
Single-cell discovery of m6A RNA modifications in the hippocampus
- Shuangshuang Feng1,2,3,
- Maitena Tellaetxe-Abete4,7,
- Yujie Zhang2,3,7,
- Yan Peng2,3,5,
- Han Zhou2,
- Mingjie Dong2,
- Erika Larrea2,3,6,
- Liang Xue2,3,
- Li Zhang2 and
- Magdalena J. Koziol1,2,3
- 1State Key Laboratory of Cognitive Neuroscience and Learning, Beijing Normal University, Beijing 100875, China;
- 2Chinese Institute for Brain Research, Beijing 102206, China;
- 3Research Unit of Medical Neurobiology, Chinese Academy of Medical Sciences, Beijing 102206, China;
- 4Intelligent Systems Group, Computer Science Faculty, University of the Basque Country, Donostia/San Sebastian 20018, Spain;
- 5Peking University, Beijing, 100871, China;
- 6Tsinghua University, Beijing 100084, China
-
↵7 These authors contributed equally to this work.
Abstract
N6-Methyladenosine (m6A) is a prevalent and highly regulated RNA modification essential for RNA metabolism and normal brain function. It is particularly important in the hippocampus, where m6A is implicated in neurogenesis and learning. Although extensively studied, its presence in specific cell types remains poorly understood. We investigated m6A in the hippocampus at a single-cell resolution, revealing a comprehensive landscape of m6A modifications within individual cells. Through our analysis, we uncovered transcripts exhibiting a dense m6A profile, notably linked to neurological disorders such as Alzheimer's disease. Our findings suggest a pivotal role of m6A-containing transcripts, particularly in the context of CAMK2A neurons. Overall, this work provides new insights into the molecular mechanisms underlying hippocampal physiology and lays the foundation for future studies investigating the dynamic nature of m6A RNA methylation in the healthy and diseased brain.
The hippocampus, crucial for learning and memory (Bird and Burgess 2008), is a focal point in neuroscience research. N6-Methyladenosine (m6A) RNA methylation, a key epitranscriptomic modification, regulates various biological processes, including neurodevelopment and learning (Li et al. 2017; Livneh et al. 2020; Jiang et al. 2021; Yu et al. 2021), with implications for neurological disorders (Lv et al. 2023).
The m6A modification is catalyzed by methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit (METTL3) and is reversible by enzymes like FTO alpha-ketoglutarate dependent dioxygenase (FTO) and ALKBH5 (Li et al. 2017). m6A-modified transcripts are bound by reader proteins such as YTH N6-methyladenosine RNA binding protein F2 (YTHDF2) (Wang et al. 2014; Du et al. 2016).
Although m6A in various tissues is well studied, despite its presence and impact in the hippocampus, it is currently unknown which cell types and transcripts have m6A and with which density (Zhou et al. 2023).
Studies on Mettl3, Ythdf2, and Fto knockout (KO) mice link m6A to hippocampal function, learning, and memory (Li et al. 2017; Engel et al. 2018; Livneh et al. 2020; Zhuang et al. 2023). Also, overexpression of Mettl3 enhances long-term memory consolidation (Zhang et al. 2018; Jiang et al. 2021). Cell-specific m6A levels have been noted in the cerebellum and cortex, suggesting potential cell type–specific functions (Chang et al. 2017).
To uncover the landscape of m6A RNA methylation in hippocampal cells, we employ single-cell sequencing. Single-cell sequencing technologies have revolutionized our ability to uncover cellular heterogeneity and identify distinct subpopulations within complex tissues (Regev et al. 2017). Applying these technologies to the study of m6A will provide unprecedented insights into the cell type–specific roles of m6A in hippocampal function, facilitating the development of future targeted therapeutics.
In this study, we aim to address the knowledge gap by performing single-cell RNA sequencing to profile m6A RNA methylation patterns in individual cells of the hippocampus. A myriad of different m6A sequencing detection methods exist, such as m6A RNA immunoprecipitation (m6A-RIP) (Dominissini et al. 2012; Meyer et al. 2012), coupled with high-throughput sequencing technologies, or m6A-SEAL (Wang et al. 2020) or MAZTER-seq (Garcia-Campos et al. 2019). However, all of them require high amounts of RNA input, currently making them incompatible with single-cell approaches in regular somatic cells, such as found in the brain (Ke et al. 2015; Linder et al. 2015; Garcia-Campos et al. 2019; Zhang et al. 2019; Shu et al. 2020; Wang et al. 2020; Yao et al. 2023; Li et al. 2024). Deamination adjacent to RNA modification targets sequencing (DART-seq), utilizing a APOBEC-YTH fusion construct in which the APOBEC1 protein is fused to the YTH m6A-binding domain of YTHDF2, enables m6A detection (Meyer 2019). In APOBEC-YTH, the YTH domain lures APOBEC1 to the close vicinity of m6A sites, where APOBEC1 deaminates cytidine into uracil (Fig. 1A; Meyer 2019). By identifying C-to-U editing events that correspond to C-to-T mutations in sequencing data, adjacent m6A sites can be identified. Ninety-seven percent of these sites disappeared in methylase METTL3-depleted cells, illustrating the specificity of this method (Meyer 2019). The feasibility of this single-cell DART-seq (scDART-seq) approach has recently also been demonstrated using a relatively homogenous HEK293T cell line (Tegowski et al. 2022). Here, we apply this single-cell approach to map m6A distribution in hippocampal cell types, enhancing our understanding of m6A modifications’ cell-specific characteristics.
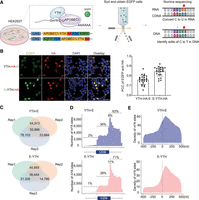
Improved m6A bulk RNA-seq detection in HEK293T cells. (A) Schematic diagram of m6A detection with bulk RNA-seq in cultured cells. The YTH protein domain binds to m6A. When bound to APOBEC1, the APOBEC1 protein converts C-to-U in the vicinity of m6A. This results in a C-to-T mutation in cDNA. C-to-T mutations detected by RNA sequencing are indicative of m6A RNA modifications. (B, left) Immunofluorescence (IF) of HEK293T cells transfected with Apobec1-Yth-HA-Egfp (Yth-HA-E) or Egfp-Apobec1-Yth-HA (E-YTH-HA). Scale bar, 20 μm. Representative images are shown. (Right) Quantification of EGFP and HA overlap. (PCC) Pearson correlation coefficient. (C) Number of C-to-T editing events identified in each bulk RNA-seq HEK293T cell replicate for Yth-E and E-Yth plasmids. Editing events identified in at least two replicates were considered for downstream analyses. The data were obtained following Apobec1-Yth-Egfp or Egfp-Apobec1-Yth transfection and EGFP FACS sorting. n = 3. (Rep) separately cultured replicate. (D) Metagene analysis showing m6A site counts along transcripts for Yth-E and E-Yth bulk RNA-seq results. Nine percent of all m6A sites occur in the first 10% of the 3′ UTR following the TTS for YTH-E and 11% for E-YTH, respectively. Shown percentage indicates number of m6A sites upstream of, within, and downstream from coding sequence (CDS). (E) Metagene analysis showing m6A density 500 nt 5′ and 500 nt 3′ from stop codon (0 nt) for YTH-E and E-YTH.
Results
Confirmation of m6A detection with bulk RNA-seq
To enable the application of DART-seq in mouse hippocampal cells in vivo, we subcloned Apobec1-Yth with an HA tag into an adeno-associated virus (AAV) plasmid under control of a chicken beta-actin (CAG) promoter. Controls included Apobec1-Ythmut, incapable of binding m6A, and Apobec1 only. Through AAV transduction, these proteins can then be expressed in different cell types of the mouse brain (Negrini et al. 2020). A P2A sequence with Egfp was added to enable isolating successfully AAV-transduced cells without interfering with APOBEC1-YTH function. We argued that FACS selection for APOBEC1-YTH (YTHmut)-positive cells is crucial to avoid false negatives owing to inadequate APOBEC1-YTH exposure.
