
Neuron-specific chromatin disruption at CpG islands and aging-related regions in Kabuki syndrome mice
- Leandros Boukas1,2,3,8,
- Teresa Romeo Luperchio2,8,
- Afrooz Razi2,
- Kasper D. Hansen2,3,4 and
- Hans T. Bjornsson2,5,6,7
- 1Department of Pediatrics, Children's National Hospital, Washington, DC 20010, USA;
- 2Department of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA;
- 3Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland 21205, USA;
- 4Department of Biomedical Engineering, Johns Hopkins University School of Medicine, Baltimore, Maryland 21218, USA;
- 5Department of Pediatrics, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA;
- 6Faculty of Medicine, University of Iceland, 101 Reykjavík, Iceland;
- 7Landspitali University Hospital, 101 Reykjavík, Iceland
-
↵8 These authors contributed equally to this work.
Abstract
Many Mendelian developmental disorders caused by coding variants in epigenetic regulators have now been discovered. Epigenetic regulators are broadly expressed, and each of these disorders typically shows phenotypic manifestations from many different organ systems. An open question is whether the chromatin disruption—the root of the pathogenesis—is similar in the different disease-relevant cell types. This is possible in principle, because all these cell types are subject to effects from the same causative gene, which has the same kind of function (e.g., methylates histones) and is disrupted by the same germline variant. We focus on mouse models for Kabuki syndrome types 1 and 2 and find that the chromatin accessibility changes in neurons are mostly distinct from changes in B or T cells. This is not because the neuronal accessibility changes occur at regulatory elements that are only active in neurons. Neurons, but not B or T cells, show preferential chromatin disruption at CpG islands and at regulatory elements linked to aging. A sensitive analysis reveals that regulatory elements disrupted in B/T cells do show chromatin accessibility changes in neurons, but these are very subtle and of uncertain functional significance. Finally, we are able to identify a small set of regulatory elements disrupted in all three cell types. Our findings reveal the cellular-context-specific effect of variants in epigenetic regulators and suggest that blood-derived episignatures, although useful diagnostically, may not be well suited for understanding the mechanistic basis of neurodevelopment in Mendelian disorders of the epigenetic machinery.
The deposition and maintenance of epigenetic marks are fundamental processes underlying development and homeostasis (Jaenisch and Bird 2003; Feinberg 2007; Meissner 2010; Allis and Jenuwein 2016). Reflecting this, the past decade has seen the emergence and rapid expansion of a group of developmental Mendelian disorders caused by disruptive de novo coding variants in genes encoding epigenetic regulators (Bjornsson 2015; Gabriele et al. 2018; Fahrner and Bjornsson 2019; Ciptasari and van Bokhoven 2020). A characteristic of these disorders is the large number of affected organ systems, which exceeds what is seen in other Mendelian disorders (Bjornsson 2015). This is consistent with the very broad expression of human epigenetic regulators across different tissues (Boukas et al. 2019), and raises the question of whether the abnormalities in the different organs are driven by the same underlying chromatin disruption in the corresponding cell types. In other words, are the chromatin effects of variants in epigenetic regulators shared between different cell types or are they primarily cell type specific?
There are two major reasons for why this question is important. First, from a basic biology standpoint, the answer would provide insights into whether the same variant, in the same epigenetic regulator, causes the same (or a highly similar) chromatin disruption in different cellular contexts. For example, is it the same genomic locations that show changes in chromatin accessibility? The degree of such similarity would serve as an indicator of how much the intra- and extracellular environments dictate the specific effect that epigenetic regulators have on chromatin, as well as in what way. Second, from a practical standpoint, answering this question is important for understanding the potential of DNA methylation–based episignatures (Aref-Eshghi et al. 2021; Levy et al. 2022a) to shed light on the pathogenesis of the disorders. Although these episignatures are almost exclusively derived from whole blood, there is increasing emphasis on their capacity to pinpoint locations of chromatin disruption responsible for the dysfunction of other tissues/cell types (Chater-Diehl et al. 2021). In particular, because intellectual disability is the most prevalent phenotypic characteristic of these disorders, a major focus is on whether the same locations that are disrupted in blood also contribute to abnormal neurodevelopment (Chater-Diehl et al. 2021; Levy et al. 2021, 2022b).
