
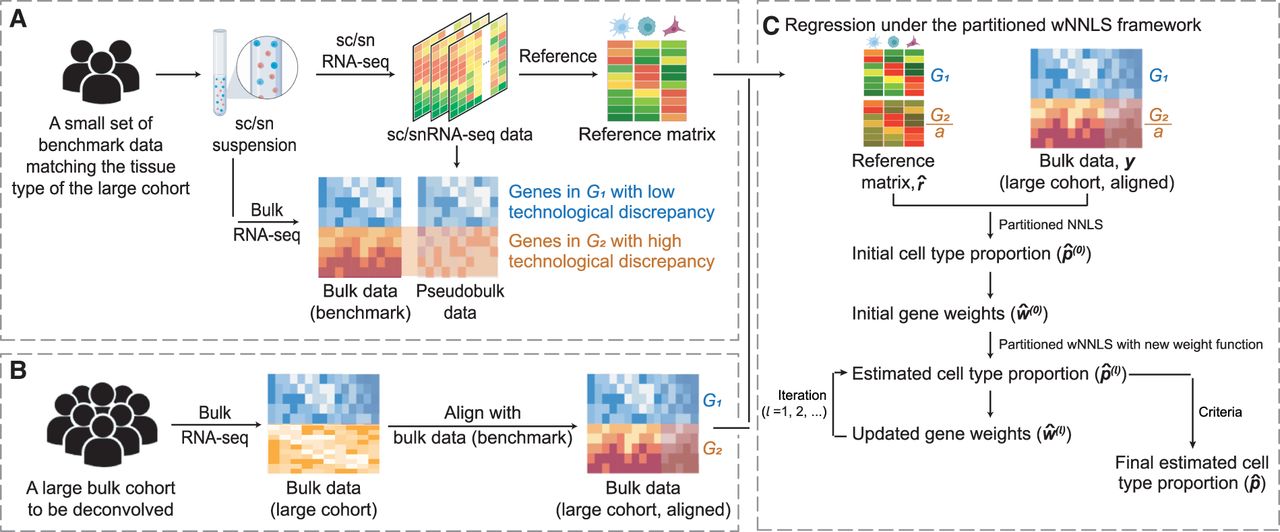
Overview of DeMixSC. The DeMixSC framework for deconvolution analysis of bulk RNA-seq data using sc/sn RNA-seq data as a reference.
(A) The framework starts with a benchmark data set of matched bulk and sc/snRNA-seq data with the same cell type proportions.
Pseudobulk mixtures are generated from the sc/sn data. DeMixSC identifies genes in G1 and G2 with the matched real-bulk and pseudobulk data. The non-DE genes are considered stably captured by both sequencing platforms
(blue), whereas the DE genes are more impacted by the technological discrepancy (orange). (B) DeMixSC then employs a normalization procedure to perform the alignment between two bulk RNA-seq data sets (e.g., with ComBat).
(C) DeMixSC estimates cell type proportions under a weighted nonnegative least square (wNNLS) framework with two improvements:
(1) partitioning and adjusting genes with high technological discrepancy and (2) a new weight function. The final estimates
are obtained when the algorithm either converges or reaches the prespecified maximum number of iterations. Here, G1 is genes with low technological discrepancy, G2 is genes with high technological discrepancy, a is a user-defined positive constant that serves as an adjustment factor,  is the reference matrix derived from the sc/snRNA-seq data, y is the observed expression in bulk RNA-seq data,
is the reference matrix derived from the sc/snRNA-seq data, y is the observed expression in bulk RNA-seq data, 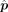 is the vector of estimated cell type proportions, and
is the vector of estimated cell type proportions, and 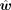 is the estimated gene weights.
is the estimated gene weights.