Testing the Apobec1-Yth-HA-P2A-Egfp (Yth-E) plasmid in HEK293T cells (subsequently referred to as the YTH-E sample) showed a weak enhanced green fluorescent protein (EGFP) signal (Fig. 1B), attributed to its location at the C-terminal end of a long RNA transcript (Zhu et al. 2023). Similarly, when Yth-E was transduced into the hippocampus, an EGFP signal could not be easily detected with FACS sorting (Fig. 1B; Supplemental Fig. S1A). Reordering the expression cassette to Egfp-P2A-Apobec1-Yth-HA (E-Yth) and Egfp-P2A-Apobec1-Ythmut-HA (E-Ythmut) resulted in enhanced EGFP signal and improved correlation with Apobec1-Yth-HA (Fig. 1B), suitable for single-cell sequencing. Both E-YTH and E-YTHmut samples exhibited EGFP in cytoplasm and nuclei (Supplemental Fig. S1B), suggesting that both could be used for single-cell sequencing.
Further evaluation involved transfecting E-Yth and Yth-E into HEK293T cells for FACS sorting and RNA sequencing (RNA-seq) (Fig. 1A). As controls, we processed E-Ythmut, Ythmut-E, E-Apobec1, Apobec1-E, and mock-transfected cells. We only considered C-to-U editing sites that were identified in at least two replicates, such as 209,256 C-to-U editing sites in YTH-E and 127,462 in E-YTH samples (Fig. 1C; Supplemental Table S1), and did similar analysis for control samples (Supplemental Fig. S1C; Supplemental Table S1). To identify m6A sites in E-YTH and YTH-E, we eliminated background C-to-U editing events detected in APOBEC1-only, YTHmut, and mock-transfected cells (for more details, see Methods). We identified 106,827 m6A sites that occur in transcripts in YTH-E and 54,395 in E-YTH (Supplemental Table S2). In both cases, we found m6A to be enriched in the 3′ UTR region, 63% in YTH-E and 71% in E-YTH, confirming previous m6A findings (Fig. 1D,E; Supplemental Table S3; Dominissini et al. 2012; Meyer et al. 2012). Nine percent of all m6A sites in YTH-E and 11% in E-YTH occur in the first 10% of the 3′ UTR (Fig. 1D,E; Supplemental Table S3). Overall, YTH-E and E-YTH confirm previous m6A findings. E-YTH, with a stronger EGFP signal and clearer correlation with APOBEC1-YTH presence, was chosen for hippocampal studies owing to its defined m6A distribution patterns and reduced background noise (Fig. 1B,D; Supplemental Fig. S1D).
Detection of m6A with bulk RNA-seq in mouse hippocampus
Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) of adult mouse brains showed higher levels of m6A in mRNAs in hippocampi than in mRNAs of the cortex, thalamus, or cerebellum. In hippocampal mRNAs, we found that m6A occurs in one per 1000 unmodified adenosines (Supplemental Fig. S2A). To identify m6A within hippocampal mRNA sequences, we tested if E-YTH can facilitate m6A detection in vivo. Thus, we packaged E-Yth and controls E-Ythmut and E-Apobec1 into AAV viruses for local hippocampal stereotaxic injection (Fig. 2A). Following successful AAV expression verification in the hippocampus (Fig. 2B), EGFP-positive cells underwent FACS sorting and RNA-seq (Fig. 2A). Wild-type (WT) hippocampal samples were included as additional controls; 2672 and 2272 C-to-U editing sites were identified in all three replicates of E-YTH and E-YTHmut, respectively. Only a few hundred C-to-U editing sites overlap between two replicates, whereas more are common between all three replicates, indicating high reproducibility (Fig. 2C; Supplemental Fig. S2B; Supplemental Table S4). To identify m6A sites, we focused C-to-U editing events present in at least two out of three replicates (Fig. 2C; Supplemental Table S4) and background from E-YTH, including those detected in WT, APOBEC1, and E-YTHmut (see Methods) (Supplemental Fig. S2B; Supplemental Table S4). In total, we identified 1578 edits in E-YTH transcripts but only 82 edits in E-YTHmut controls (Supplemental Table S5). To further verify the feasibility of m6A detection with E-YTH in the mouse hippocampus, we detected 8220 m6A regions that correspond to 5431 genes with m6A-RIP (Supplemental Fig. S2C; Supplemental Table S6). Thirty-two percent of the E-YTH m6A sites were also detected and corroborated with m6A-RIP (Supplemental Fig. S2D; Supplemental Table S7). In accordance to previous studies using m6A-RIP and DART-seq in cells (Meyer et al. 2012), our E-YTH identified m6A enrichment in the 3′ region (80.8%) (Fig. 2D; Supplemental Fig. S2E), particularly near the TTS (Fig. 2E). We detected that most m6A occur 118 nt downstream from the TTS site (Fig. 2F). To assess the frequency of m6A occurrence across multiple transcript copies from a single gene (referred to as RNA replicates), we calculated the mutation per read ratio (m/k) for each editing site, excluding sites with fewer than 10 reads (see Methods). A m/k ratio of one indicates that all copies of a specific transcript have m6A, notably at the same site, whereas a m/k ratio of 0.25 stipulates that 25% of them share m6A. Our bulk sequencing data have low m6A density at each position. Although some transcripts exhibit m6A on all their RNA copies (m/k = 1), the majority have m6A in <10% (Fig. 2G; Supplemental Fig. S1D). The bulk sequencing data suggest that m6A, although functionally crucial in the hippocampus (Zhang et al. 2018; Du et al. 2021; Yin et al. 2023), is predominantly heterogenous at specific sites. However, certain cells have sites where m6A occurs on many RNA replicates. To investigate this, we next applied our system to detect m6A sites on a single-cell level in the mouse hippocampus.
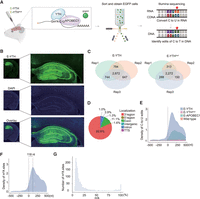
Detection of m6A with bulk RNA-seq in mouse hippocampus. (A) Schematic diagram of bulk RNA-seq in the mouse brain. The Egfp-Apobec1-Yth is packaged into AAV viruses to infect brain cells. EGFP-positive cells are isolated from the hippocampus and processed for C-to-U edit and m6A site identification. (B) Confocal image of mouse hippocampus after AAV infection. Representative image of E-YTH is shown. Half-brain image: Scale bar, 1 mm. Hippocampus image: Scale bar, 400 μm. (C) Number of overlapping C-to-U editing events identified by RNA-seq in hippocampus following Egfp-Apobec1-Yth and Egfp-Apobec1-Ythmut AAV virus injection and EGFP FACS sorting. Editing events identified in at least two replicates were considered for downstream analyses. n = 3, (Rep) Biological replicates from different animals. (D) Pie chart showing m6A localization identified by E-Yth in mouse hippocampus. (TTS) Transcription termination site. (E) Metagene analysis showing C-to-U edit scaled density 500 nt 5′ and 500 nt 3′ from stop codon (0 nt) in E-YTH, E-YTHmut, E-APOBEC1, and wild-type (WT) samples. The peak value for E-YTH is 2716 editing events. (F) Metagene analysis showing m6A density 500 nt 5′ and 500 nt 3′ from stop codon (0 nt). m6A sites were obtain after eliminating background from E-YTH editing sites. m6A peak density occurs 118 nt downstream from the stop codon. (G) Histogram of m6A site counts over mutation per read (m/k) ratio. Minimum threshold: 5%.