One of the prototypical Mendelian disorders of the epigenetic machinery (EM) is Kabuki syndrome (KS). Approximately 70% of cases of KS are caused by variants in either the histone methyltransferase KMT2D (KS type 1 [KS1]) or the histone demethylase KDM6A (KS type 2 [KS2]) (Ng et al. 2010; Miyake et al. 2013). Several studies have recently investigated the molecular basis of KS, uncovering defects in different cell types (Zhang et al. 2015; Ang et al. 2016; Lindsley et al. 2016; Lin-Shiao et al. 2018; Carosso et al. 2019; Fahrner et al. 2019; Pilarowski et al. 2020; Gabriele et al. 2021; Luperchio et al. 2021; Tian et al. 2021; Ma et al. 2022; Jung et al. 2023; Potter et al. 2024). In addition, KS was one of the first disorders for which episignatures were derived and shown to have diagnostic utility (Aref-Eshghi et al. 2017; Butcher et al. 2017; Sobreira et al. 2017). Therefore, KS is ideally suited for understanding the extent to which chromatin abnormalities caused by the disruption of the same epigenetic regulator are similar in different cell types.
Here, we investigate this using mouse models of KS1 and KS2 and profiling genome-wide chromatin accessibility in sorted hippocampal neurons, as well as sorted B and T cells from peripheral blood.
Results
Widespread disruption of chromatin accessibility in hippocampal neurons, B and T cells in KS1 and KS2 mouse models
We performed ATAC-seq in order to characterize the genome-wide chromatin accessibility landscape in KS mice (both type 1 and type 2) and wild-type littermates (Methods). We examined sorted hippocampal neurons from the dentate gyrus (henceforth, neurons) and sorted peripheral T cells (Methods). We also used previously generated ATAC-seq data from sorted peripheral B cells (Luperchio et al. 2021). These cell types were chosen because (1) there is convincing evidence supporting their involvement in KS pathogenesis (Lindsley et al. 2016; Carosso et al. 2019; Pilarowski et al. 2020; Luperchio et al. 2021; Potter et al. 2024); (2) two of them (B and T cells) have similar functionality and common developmental origins, whereas neurons are functionally, morphologically, and developmentally distinct; and (3) they allow us to assess if we should expect blood episignature locations to provide information about the epigenetic basis of abnormal neurodevelopment in KS.
We started with a differential accessibility analysis in neurons, after validating our detected ATAC peaks (Methods). Comparing eight KS1 mice to five wild-type littermates (Methods), we discovered extensive chromatin accessibility disruption in mutant mice. Specifically, 20,683 (13.4%) of the 154,560 tested regulatory elements (ATAC peaks) show differential accessibility (Methods) (Supplemental Table 1). Among the top disrupted elements (Q-value ≤ 0.1), the vast majority (87.6%) have a more open chromatin state in mutants. Focusing on regulatory elements at the promoters (±2 kb from the transcriptional start site [TSS]; 19,013 peaks total), we estimated that 63.6% are disrupted; almost all of the top promoter regulatory elements (99.3% among those with Q-value ≤ 0.1) are more open in mutants. Turning our attention to KS2, we found similar results after comparing four mutant mice to six wild-type littermates (different from wild-type mice compared against the KS1 mutants). Specifically, we detected disruption of normal accessibility in 49,625 (31.7%) of the 156,309 tested neuronal regulatory elements (Methods) (Supplemental Table 2), with 68.2% of the promoter regulatory elements affected. Although the genome-wide disruption was largely evenly balanced (58% of elements with Q-value ≤ 0.1 are more open in mutants), 96.3% of the promoter elements with Q-value not exceeding 0.1 again have more open chromatin in mutants. This systematic shift toward greater accessibility of promoter regulatory elements is also seen in B and T cells (82.3% and 83.5% of the top disrupted promoter elements show increased accessibility in mutant B and T cells, respectively, in KS1 and 91.3% and 77.3% in KS2) (see also Luperchio et al. 2021; Jung et al. 2023). This is notable because both KMT2D and KDM6A are believed to promote transcriptional activity.
