peripheral retina tissues from donors aged between 55 and 94, with a mean age of 80 ± 9.95 years. Reference proportions were calculated based on snRNA-seq profiling of the peripheral retina from three human donors aged 60 to 80 years, matching the non-AMD samples by age. Seven major cell types were identified with the following proportions: ACs, 7.74%; BCs, 15.6%; cones, 4.61%; HCs, 3.61%; MGs, 14.08%; RGCs, 1.07%; and rods, 53.29% (Supplemental Table S3). To account for the additional three minor cell types (RPE, astrocyte, and microglia) estimated in the non-AMD samples but not measured in the reference, we rescaled the proportions of the seven major cell types so that they sum to one by dividing their total.
Accounting for the total cell loss in the AMD cohort
The decrease in overall cell count induced by photoreceptor (cone and rod cells) loss likely amplifies the cell type proportions in the AMD samples (Ambati et al. 2013; Menon et al. 2019; Fleckenstein et al. 2021). The mean estimated fraction of photoreceptors is 55.21% (3.43% cone + 51.78% rod) in non-AMD and 51.88% (2.82% cone + 49.06% rod) in AMD (Fig. 4D). We use “x” to represent the mean percentage of lost photoreceptors in the AMD condition and derive the relation: 0.5521(1 − x)/(1 − 0.5521x) = 0.5188. Solving for x shows a 12.53% reduction in photoreceptors in the peripheral AMD retina, which aligns well with biological evidence that the peripheral retina experiences a more modest photoreceptors loss (10%–20%) compared with the macular region (>30%) (Curcio et al. 1996). Next, we investigated whether the observed increases in BCs, MGs, and astrocytes were driven by the death of photoreceptors or cell proliferation. Using our photoreceptor loss metric, we estimated the expected cell fractions in AMD: (estimated non-AMD proportion)/(1 − 0.5521× 0.1253). The expected fractions were 19.00% for BCs (from 17.69% in non-AMD), 8.31% for MGs (from 7.74%), and 4.69% for astrocytes (from 4.37%). The expected fraction of BCs closely matches DeMixSC's estimate (18.73%), suggesting the increase of BCs was because of photoreceptor loss. For glial cells, DeMixSC's estimates (9.19% for MGs and 5.52% for astrocytes) exceeded the expected fractions, suggesting an increase of these cells in the AMD condition.
Deconvolution analysis on the human primary HGSC benchmark data set
The HGSC benchmark data set, obtained from GEO under accession number GSE217517 (Hippen et al. 2023), comprised seven primary HGSC samples. For each of these samples, three types of bulk RNA-seq data were generated, with three technical replicates for each data type: (1) dissociation with poly(A) enrichment (Disso&poly(A)+, n = 21), (2) dissociation with rRNA depletion (Disso&rRNA−, n = 21), and (3) tissue chunk with rRNA depletion (Chunk&rRNA−, n = 21). For the matched single-cell data, we followed the cell type annotation as described in the original paper. Thirteen cell types were identified: epithelial cells, endothelial cells, fibroblasts, B cells, plasma cells, natural killer (NK) cells, innate lymphoid cells (ILCs), monocytes, macrophages, mast cells, dendritic cells (DCs), plasmacytoid dendritic cells (pDCs), and T cells. All deconvolution methods were performed using the same single-nucleus reference derived from the HGSC benchmark data set.
Deconvolution analysis on the unmatched human primary HGSC cohort
Data acquisition and quality control
The unmatched human primary HGSC cohort was obtained from the study of Lee et al. (2020) from the European Genome-phenome Archive (EGA; https://ega-archive.org) under accession number EGAD00001005238. This cohort contains 30 primary HGSC samples categorized into three treatment response groups: complete gross resection (R0) and received neoadjuvant chemotherapy with an excellent (ER) or a poor (PR) response.
Computational deconvolution with consensus reference
We performed deconvolution with four methods, including DeMixSC, MuSiC, CIBERSORTx, and SQUID. Unlike the AMD study, we used MuSiC rather than MuSiC2 because MuSiC and MuSiC2 shared the same computational framework, but MuSiC2 was specifically designed to use normal references for deconvolving disease samples, which was not applicable in the HGSC study. To run each method, we generated a consensus reference by integrating all seven scRNA-seq samples from the HGSC benchmark data set. For each sample, we randomly selected up to 1000 cells per cell type or selected all available cells if fewer than 1000 were present. The methodology for generating the consensus reference matrix followed the same approach described in the section Deconvolution Analysis on the Human Diseased Retina Cohort (AMD).
Comparing with the immunostaining results
Out of the 30 primary HGSC samples, 21 had both RNA-seq and immunostaining data for macrophage, as measured using CD68 and CD163 antibodies. The detailed data description and immunostaining results were obtained from the original study (Lee et al. 2020).
Software availability
DeMixSC is freely available as an R package and can be downloaded from our GitHub repository (https://github.com/wwylab/DeMixSC). A tutorial for DeMixSC is available at GitHub (https://wwylab.github.io/DeMixSC/). The DeMixSC source code is also available as Supplemental Code.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE175937. The human retina tissue snRNA-seq data in this study have been submitted to the Human Cell Atlas Data Portal (https://explore.data.humancellatlas.org/projects/9c20a245-f2c0-43ae-82c9-2232ec6b594f).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
S.G. is supported by Human Cell Atlas Seed Network-Breast, Chan Zuckerberg Institute, MD Anderson Colorectal Cancer Moon Shot Program, and U.S. Department of Defense (DoD) (PC210079). X.L. is supported by the National Institutes of Health (NIH) (R01CA239342). A.K. is supported by NIH (5T32CA096520-15). J.P.S. is supported by the Cancer Prevention and Research Institute of Texas (CPRIT) as a CPRIT Scholar in Cancer Research and by NIH (K22CA234406). R.C. is supported by Human Cell Atlas Seed Network-Retina, Chan Zuckerberg Institute (CZF2019-02425), National Eye Institute (R01EY022356 and R01EY018571), and the Retinal Research Foundation. W.W. is supported by Human Cell Atlas Seed Network-Retina, Chan Zuckerberg Institute, NIH (R01CA268380), and DoD (PC210079; P30CA016672). This work is also supported by CPRIT Single Core grant (RP180684), the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), NIH (CA125123 and RR024574), and the assistance of Joel M. Sederstrom. Single-nucleus RNA sequencing was performed at the Single Cell Genomics Core at BCM partially supported by NIH shared instrument grants (S10OD018033, S10OD023469, and S10OD025240), P30CA125123, P30EY002520, and CPRIT Comprehensive Cancer Epigenomics Core Facility (RP200504).
Author contributions: W.W. and R.C. supervised the research. W.W. and R.C. conceived the ideas and designed the study. S.G. and X.L. developed the method, implemented the R package, and conducted all the analysis. X.C. conducted the sequencing experiments and generated the paired bulk and single-nucleus RNA-seq data for benchmarking purposes. Y.J. and S.J. helped with the initial development of the method and evaluation. Q.L. and Y.L. helped with the sequencing experiments. L.A.O., I.K.K., A.A., S.K., J.P.S., M.M.D., and R.C. provided samples, advised on the study design, and assisted with the interpretation of results. A.K. performed the initial benchmarking analysis. J.N.W. and R.C. contributed technical suggestions. S.L. and A.K.S. helped with ovarian cancer data analysis. W.W., S.G., and X.L. wrote the paper, with input from all authors. All authors reviewed and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278822.123.
-
Freely available online through the Genome Research Open Access option.
- Received December 5, 2023.
- Accepted November 19, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