Single-cell sequencing of E-YTH-transduced hippocampus
We administered AAV containing E-Yth or E-Ythmut into the hippocampi of 3-month-old mice (Fig. 3A) and optimized the protocol for dissociating the mouse hippocampus into single cells owing to their fragility and variability in shape and size (see Methods). We next FACS-sorted for EGFP to isolate cells successfully transduced with E-Yth or E-Ythmut. This selection process is crucial to exclude cells with insufficient E-YTH or E-YTHmut expression, thereby preventing m6A false negatives that could lead to overestimations of m6A heterogeneity in RNA replicates. Following stringent EGFP selections, cells were then loaded into microwells (Rhapsody, BD Biosciences) for barcoding, leading to library generation and high-throughput sequencing (Fig. 3A; Supplemental Fig. S3A). Our evaluation confirmed the high quality and adequate read counts in the E-YTH and E-YTHmut single-cell libraries (Supplemental Fig. S3B; Supplemental Table S8). We identified 11,561 cells with barcodes and UMIs in the E-YTH samples and 16,243 in the E-YTHmut samples (Supplemental Table S8) and conducted clustering on the integrated E-YTH and E-YTHmut single-cell data sets (Fig. 3B) using the uniform manifold approximation and projection (UMAP) dimension reduction method, revealing 28 clusters. Despite the absence of cluster 23 in our E-YTH data, all other clusters exhibited similar cell numbers between E-YTH and E-YTHmut (Fig. 3C; Supplemental Fig. S3C). The absence of cluster 23 was unexpected, as the number of genes detected in all other clusters is very similar, with an average of 2800 for E-YTH and 2373 for E-YTHmut (Fig. 3D). Also, few transcriptional changes were detected in E-YTH versus E-YTHmut single-cell data (Supplemental Fig. S4A,B). Because clusters 21, 22, and 24–27 have fewer cell numbers than cluster 23 of E-YTHmut (Supplemental Fig. S3C), the absence of cluster 23 in E-YTH cannot be attributed to not enough sequencing depth. Thus, the absence of cluster 23 seems E-YTH specific. Cluster 23, identified as neurons through automatic gene annotation (Supplemental Fig. S4C), was specifically enriched in CAMK2A (Supplemental Table S10), a gene expressed in a subgroup of excitatory neurons in the hippocampus and cortex, known for its role in long-term memory consolidation (Zhang et al. 2018; Yasuda et al. 2022). We next asked if we can validate our single-cell data and the absence of CAMK2A-expressing cells in E-YTH but not in E-YTHmut hippocampi. Although our single-cell data did not reveal any CAMK2A-expressing cells following E-Yth injections, western blot analysis still detected a substantial, albeit reduced amount of CAMK2A proteins in E-YTH (Supplemental Fig. S4D). This discrepancy stems from our single-cell experiments, in which we FACS-sorted E-Yth transduced cells. In contrast, western blot analysis encompassed the entire hippocampus owing to material limitations, encompassing both transduced and nontransduced cells. To corroborate our findings, we employed pAAV-Camk2a-mCherry to visualize CAMK2A-expressing cells, identifiable by their red appearance. This virus was mixed with equal amounts of EGFP-expressing E-Yth or E-Ythmut AAVs along with Camk2a-mCherry alone and subsequently injected into mouse hippocampi. When we coinjected E-Ythmut and pAAV-Camk2a-mCherry, EGFP and mCherry signals overlapped, indicating the presence of CAMK2A-expressing cells. However, when coinjecting E-Yth and pAAV-Camk2a-mCherry, the overlap was reduced, suggesting fewer or no CAMK2A-expressing cells in our E-YTH single-cell data, and reinforces our single-cell findings, suggesting the importance of m6A containing transcripts in CAMK2A neurons (Supplemental Fig. S4E).
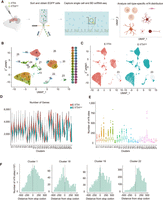
Hippocampal single-cell identification following AAV transduction. (A) Schematic diagram of detecting m6A RNA modification in the mouse hippocampus on a single-cell level through AAV transduction with E-Yth and E-Ythmut controls. (B) Integration of uniform manifold approximation and projection (UMAP) of 27,804 single-cell transcriptomes. Cluster numbers from zero to 27 are indicated. (C) Separate UMAP for E-YTH and E-YTHmut: 11,561 single cells in E-YTH and 16,243 in E-YTHmut. (Circle) Cluster 23 is missing in E-YTH. (D) Violin plot visualizing the number of genes per cluster. No genes were detected in cluster 23 in E-YTH samples. (Red) E-YTH, (blue) E-YTHmut. (E) m6A counts per cluster. (F) Metagene analysis of individual cell clusters identified by single-cell sequencing. The m6A number surrounding the stop codon (position 0) is shown. m6A sites were obtained after eliminating background editing sites. Clusters 1, 18, 19, and 22 are shown, which represent different m6A distribution patterns.
Identification of m6A sites in single-cell clusters of the hippocampus
We identified C-to-U editing sites in our mouse hippocampus single-cell sequencing data and isolated m6A sites by filtering out bulk background C-to-U editing events detected in the WT, E-APOBEC1, and E-YTHmut controls (see Methods). This process yielded 2,566,141 potential m6A sites. To address unequal background distributions in clusters, we also removed cluster-specific E-YTHmut background for each cell in the E-YTH single-cell data, resulting in 923,249 of m6A sites in transcripts on a single-cell level (see Methods) (Supplemental Table S9). Cluster 22 exhibited the highest average number of m6A sites per cell (178), whereas cluster 18 displayed the lowest (18) (Fig. 3E; Supplemental Table S9). To validate our single-cell m6A data, we compared the data with m6A sites identified by m6A-RIP and our bulk E-YTH data (Supplemental Fig. S3D). Although we find m6A overlaps across all methods, more m6A sites were detected in the single-cell data owing to their enhanced sensitivity.
We then examined the m6A distribution near the TTS. Aggregating all clusters, we observed a m6A distribution near the TTS like bulk m6A sequencing data (Supplemental Fig. S5A). Although most m6A distribution patterns aligned with bulk m6A detection, cluster-specific variations were evident (Fig. 3F; Supplemental Fig. S5B). For instance, cluster 1 exhibited a common m6A distribution pattern with heightened enrichment post-TTS and a minor peak within 200 bp before the TTS, consistent with bulk m6A distribution (Fig. 3F; Supplemental Fig. S5A,B). In contrast, clusters like 22 and 5 lacked the pre-TTS enrichment peak, whereas cluster 19 showed the m6A peak at the TTS and cluster 18 upstream of the TTS (Fig. 3F; Supplemental Fig. S5B). These distinct m6A distribution patterns, often concealed in bulk data, may indicate regulatory or functional differences in m6A within specific cell types, emphasizing the benefits of single-cell resolution for modification detection.
Identification of five major cell types in hippocampal m6A single-cell data
Using automatic annotation (see Methods) and validating with published work (Ximerakis et al. 2019), we identified cell type clusters based on specific gene markers (Fig. 4A–C; Supplemental Table S10). Clusters 2, 3, 5, 6, 7, 12, and 13 were classified as oligodendrocytes, with cluster 16 as oligodendrocyte precursor cells, confirmed by Sox10 expression and all referred to as the oligodendrocyte cell lineage (OLG) (Ximerakis et al. 2019). Cluster 22 was designated as astrocytes (ASCs) owing to exclusive Gja1 expression (Fig. 4B; Cid et al. 2021). Neuronal lineage cells (NEUs) were found in clusters 11 and 23 expressing Snap25 (Ximerakis et al. 2019). Endothelial cells (ECs) were identified in cluster 26 expressing Esam (Ximerakis et al. 2019). Immune cell lineage (IMC) encompassed clusters 0, 1, 4, 8–10, 14–15, 17–21, 24–25, and 27 expressing Lcp1, subdivided into microglia (Aif1 [also known as Iba1]), Tmem119, and P2ry12 expression; clusters 1, 4, 14, 20) (Jurga et al. 2020), myeloid cells (Cd74 expression; clusters 0, 9–10, 15, 17, 27) (Ximerakis et al. 2019), B cells (Ly6d expression; cluster 21, 25) (Blumberg et al. 1990; Ximerakis et al. 2019), and T cells (Cd3e expression; clusters 8, 18–19, 24) (Blumberg et al. 1990) based on marker expression (Fig. 4A–C; Ximerakis et al. 2019).