Finally, like neurons, B and T cells show widespread chromatin accessibility disruption (24.5% and 17.5% of all regulatory elements disrupted in mutant B and T cells, respectively, in KS1 and 15.8% and 13.3% in KS2). Looking at enhancers near disrupted promoters (ATAC peaks within 100 kb from the TSS; Methods), we found that enhancer accessibility tends to be disrupted toward the same direction as nearby promoter accessibility (Supplemental Fig. S1); this phenomenon is especially prevalent in KS2 neurons and B cells and suggests a coordinated disruption of nearby regulatory elements. Some of these enhancer accessibility changes may exacerbate the effect of disrupted promoter accessibility on target gene expression; other enhancer changes could have a compensatory effect, even if codirectional with promoter accessibility changes, in cases of enhancers bound by repressors.
P-value distributions reveal no preferential chromatin accessibility disruption of the same regulatory elements in hippocampal neurons versus B/T cells
We next turned to our main question of interest: whether the same regulatory elements are preferentially disrupted in neurons and immune cells. The most straightforward way to assess this would be to select a significance threshold and examine the list of regulatory elements whose differential accessibility P-value is below that threshold in both neurons and B or T cells. However, the arbitrary nature of such significance thresholds is a serious disadvantage of this approach. We have previously shown (Luperchio et al. 2021) that in simulations with known ground truth, this approach severely underestimates the number of elements with shared disruption. We expect this problem to be especially pronounced here, as the majority of accessibility changes in the three cell types are of relatively small magnitude (95% of absolute log-fold changes less than 0.49, 0.52, 0.54 in neurons, B cells, and T cells, respectively, in KS1 and less than 0.8, 0.47, 0.55 in KS2); this implies that many biologically true changes will not exceed traditional significance thresholds. We thus used a method we previously developed to address this issue (for details, see Methods) (Fig. 1A,B; Luperchio et al. 2021). This method starts with one cell type and asks if regulatory elements differentially accessible between mutants and wild type in that first cell type have differential accessibility P-values overrepresented close to zero in the other cell type of interest. Whether such an overrepresentation is present and exceeds what is expected by chance can be rigorously assessed from the P-value distribution in that other cell type. If an overrepresentation is indeed present, it means that the same regulatory elements are preferentially disrupted in both cell types (Fig. 1A,B). The advantages of this approach are as follows: (1) It has less reliance on significance thresholds; (2) it leverages information from the entire P-value distribution of the second cell type and thus has the ability to answer whether the same regulatory elements are disrupted in the two cell types beyond a chance expectation, separately from identifying individual shared elements (Storey 2003; Storey and Tibshirani 2003); (3) it has increased power (Luperchio et al. 2021); and (4) it allows for an intuitive visual assessment (Methods) (Fig. 1A,B).
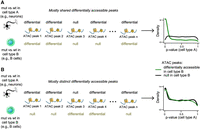
Testing whether disruption of chromatin accessibility preferentially occurs at the same regulatory elements in two cell types using conditional P-value distributions; illustration of the basis of our approach. (A) Hypothetical example of two cell types showing disruption of chromatin accessibility primarily at the same regulatory elements when comparing mutant and wild-type mice in each cell type. There are also some regulatory elements differentially accessible between the mutant and wild type in only one of the two cell types (such as peak 4), but the overlap between the two cell types exceeds what is expected by chance. The resulting distribution of P-values in cell type A, when restricting to regulatory elements that are differentially accessible in cell type B, shows an overrepresentation of P-values close to zero. This overrepresentation is not observed when restricting to regulatory elements that are not differentially accessible in cell type B. (B) Like A, but with different regulatory elements disrupted in cell types. There are also some regulatory elements differentially accessible between the mutant and wild type in both cell types (such as peak 4), but the overlap between the two cell types does not exceed what is expected by chance. The resulting distribution of P-values in cell type A shows the same overrepresentation close to zero regardless of whether we restrict to regulatory elements that are differential in cell type B or regulatory elements that are not differential in cell type B.