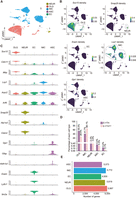
Identification of cell types at the single-cell level in the hippocampus. (A) UMAP with five main cell type populations were annotated and color coded based on cell type identifications. (NEUR) Neuronal cell lineage, (OLG) oligodendrocyte cell lineage, (IMC) immune cell lineage, (ASC) astrocyte cell lineage, (EC) endothelial cell lineage. (B) UMAP with expression levels of cell type–specific marker genes identifying all five major cell populations. Legend color represents RNA density. Circles were added to visualize grouped cell populations. OLGs have high expression of Sox10; NEURs have high expression of Snap25; ECs have high expression of Esam; IMCs have high expression of Lcp1; and ASCs have high expression of Gja1. (C) Violin plot showing the distribution of expression levels of well-known representative cell type–enriched marker genes across five cell types, 27,804 cells in total. (D) Percentage of each cell type. (E) Number of detected genes per cell type.
In total, we analyzed 27,804 cells, categorizing them into five main cell type lineages: NEUR, OLG, IMC, ASC, and EC (Fig. 4A). OLG cells dominated, followed by IMC, NEUR, ASC, and EC (Fig. 4D,E; Supplemental Fig. S6A; Supplemental Table S11). Notably, E-YTHmut exhibited more IMC cells compared with E-YTH, without missing clusters in either group.
Hippocampal m6A cell type characteristics
We investigated cell type–specific m6A characteristics, identifying the following average m6A site counts per cell: 612,705 m6A sites in OLG (5972 cells, 102 sites/cell), 274,170 m6A sites in IMC (4487 cells, 61 sites/cell), 28,112 m6A sites in NEUR (232 cells, 121 sites/cell), 7667 m6A sites in ASC (43 cells, 178 sites/cell), and 595 m6A sites in EC (23 cells, 25 sites/cell) (Fig. 5A,B; Supplemental Table S12). UMAP plots revealed differential m6A site expression for various genes (Fig. 5C).
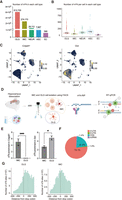
m6A single-cell distribution per mouse hippocampal cell type. (A) m6A counts per cell type. (OLG) oligodendrocyte cell lineage, (IMC) immune cell lineage, (NEUR) neuronal cell lineage, (ASC) astrocyte cell lineage, (EC) endothelial cell lineage. (B) m6A counts per cell for each cell type. Number reflects average. (C) UMAP illustrating the m6A density on RNA transcribed from one gene, per cell. Plots for genes Colgalt1 and Gsn are shown. Legend color represents density of m6A on RNA transcribed. (D) Schematic diagram of approach confirming differential m6A RNA modifications in different cell populations. Relevant cell groups from hippocampi are isolated by FACS sorting, such as IMC versus OLG, followed by m6A RNA immunoprecipitation (m6A-RIP), reverse transcription, and qPCR (RT-qPCR). m6A-RIP enrichment represents relative m6A abundance in different cell populations. (E) m6A transcript enrichment quantifications in IMC and OLG populations following m6A-RIP and RT-qPCR versus input control samples. Transcript m6A enrichments represent m6A abundance in IMC and OLG cells. Both transcripts of Colgalt1 and Gsn were detected in all input control samples. No Colgalt1 transcripts were detected following m6A-RIP in OLG. Unpaired t-test (two-tailed) was used to test the difference between OLG compared with IMC. (***) P ≤ 0.001, (*) P ≤ 0.05. (F) m6A distribution within RNA. Data represent pooled single-cell data. (CDS) coding site, (ncRNA) noncoding RNA. (G) m6A distribution surrounding the stop codon (0 nt) identified by single-cell sequencing for OLG and IMC.
To validate the differential m6A patterns in our single-cell data, we targeted the abundant IMC and OLG cell types. Colgalt1 and Gsn were identified as genes with distinct m6A patterns in IMC and OLG (Fig. 5C). Isolating these cell groups from WT hippocampi, we conducted m6A-RIP followed by RT-qPCR to quantify Colgalt1 and Gsn enrichment (Fig. 5D). Colgalt1 displayed m6A sites in IMC but not OLG, whereas Gsn exhibited higher m6A enrichment in IMC than OLG (Fig. 5E). This m6A-RIP experiment validated our single-cell m6A findings, bolstering the reliability of our conclusions (Fig. 5C–E).
To determine if the m6A differential distribution could be explained by different levels of m6A regulatory enzymes, we also evaluated m6A regulatory enzymes at the single-cell level (Supplemental Fig. S6B). Although we observed a correlation between certain m6A methylases and increased m6A density, we also noted elevated levels of the m6A demethylase Alkbh5 (Supplemental Fig. S6B). This implies that m6A regulation extends beyond the primary regulatory enzymes, suggesting a complexity in m6A regulation that surpasses current understanding, indicating tightly controlled, cell-specific mechanisms at play.
Although our pooled single-cell data confirmed previously reported m6A mRNA distribution patterns (Fig. 5F; Supplemental Fig. S5A; Supplemental Table S13), cell type–level analysis demonstrates specific m6A distribution patterns: OLG, NEUR, ASC, and EC exhibited a peak post-TTS, whereas IMC displayed an additional peak pre-TTS, indicating potential regulatory or functional differences between cell types (Fig. 5G; Supplemental Fig. S6C).
Heterogeneous and homogeneous m6A sites in single cells
We investigated m6A variations across clusters and cell types in our single-cell data by analyzing m/k ratios per gene per cell. Despite heterogenous m6A sites, many homogenous m6A sites (m/k = 1) were identified across all clusters and cell types (Fig. 6A; Supplemental Fig. S7). Comparing m/k distributions with E-YTHmut controls supports m6A homogeneity (Fig. 6A; Supplemental Fig. S7). To exclude the possibility that the observed homogeneous m6A sites were not mouse-specific SNPs, we isolated m/k = 1 m6A sites per cell type, retaining only those that are present in other cells with a lower m/k ratio (m/k < 0.9) (Supplemental Table S14). To authenticate m6A sites and their stoichiometry, we generated Mettl3 KO (Mettl3−/−:Emx1-Cre) mice to reduce m6A levels in hippocampi (Fig. 6A; Supplemental Fig. S8A–C, Supplemental Table S15). We next conducted m6A single-cell sequencing, as previously performed for WT mice (Supplemental Fig. S8D–F). In our WT m6A single-cell data, we could identify CAMK2A neurons in E-YTHmut samples but not in E-YTH samples (Fig. 3C; Supplemental Fig. S4C–E). However, in our METTL3-depleted m6A single-cell data, we could identify CAMK2A neurons in both E-YTHmut and in E-YTH samples (Supplemental Fig. S8G). The rescue of CAMK2A neurons in E-YTH samples following Mettl3 depletion indicates that CAMK2A neurons are lost in E-YTH WT samples owing to the binding of the E-YTH construct to actual m6A sites, further corroborating our approach and CAMK2A findings. Furthermore, a considerable reduction in m6A sites was detected in Mettl3 KO mice compared with those identified in WT hippocampi, thereby validating the authenticity of WT m6A sites (Fig. 6A; Supplemental Fig. S8H, Supplemental Table S15). This was further supported by the m/k ratio analysis, in which m6A sites with m/k = 1 in WT exhibited either no or reduced methylation (m/k = 0 or 0 < m/k < 1, respectively) in METTL3-depleted samples, corroborating the m6A homogeneity observed in single cells (Fig. 6A; Supplemental Fig. S8I).