To apply our method, we started with regulatory elements differentially accessible in B cells (ATAC peaks with Q-value ≤ 0.1). In both KS1 and KS2, we discovered that these regulatory elements are not preferentially disrupted in neurons when focusing at promoters. When looking genome-wide, there is a small, but not robust, preferential disruption signal (Methods) (Fig. 2A,C,E,G; Supplemental Fig. S2A–D). In stark contrast, when starting with disrupted elements in B cells, there is pronounced signal indicating their preferential disruption in T cells at both the level of the promoters and genome-wide. This is evident in both KS1 (Fig. 2B,F; Supplemental Fig. S2A,C) and KS2 (Fig. 2D,H; Supplemental Fig. S2B,D). These results hold true regardless of whether the starting peaks in B cells are more open or more compact in mutants (Fig. 2I; circles vs. squares). They are also recapitulated when starting with differentially accessible regulatory elements in neurons or T cells and comparing to B cells, as well as when comparing neurons to T cells (Methods).
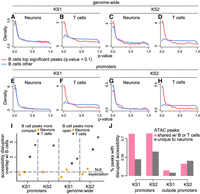
Evaluating whether neurons, B cells, and T cells show preferential disruption of chromatin accessibility at the same regulatory elements in Kabuki syndrome (KS) types 1 and 2. (A–H) P-value distributions from mutant (KS1 or KS2) versus wild-type differential accessibility analyses in either neurons or T cells. Shown both for ATAC peaks genome-wide and for promoters only. The red densities correspond to ATAC peaks overlapping peaks disrupted in B cells (Q-value < 0.1), whereas the blue densities correspond to ATAC peaks overlapping peaks not disrupted (but present) in B cells. (I) The ratio (y-axis) of the proportion of ATAC peaks that overlap differentially accessible peaks in B cells and are also differentially accessible in either neurons (orange dots/squares) or T cells (black dots/squares) over the average proportion of ATAC peaks that overlap randomly sampled B cell peaks and are also differentially accessible in either neurons or T cells. The ratio is computed starting either with B cell peaks more compact in mutants (dots) or with B cell peaks more open in mutants (squares). Only squares are depicted for KS2 promoters, because there are too few B cell promoter peaks that are significantly more compact in mutants (17 peaks; see also Results) to do a meaningful analysis. The dashed horizontal line at one corresponds to the null expectation when there is no significant overlap between the ATAC peaks with disrupted chromatin accessibility in the two cell types tested. (J) The percentage of differentially accessible ATAC peaks in neurons (1 − π0; Methods), among ATAC peaks that are specific to neurons (gray bars) or ATAC peaks that overlap peaks that are also present in B or T cells (pink bars).
We note that all pairwise comparisons between cell types were performed after restricting to ATAC peaks detected in both cell types (Methods). This ensures that the presence of the preferential disruption signal when comparing B cells to T cells, and its absence when comparing B cells to neurons, is not explained by a greater overall similarity of the regulatory landscape between B and T cells. Related to this, we found that, with the exception of nonpromoter regulatory elements in KS2, the neuronal chromatin disruption occurs preferentially at regulatory elements shared with either B or T cells and not at elements uniquely active in neurons (Fig. 2J; Supplemental Fig. S3).