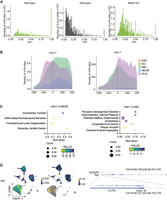
Heterogeneous and homogeneous m6A sites in single cells. (A) m6A site counts over mutation per read (m/k) ratio (left) and C-to-U edit E-YTHmut background over mutation per read (m/k) ratio (middle) for WT mice. m6A site counts over m/k ratio for Mettl3 knockout (KO) mice (Mettl3−/−:Emx1-Cre; right). Minimum threshold: 5%. (B) Normalized m6A scaled density 500 nt 5′ and 500 nt 3′ from stop codon (0 nt) for ASC, EC, IMC, NEUR, and OLG. (Left) m6A sites with m/k = 1 (homogenous m6A). (Right) m6A sites with m/k < 1 (heterogenous m6A). (ASC) astrocyte cell lineage, (EC) endothelial cell lineage, (IMC) immune cell lineage, (NEUR) neuronal cell lineage, (OLG) oligodendrocyte cell lineage. (C) Disease gene enrichment analyses (GO). GO terms with an adjusted Q-value < 0.05 and P-value < 0.05 are shown for NEUR and ASC. (D) Localization of m6A for gene. (Left) UMAP plot of single-cell expression for one gene. Legend color represents transcript density. (Middle) UMAP density plot of m6A on RNA transcribed from one gene, per cell. Legend color represents m6A density on RNA transcribed from one gene. (Right) IGV RefSeq gene annotation with editing sites representing adjacent m6A sites is shown. Last exon with 3′ UTR region is illustrated with higher magnitude.
Examining the distribution of conserved homogenous (m/k = 1) m6A sites, we observed distinct patterns compared with more heterogeneous (m/k < 1) sites (Fig. 6B). In OLG, homogenous m6A sites were enriched pre-TTS, with an additional peak at the TTS, whereas heterogeneous sites were mainly enriched shortly post-TTS. In IMC, heterogeneous m6A sites showed enrichment post-TTS, with a single major peak, whereas homogenous sites were distributed more evenly along the 3′ UTR, also occurring post-TTS (Fig. 6B). These differential distribution patterns suggest cell type–specific m6A regulation and potential functional implications.
To gain insights into the transcripts and pathways potentially regulated by homogeneous m6A sites, we conducted Gene Ontology (GO) analysis (Fig. 6C; Supplemental Fig. S9A). Our analysis revealed lineage-specific pathways, such as axon-related pathways for NEUR, oligodendrocyte-specific pathways for OLG, and postsynaptic pathways for ASC (Supplemental Fig. S9A). Additionally, disease pathways such as amyloidosis, dementia and degeneration for NEUR and hydrocephalus and amyloid plaque for ASC were identified (Fig. 6C; Supplemental Fig. S9B).
Focusing on genes with m6A sites at m/k = 1 in at least one cell, we observed m6A occurrences on transcripts encoding the m6A demethylase ALKBH5, suggesting a regulatory feedback mechanism (Supplemental Table S14). Moreover, m6A was found on Fos and Jun in OLG and IMC, with instances of multiple m6A sites on a single transcript, such as Stat1 (Supplemental Table S14). We also detected m6A sites on Smarcc2 (Supplemental Fig. S10), a regulator of chromatin structure implicated in neural stem cell proliferation and neuronal differentiation (Nguyen et al. 2018), hinting at a potential regulatory role of m6A via SMARCC2. Furthermore, several genes associated with Alzheimer's disease (AD) exhibited multiple cells with an m/k = 1 ratio, including App, Apoe, Aplp1, Ctsb, and Itm2b (Fig. 6D; Supplemental Fig. S10; Supplemental Table S14; Turner et al. 2003; Priller et al. 2006; Hook et al. 2023), suggesting potential m6A regulation. Additionally, other m6A transcripts with m/k = 1 are linked to diseases, such as Mecp2 with Rett syndrome (Guy et al. 2007), Syt11 with schizophrenia and Parkinson's disease (Inoue et al. 2007; Lill et al. 2012), Lamp1 as a Lassa virus receptor (Enriquez et al. 2022), and Brd2 with epilepsy (Supplemental Fig. S10; Supplemental Table S14; Pal et al. 2003), highlighting a broader impact of m6A modifications on disease-associated genes.
In summary, our single-cell data from the hippocampus unveiled various transcripts, cell clusters, and cell types likely regulated by m6A modifications. This comprehensive exploration of m6A dynamics within individual cells offers novel insights into the molecular underpinnings of hippocampal physiology, setting the stage for future investigations into the dynamic landscape of m6A RNA methylation in the brain.
Discussion
In this study, we optimized the DART-seq system (Meyer 2019) to identify m6A sites in single-cell data from the mouse hippocampus. Initially, we generated E-Yth and Yth-E constructs and their controls, testing their efficacy in detecting m6A sites in RNA from HEK293T cells. Our findings align with previous m6A studies, validated through functional investigations (Meyer 2019). The E-Yth construct showed superior performance, improving the detection of E-Yth-transfected cells and correlating with APOBEC1-YTH presence. Using the E-Yth construct, we achieved a distinct m6A distribution pattern by excluding cells with low E-YTH expression, reducing background noise. Subsequently, we packaged the E-Yth construct and its E-Ythmut control into AAVs and injected them into mouse hippocampi, isolating transduced cells for bulk and single-cell sequencing analysis to identify m6A sites.
We identified 923,249 m6A sites within transcripts at single-cell resolution in the mouse hippocampus. CAMK2A-expressing cells were absent after APOBEC1-YTH treatment, suggesting a potential interference with essential m6A transcripts in these neurons. This interference may be owing to C-to-U near m6A sites, which are mutations, introduced by APOBEC1-YTH, potentially affecting m6A-containing transcripts or competing with m6A reader proteins like YTHDF2. Although APOBEC1-YTH have been previously reported not to interfere with transcriptional changes or cell viability (Meyer 2019), occasionally, such mutations could result in changes, which might explain the E-YTH-specific absence of CAMK2A neurons. The absence of CAMK2A neurons in E-YTH data but not in E-YTHmut controls underscores the importance of m6A in these cells, which has been observed previously (Engel et al. 2018; Zhang et al. 2018; Xu et al. 2022; Yasuda et al. 2022).
Our study also reveals homogeneity of m6A in individual cells, contrasting with previous reports of heterogeneity (Tegowski et al. 2022). Differential distribution patterns near TTS between homogenous and heterogeneous m6A sites suggest regulatory and functional differences. Transcripts with high m/k ratios, like App and Smarcc2, may be particularly sensitive to m6A regulation. We discovered many such m6A transcripts in specific cell types associated with brain diseases, indicating potential implications for conditions like AD. Because an earlier study highlighted a decline in m6A levels with age and in AD (Castro-Hernández et al. 2023), our findings suggest that specific m6A transcripts like App, Apoe, Aplp1, Ctsb, and Itm2b, alongside m6A-modulated transcripts in CAMK2A neurons of the hippocampus, not only exhibit sensitivity to m6A regulation but are also likely implicated in the aging process and the pathology of AD. By uncovering novel insights into the hippocampal m6A transcriptome at the single-cell level, our work paves the way for future therapeutic targets and studies on m6A dynamics in brain health and disease.
Methods
Cell lines
HEK293T cells were purchased from the Beijing Xiehe Cell Bank and cultured at 37°C with 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM, Gibco C11965500BT) containing 10% FBS (VisTech SE100-B) and 1% penicillin/streptomycin. Cells were passaged for fewer than 20 times and have been regularly tested for mycoplasma.
Mouse strains
Animals were maintained and handled following the guidelines of the Chinese Institute for Brain Research (CIBR). All experimental methods were approved and adhered to the regulations of the Welfare and Ethics Review Committee for Laboratory Animals. WT C57BL/6J mice were made available through CIBR's animal facility. Mettl3-floxed (Mettl3f/f) mice from GemPharmatech were bred with Emx1-Cre mice (JAX stock 005628) (Gorski et al. 2002) to produce homozygous Mettl3 KO (Mettl3−/−:Emx1-Cre) mice. The genotyping primers for Mettl3f/f facilitate detection of the 5′ flox region (forward primer: ATAACCCTGGCTGTCCCG; reversed primer: ATAACCCTGGCTGTCCCG) and the 3′ flox region (forward primer: CCTTTGGAATGGCTACTGC; reversed primer: ATCAGAAAGCCCATCCTCA). Adult WT and homozygous Mettl3 KO adult mice, aged 8–12 weeks, were utilized for single-cell RNA sequencing. The mice were housed in a 12:12 light–dark cycle, maintained under controlled climate conditions, provided with enrichment environments, and had ad libitum access to sterile food and water.
Vector cloning and AAVs
An Adeno-associated plasmid pAAV-Cag-Egfp-Wpre-Sv40 (gift from Minmin Luo) was used as the backbone to generate the viral expression constructs pAAV-Cag-Apobec1-Yth-Egfp (Yth-E, Addgene 209322), pAAV-Cag-Apobec1-Ythmut-Egfp (Ythmut-E, Addgene 209323), pAAV-Cag-Apobec1-Egfp (Addgene 209324), pAAV-Cag-Egfp-Apobec1-Yth (E-Yth, Addgene 209319), pAAV-Cag-Egfp-Apobec1-Ythmut (E-Ythmut, Addgene 209320), and pAAV-Cag-Egfp-Apobec1 (Addgene 209321). To clone these vectors, the cassette pCMV-Apobec1-Yth (Addgene 131636) and pCMV-Apobec1-Ythmut (Addgene 131637) were inserted into the backbone of pAAV-Cag-gfp-Wpre-Sv40 using in-fusion cloning. pAAV-Camk2a-mCherry was also used (gift from Fei Zhao). The plasmid to be packaged was cotransfected into HEK293T cells with a rep/cap-containing plasmid pUCmini-iCAP-PHP.eB (Addgene 103005) and the helper plasmid pAdDeltaF6 (Addgene 112867), in the presence of polyethyenimine. After 72 h, the AAV virus was harvested, purified by chloroform, titrated, and quantified by qPCR (Negrini et al. 2020). AAV stereotaxic injections were performed targeting the hippocampus of adult mice, with X/P 1.94 mm, M/L 1.5 mm from the bregma point, and 2 mm depth for C57BL/6J mice. Because of the reduced brain volume observed in Mettl3 KO mice, a depth of 0.7 mm was applied in these mice, followed by an additional injection into the retro-bulbar sinus.
Immunostaining and microscopy
HEK293T cells were seeded on a 35-mm-diameter dish (WPI's FluoroDish FD35-100); 5 µg plasmids were transfected; and images were taken after 24 h. Cells were fixed with 4% paraformaldehyde for 10 min and washed with PBS followed by 15 min permeabilization with 0.1% Triton X-100. Following PBS washes, samples were blocked for 1 h at RT in PBS with 1% BSA and incubated with HA antibody (Alexa Fluor 555, Invitrogen 26183-A555). After PBS washes and 0.1% DAPI staining, fluorescent images were captured using the Zeiss inverted confocal microscope (ZEN blue software; DAPI: excitation 365, BS FT 395, emission BP 445/50; GFP: excitation BP 470/40, BS FT 495, emission BP 525/50; CY3: excitation BP 545/25, BS FT 570, emission BP 605/70).
Figure 2B was imaged with the Olympus VS120 virtual slide microscope (OlyVIA software; DAPI: excitation 365/10 nm, emission 440/40 nm; GFP: excitation 472/30 nm, emission 520/35 nm). Supplemental Figure S4 was captured using the Olympus VS200 virtual slide microscope (OlyVIA software; Cy3: excitation 555/20 nm, emission 595/33 nm; GFP: excitation 480/30 nm; emission 519/26 nm; DAPI: excitation 395/25 nm, emission 434/32 nm).
Bulk m6A sequencing in HEK293T cells
HEK293T cells were cultured and processed independently of each other to generate independent biological replicates. Three independent replicates were processed for Yth-E- and Ythmut-E-expressing cells. Twenty-four hours after plasmid transfections, cells were rinsed with DPBS and digested using 0.5% trypsin-EDTA. Cells were pelleted, washed with DPBS, and filtered through a 40 µm cell filter prior to FACS analysis. Cells were loaded onto a custom FACS ARIA III flow sorter (BD Biosciences) equipped with a 100 µm nozzle. Particles smaller than cells (dots) were eliminated using the forward-scatter (FSC-PMT-A) versus side-scatter (SSC-A). Cell-sized particles were gated (box). Plots of height versus width in the forward-scatter and side-scatter channels were used to exclude aggregates of two or more cells. Live cells were selected by gating the non-DAPI signal (405 nm laser, violet DAPI). GFP cells from C57BL/6J mice were isolated by green excitation light (488 nm laser, green FITC). Given the reduced hippocampal volume in Mettl3 KO mice, no gating was applied to obtain enough Mettl3 KO hippocampal cells. Total RNA was isolated with the micro total RNA isolation kit (Invitrogen AM1931) according to the manufacturer's instructions. After treatment with DNase I (Tiangen RT411) for 15 min at room temperature, sequencing libraries were generated from 1–10 ng of total RNA from each replicate using the single-cell full-length mRNA kit (Vazyme N712) and TruePrep DNA library prep kit V2 (Vazyme TD502), according to the manufacturer's instructions. Quality control was performed using the fragment analyzer systems capillary arrays (AATI, FA12) and quantified with a Qubit 1 × dsDNA HS kit (Invitrogen Q33231); 150 bp paired-end sequencing was performed on a NovaSeq 6000 (Illlumina) using a S4 flow cell.
Identification of m6A sites in bulk RNA sequencing data
Low-quality bases (<Q20) and adaptor sequences were trimmed using Trimmomatic (0.39) (Bolger et al. 2014), and reads with fewer than 36 nucleotides were subsequently discarded. The cleaned reads were aligned to the mm39 reference genome using BWA-MEM (0.7.17) (Li and Durbin 2009). Duplicate reads were identified using the MarkDuplicates tool from Picard (https://broadinstitute.github.io/picard/). Subsequently, the CLIP tool kit (CTK) was employed to collapse PCR duplicates and to identify C-to-U editing events, following default parameters (Shah et al. 2017).
To identify C-to-U and m6A sites, we used an approach as described by Meyer (2019), using the following filters: (1) Only C-to-T mutations with a false-discovery rate (FDR) of less than 0.01 were kept for any downstream analyses; (2) only C-to-T mutations with two or more editing events (m ≥ 2) and a coverage of at least 10 (k ≥ 10) were considered for downstream analyses; (3) C-to-T mutations with a ratio of m/k (number of reads with a C-to-T mutation/total reads per site) >5% were kept for downstream analyses; (4) C-to-T mutations sites that were found in the single-nucleotide polymorphism (SNP) databases Mouse Genome Project (mgp_REL2021_snps) and Genome Reference Consortium Mouse Build 39 (GCA_000001635.9) were discarded; and (5) only C-to-T mutation sites that were found in at least two out of three replicates were considered for downstream analyses.
To identify m6A sites in the C-to-T-converted APOBEC1-YTH sequencing data, C-to-T conversion sites that were also present in the WT and APOBEC1 overexpression background control were removed. Only C-to-T conversion sites in the APOBEC1-YTH sequencing data that occurred at least 1.5 times more frequently than in the APOBEC1-YTHmut negative control sequencing data were retained. By implementing these criteria, false positives were eliminated, resulting in a set of stringent C-to-T conversion sites in the APOBEC1-YTH data sets. These remaining C-to-T conversion sites in the APOBEC1-YTH data were identified as m6A sites.
Analyses and plotting
Biorender (https://www.biorender.com/) was used for some illustrations, as well as the Integrative Genomics Viewer (IGV) (Robinson et al. 2011) and GraphPad Prism (version 9.5.1; RRID:SCR_002798). All experiments were carried out with three technical and biological replicates, indicated by n. Statistical analyses and plots were performed using R (version 3.6.2) (R Core Team 2024). The VennDiagram package was used for Venn diagrams, Seurat (Stuart et al. 2019) for UMAP plots, and ggplot2(version 3.3.5) (Wickham 2016) for the rest of the plots. metaPlotR (Olarerin-George and Jaffrey 2017) was used to generate m6A metagene plots, such as histograms along simplified transcript models, of the C-to-U conversion (Olarerin-George and Jaffrey 2017). When multiple transcript isoforms could potentially contain the C-to-U site, the longest isoform was chosen.