We next looked at mouse orthologs of human genes whose promoters are differentially methylated in KS1 patients versus controls in whole blood (Aref-Eshghi et al. 2017); these are the promoters used to derive the KS1 episignature. Consistent with our other results, we found that these promoters are no more likely to be disrupted in neurons than are randomly selected promoters (Methods) (Supplemental Fig. S4). In contrast, in B and T cells, these mouse ortholog promoters show clear evidence of preferential disruption compared with other promoters (Supplemental Fig. S4), which suggests that the null result in neurons is not owing to human–mouse differences.
Promoter regulatory elements with disrupted accessibility in KS neurons, but not B or T cells, are concentrated at CpG island promoters
We set out to investigate the nature of the discrepancy between neurons and B/T cells. We focused on promoters, because their unambiguous association with the downstream gene can help connect that discrepancy to the underlying biology. A major distinction of mammalian promoters is between those that are associated with CpG islands and those that are not (Saxonov et al. 2006). We thus first asked if any systematic differences exist at this scale.
Ranking promoter ATAC peaks based on how strong the evidence for disruption of their accessibility is in KS1 neurons, we discovered a clear pattern: The stronger the evidence for accessibility disruption, the higher the probability of the promoter peak overlapping a CpG island (Fig. 3A). Of the most confidently disrupted promoter peaks (smallest 10% of P-values), 94% overlap CpG islands compared with 17% of the least confidently disrupted ones (highest 10% of P-values). In KS1 B or T cells, this relationship with CpG islands is completely absent (Fig. 3B,C), even though the number of CpG islands containing ATAC peaks is—as expected—very similar in the three cell types (12,171 in B cells and 12,397 in T cells vs. 11,377 in neurons). This indicates a fundamentally different localization of the most statistically robust chromatin alterations and sheds further light on our observation that broadly active regulatory elements show neuron-specific disruption. An almost identical picture is seen in KS2, with preferential disruption of CpG island promoters in neurons but not in B or T cells (Fig. 3D–F). Further dissecting the neuronal chromatin disruption, we saw that, in KS1 as well as KS2 neurons, both of the main CpG island promoter classes (Lenhard et al. 2012) are preferentially disrupted: (1) CpG island promoters bound by Polycomb Repressive Complex 2 (PRC2) and associated with tissue-specific genes (Methods) and, even more so, (2) non-PRC2-bound CpG island promoters that are associated with genes widely expressed across tissues (Fig. 3G). As expected based on our earlier results, disrupted promoter regulatory elements overlapping CpG islands almost always have increased accessibility relative to the wild type in mutant neurons (Fig. 3H).
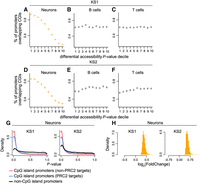
Assessing the relationship between promoter chromatin accessibility disruption and promoter CpG-richness in neurons, B cells, and T cells in KS1 and KS2. (A–F) The proportion of promoter ATAC peaks overlapping CpG islands, stratified according to P-value decile. The horizontal lines of the crosses correspond to these proportions across the deciles, and the vertical lines of the crosses depict the associated 95% binomial confidence intervals. Shown separately for neurons, B cells, and T cells in KS1 (first row) and KS2 (second row). The P-values for these promoter peaks are derived from the corresponding differential accessibility analyses (Methods). (G) The P-value distributions of promoter peaks, stratified according to whether these peaks overlap CpG islands targeted by Polycomb Repressive Complex 2 (blue densities), overlap other CpG islands (red densities), or are located outside CpG islands (black densities). Shown for KS1 and KS2 neurons. (H) The distribution of the magnitude of accessibility changes (log2(fold change)) of the most disrupted CpG island–overlapping promoter peaks (bottom 10% P-value) in KS1 and KS2 neurons.