Mass spectrometry
Analysis of global levels of A and m6A was performed on a TSQ Altis triple quadrupole mass spectrometer (Thermo Fisher Scientific) coupled to a Vanquish flex UHPLC system (Thermo Fisher Scientific) fitted with an acquity UPLC HSS T3 column (2.1 × 100 mm, 1.8 μm particle size, Waters). The mobile phase consisted of 0.5% aqueous formic acid (solvent A) and 0.5% formic acid in acetonitrile (solvent B) at a flow rate of 300 µL/min. Calibration curves were generated using serial dilutions of synthetic standards for adenosine (A; Sigma-Aldrich) and N6-methyl-2′-adenosine (m6A; Sellechchem). The mass spectrometer was set in a positive ion mode and operated in selective reaction monitoring. The precursor ions of A (m/z 268.1) and m6A (m/z 282.1) were fragmented, and the product ions of A (m/z 136.1) and m6A (m/z 150.1) were monitored. The EIC of the base fragment was used for quantification. Accurate mass of the corresponding base fragment was extracted using the XCalibur qual browser and XCalibur quan browser software (Thermo Fisher Scientific) and used for quantification. m6A presentage was calculated according to the following equation: m6A(%) = 100 × m6A/[A]. Differences in m6A percentage abundance were considered significant when P ≤ 0.05.
m6A RNA immunoprecipitation
Total RNA was extracted from the hippocampus of adult mice (C57BL/6J) using TRIzol (Thermo Fisher Scientific 15596018) reagent. After removing genomic DNA, a RNeasy mini kit (Qiagen 74106) was used for RNA purification, resulting in ∼20 μg of total RNA per mouse. The integrity of RNA was assessed using the fragment analyzer system capillary arrays (AATI F12), whereas the concentration and purity were determined using a spectrophotometer (Thermo NanoDrop one). For RNA fragmentation, the samples were incubated for 4 min at 94°C in a fragmentation buffer (containing 10 mM ZnCl2, 10 mM Tris-HCl at pH 7), followed by standard isopropanol precipitation. For m6A-RIP, an existing protocol was adjusted (Dominissini et al. 2012). Protein A Dynabeads (Invitrogen 10001D) were washed three times with IP buffer (containing 150 mM NaCl, 0.1% NP-40, 10 mM Tris-HCl at pH7.4 and 6 µg/µL BSA) and incubated with rotation in IP buffer for 2 h at 4°C. Each RNA sample was divided to obtain an input control sample (10%), and the 90% was incubated with an anti-m6A antibody (Synaptic Systems 202011, 2.5 μg). The RNA–antibody mixture was incubated for 2 h at 4°C with rotation. The magnetic beads were then conjugated with the antibody–RNA solution, which selectively captures RNA fragments containing m6A modifications. After three washes with IP buffer, the antibody-captured RNA was eluted for 1 h at 4°C with shaking rotation using elution buffer (containing IP buffer and 6.6 mM m6A) and was then concentrated by isopropanol precipitation. To generate RNA-seq libraries for input control and m6A-RIP pulldown samples, the RNA was processed using the SMARTer stranded total RNA-seq kit v2 (Takara 634411). The fragment length of the libraries was verified using the fragment analyzer 12 (AATI). Paired-end 150 bp reads were sequenced with the NovaSeq 6000 platform (Illumina).
m6A-RIP sequencing data analysis
rRNAs were removed using the mouse rRNA reference (GCF_000001635.27_GRCm39_rna_from_genomic.fna) from NCBI. Adapters were eliminated with cutadapt (version 2.8) (Martin 2011). The first 10 5′ nucleotides were eliminated from the ends owing to the presence of potentially low-quality nucleotides. PCR duplicates were removed from the aligned data sets. Mapping and alignment was done by HISAT2 (version 2.2.1) (Kim et al. 2019), followed by peak calling using MACS2 (version 2.2.6) (Gaspar 2018).
Hippocampus dissociation
Two weeks after AAV brain stereotaxic injections, mice were anesthetized and then perfused with Dulbecco's phosphate buffer saline (MacGene CC010). Hippocampi were extracted in cold DPBS solution containing calcium, magnesium, and glucose and were dissociated into single cells using the adult brain dissociation kit (Miltenyi Biotex 130-107-677), under the following conditions: (1) ∼25 mg of adult hippocampi was used as starting material per sample; (2) the MACS program >100 mg:37°C_ABDK_01 was chosen; (3) debris was removed following the manufacturer's manual; (4) 10 mL PB buffer was used to suspend cells with cold 1× red blood cell removal solution; and (5) PB buffer was used for FACS sorting.
Single-cell m6A sequencing
Eight hippocampi from adult mice were pooled for each sample to ensure an adequate number of cells. E-YTH and E-YTHmut EGFP-positive cells were isolated through FACS sorting and converted into cDNA libraries. Thirty thousand cells per sample were loaded into a Rhapsody cartridge (BD Biosciences 633733) and processed following the manufacturer's instructions. Single-cell RNA sequencing libraries were generated using the Rhapsody WTA kit (BD Biosciences 633801). The Rhapsody platform by BD allows cells to settle naturally on a chip for gentle capture without disruption. After pooling the libraries, sequencing was performed using NovaSeq 6000 (Illumina).
Single-cell sequencing gene expression analysis
The Rhapsody docker image (BD Biosciences) was used to perform barcode processing and single-cell gene-UMI counting, following manufacturer's instructions (BD Rhapsody sequence analysis setup). A digital expression matrix was obtained for each experiment with default parameters and was mapped to the mouse reference genome mm39. Matrices containing RSEC-corrected molecules per bioproduct per cell numbers were loaded into Seurat (version 3.1.5) (Butler et al. 2018). Low-quality cells with fewer than 500 and more than 6000 detected genes and cells with a high mitochondrial content (>10%), indicative of poor cell quality, were excluded. In Mettl3 KO mice, given that Emx1-Cre expression is not ubiquitous (Gorski et al. 2002), it is possible that METTL3 may persist in a subset of Mettl3 (Mettl3−/−:Emx1-Cre) KO cells. To ensure the removal of data from cells retaining METTL3 expression, any cells with Mettl3 transcript counts of one or more were filtered out from all subsequent analyses in the Mettl3 KO samples. The data from each individual single-cell sample were log-normalized. The top 2000 more variable genes within each sample were identified using the FindVariableFeatures function in Seurat and were used as integration anchors for the integration of E-YTH and E-YTHmut samples. This was done using the FindIntegrationAnchors and IntegrateData functions in Seurat. Integrated data were subsequently scaled, and principal component analysis (PCA) was performed to reduce the dimensionality of the data. The top 30 and 21 principal components were used to identify cell populations for WT and Mettl3 KO mice, respectively, using the FindNeighbors and FindClusters functions. The resolution parameter in FindClusters function was set to 0.8. UMAP was subsequently applied to visualize the clustered cells in a two-dimensional space.
To determine cluster-specific marker genes, the FindConservedMarkers function in Seurat was employed, comparing each cluster against all other clusters within the integrated data set. For each run of this function, only genes detected in at least 10% of the cells in either of the two populations were considered for analysis. Genes with a log-fold change greater than 0.3 were considered as cluster-specific markers. To help annotate the identified clusters, we followed two automatic cell type annotation approaches. First, we used the scMCA function in the scMCA package (0.2.0) (Sun et al. 2019). This function assigns mouse cell types to cell clusters based on expression profiles. Second, we used SciBet (1.0) cell type classifier with the Tabula Muris brain nonmyeloid model (Li et al. 2020).
Seurat was used for UMAP plots (Hao et al. 2021) and ggplot2 for the rest of the plots (Wickham 2016). UMAP plots were generated with Nebulosa, using the plot_density function (Alquicira-Hernandez and Powell 2021).
Differential gene expression analysis
Gene-level read counts were obtained from the aligned files using featureCounts (2.0.0) (Liao et al. 2014). Differential expression analysis between E-YTH and E-YTHmut cells was performed using DESeq2 (Love et al. 2014). After filtering out genes with low expression levels (number of reads <10) and adjusted P-value > 0.1, volcano plots were plotted. Adjusted P-value < 0.05 and a Log2FC > 1 and Log2FC < –1 are considered significant. Genes were identified as differentially expressed between the two conditions with a fold change of Log2FC > 1 and Log2FC < –1 (adjusted P-value < 0.05).
Western blot
Hippocampi were homogenized in 500 µL of RIPA medium lysis buffer (Beyotime P0013E-2) in the presence of protease inhibitor cocktail (Roche 11836170001). Samples were run on 10% PAGE gels (Vazyme E303-01) and transferred onto activated PVDF transfer membranes (Immobilon IPVH00010). Membranes were washed with TBST (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, 0.05% Tween 20), followed by blocking in 5% nonfat dried milk and incubation with antibodies such as CAMK2A (Thermo Fisher Scientific MA1-048) and GAPDH (Abcam ab9485). After TBST washes, membranes were incubated with HRP-conjugated secondary antibodies (Abcam Ab205718 & Ab205719) and washed, and proteins were visualized with a HRP substrate peroxide solution and luminol reagent (Immobilon WBKL5S0500). iBright 1500 (Invitrogen) was used to quantify protein presence (background corrected volume [Local Bg. Corr. Vol.]) versus GAPDH.
Identification of m6A sites in single-cell sequencing data
The software Bullseye was used to identify m6A sites in single-cell sequencing data (Tegowski et al. 2022). We applied the same conditions for single-cell analysis as we did for bulk data, except for the following.
We excluded any C-to-U editing sites identified in the APOBEC1 and WT bulk sequencing analysis from the E-YTH single-cell data. Only sites showing a 1.5-fold increase over bulk E-YTHmut controls were retained. Given that C-to-U background edits are not evenly distributed across all clusters, it was crucial to eliminate the E-YTHmut cluster-specific background. We computed the average C-to-U editing events in E-YTHmut per cluster. Although the ideal scenario would involve removing background at the single-cell level, this is unfeasible owing to the uniqueness of each cell. Consequently, we calculated the average C-to-U editing events in E-YTHmut per cluster. Subsequently, for each cell in our E-YTH single-cell data, we subtracted the corresponding cluster-specific background average. This approach was vital for accurately capturing cluster-specific m6A characteristics.
Comparison of bulk, single-cell, and m6A-RIP data sets
The m6A overlap among single-cell RNA-seq, bulk RNA-seq, and m6A-RIP was determined by identifying the intersection of genes in which a m6A position (or region for m6A-RIP) was identified.
Confirmation of differential m6A methylation
Four WT mouse hippocampi were isolated and dissociated into single cells. OLG and IMC cells were isolated by FACS sorting using the oligodendrocyte marker O1 monoclonal antibody (O1), eFluor 660 (eBioscience 50-6506-80), the CX3CR1 monoclonal antibody (2A9-1), and Alexa Fluor 488 (eBioscience 53-6099-42), respectively. Following RNA isolation, m6A-RIP, and reverse transcription, qPCR was performed with the following primers: Colgalt1 (F:AAGAACTCAGATGTGCTCCAG; R:CTATAGTCCCAGGCAAGCAC) and Gsn (F:CATCACAGTCGTTAGGCAGG; R:TGATGGCTTTGGTCCTTACTC). m6A-RIP versus matching input control samples were calculated.
Identification of homogenous m6A sites in single-cell sequencing data
To identify homogenous m6A sites, the m/k ratio was first calculated, as described above for bulk m6A sites. To identify truly homogenous m6A sites (m/k = 1) in the single-cell data, we excluded the possibility that any homogenous (m/k = 1) sites might still represent mouse-specific SNPs. Thus, we identified all m/k = 1 m6A sites per cell for all cells and then only kept those m/k = 1 m6A sites that occurred in at least five other cells with a lower m/k ratio (m/k < 0.9) and a minimum read overage of 10 per cell.
GO analysis
Prior to GO analysis, conserved homogenous m6A sites were identified. Such sites for each cell types consist of m/k = 1 sites identified in our merged single-cell RNA sequencing data that occur with an m/k < 0.9 ratio in at least five cells of other cell types. For each cell type, GO biological process, molecular function, and cellular component enrichment analyses were carried out on the set of genes in which these conserved homogenous sites occur, using cluster Profiler (version 3.14.3) (Yu et al. 2012). Terms with an adjusted Q-value < 0.05 and P-value < 0.05 were considered statistically significant. The top five GO terms were plotted, in addition to five selected terms that are statistically significant. Additionally, disease enrichment analysis was conducted on the human orthologue genes corresponding to the genes with conserved homogenous sites, using DOSE (version 3.12.0) and DisGeNET (Yu et al. 2015; Piñero et al. 2017). Terms with an adjusted Q-value < 0.05 and P-value < 0.05 were considered statistically significant and plotted. Prism (V9.5.1) was used to plot any GO terms. The Rich factor is the ratio of gene numbers with m/k = 1 m6A in a pathway term to all gene numbers annotated in this pathway term.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE240863. All plasmids generated for this study can be obtained through Addgene (Addgene 209319 to 209324; https://www.addgene.org/Magdalena_Koziol). Codes used for this work are available through GitHub (https://github.com/KoziolLaboratory/sc-m6a-hippocampus) and as Supplemental Code. Single-cell RNA and m6A density UMAP visualizations can be accessed via our interactive website (https://scm6a.cibr.ac.cn/). This website is accompanied by a navigation guide and summary. Within the scm6A-seq menu, all Gene_Density and m6A_Density UMAP plots generated can be accessed. Gene names can be searched (the first letter needs to be capitalized, followed by enter key) within the Gene_Density and m6A_Density sections. The corresponding Gene_Density or m6A_Density UMAP plots will be displayed for the gene of interest and can be interpreted through legends provided.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank all the members of the Koziol laboratory, Susu Qu, Miao Jing, Fei Zhao, and Juan Huang for valuable discussions and critical comments. We thank the Genomics Center, Vector Core, Optical Imaging, HPC Facility, the Laboratory Animal Resource, and Mass Spectrometry Center Facilities at the Chinese Institute for Brain Research (CIBR) for their generous support. We specifically thank Tong Guo and Wenlong Li from our Laboratory Animal Resource Facility who supported us during the review process of this manuscript. We appreciate Xiangyu Zhou for creating the digital single-cell expression matrix and Xinshuang Zhang for contributing to the bulk RNA sequencing library. Dongli Fan drew and provided artistic images that accompany our manuscript and interactive UMAP website. Jiayue Li helped with the illustration of Figure 5D. We thank Minmin Luo's laboratory for their pAAV-CAG-EGFP-WPRE-SV40 plasmid. We thank Yuyu Sheng from the BD Biosciences company for technical support. We also thank the China Infrastructure of Cell Line Resource for providing us with the HEK293T cell line. This project was supported by CIBR’s core grant, the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2019-I2M-5-015), and the Beijing Natural Science Foundation (IS23091). M.T.-A. contribution toward this work was funded by the Koziol laboratory. Y.P. and E.L. are supported by the Beijing Postdoctoral Research Foundation fellowship (2020-YJ-002 and 2020-YJ-004, respectively) and by the International Postdoctoral Exchange Fellowship Program. We thank the CIBR Institute community for their generous support.
Author contributions: S.F. designed and performed experiments, developed ideas, analyzed the data, supervised all experiments, assembled all the data, and wrote the paper. M.T.-A. performed the bioinformatic analyses, critically evaluated data, and reviewed the paper. Y.Z. helped with all animal experiments. Y.P. performed all UHPLC-MS/MS experiments and analyses. M.D. assisted with bioinformatic data processing and analyses. E.L. contributed through discussions and ideas and reviewed the paper. H.Z. developed the single-cell m6A website. H.Z. was supported by L.Z. L.X. supported experimental and mouse work. M.J.K. conceived the study and designed experiments, analyzed the data, wrote the paper, and supervised all research.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278424.123.
-
Freely available online through the Genome Research Open Access option.
- Received August 21, 2023.
- Accepted June 11, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution 4.0 International), as described at http://creativecommons.org/licenses/by/4.0/.