Promoters of epigenetic machinery genes are preferentially disrupted in neurons but not B or T cells
It is known that the chromatin accessibility of CpG islands is related to the absence of DNA methylation and the presence of specific histone modifications (Thomson et al. 2010; Bian et al. 2011; Wachter et al. 2014; Li et al. 2016b; Kurup et al. 2019; Hughes et al. 2020). We thus wondered if changes of such modifications are driving the observed changes in CpG island accessibility. Although we did not directly measure histone modifications or DNA methylation, we reasoned that we can indirectly gain insight into this question by examining whether there is preferential chromatin disruption at individual promoters of EM genes compared with promoters of other genes. If this is true in neurons, but not in B or T cells, then chromatin disruption could be driving the CpG island accessibility changes by causing changes in the expression of EM genes and, thus, changes in histone modification and DNA methylation patterns at CpG islands. In both types of KS, we found that regulatory elements at the promoters of EM genes are preferentially disrupted compared with regulatory elements at the promoters of other genes in neurons (Supplemental Fig. S5A,B; Supplemental Tables 3, 4). However, this is not true in B or T cells (Supplemental Fig. S5A,B). The neuronal preferential disruption signal is especially strong at promoters of EM genes whose human homologs have previously been shown to be highly intolerant to loss-of-function variation and coordinately expressed with one another in multiple tissues, and are thought to be particularly important in neurodevelopment (Methods) (labeled as highly coexpressed EM genes in Supplemental Fig. S5A,B; Boukas et al. 2019). In KS1, among the EM genes with disrupted promoters is Kdm6a, the histone demethylase causing KS2 (P = 0.001) (Supplemental Fig. S5C). The increased accessibility of its promoter is consistent with its transcriptional up-regulation in human iPSC-derived neurons (Gabriele et al. 2021). Other notable genes include Kmt2a (histone methyltransferase causing Wiedemann–Steiner syndrome; P = 0.004) (Supplemental Fig. S5C), Tet3 (DNA demethylase causing Beck–Fahrner syndrome; P = 0.007), and Chd1 (chromatin remodeler causing Pilarowski–Bjornsson syndrome whose targets have been implicated in neural crest stem cell transcriptional dysregulation upon KMT2D hapoinsufficiency; P = 0.005) (Supplemental Fig. S5C; Gabriele et al. 2021). In KS2, epigenetic regulators with disrupted promoters range from Kmt2d (histone methyltransferase causing KS1; P = 3.95 × 10−6) (Supplemental Fig. S5D) and Suz12 (PRC2 subunit causing Imagawa–Matsumoto syndrome; P = 4.08 × 10−5) (Supplemental Fig. S5D) to Kat6a (histone acetyltransferase causing KAT6A syndrome; P = 0.001) (Supplemental Fig. S5D). These findings indicate broad epigenetic dysregulation in KS1 and KS2 that extends beyond what would be expected on the basis of the function of their respective causative genes, and could explain the disruption of CpG island chromatin. In addition, they may help explain the high degree of neurodevelopmental/neurological phenotypic overlap among the different Mendelian disorders of the EM (Fahrner and Bjornsson 2019).
Transcriptome profiling in B and T cells reveals concordance between promoter chromatin accessibility disruption and gene expression dysregulation
To assess whether the chromatin accessibility changes we detect have functional consequences, we examined the B and T cell transcriptomes, using RNA-seq (Methods). We examined both KS1 and KS2 mutants, as well as their corresponding wild-type littermates. In all cases, RNA was isolated from the same mice and at the same time point as chromatin. The overlap between genes with dysregulated expression in B and T cells was very pronounced (Supplemental Fig. S6), like the overlap at the chromatin accessibility level.
Using a sliding rank threshold, we obtained promoter ATAC peaks with a differential accessibility P-value rank below that threshold, and asked if the expression of the associated genes is dysregulated (Methods). We found a very clear relationship: The more stringent the threshold for promoter differential accessibility, the greater the percentage of differentially expressed genes (Fig. 4A–D). This is true in both KS1 and KS2, and in B as well as T cells. In ∼90% of cases in which both gene expression and promoter accessibility change in mutants, there is concordance between the direction of promoter accessibility disruption and gene expression dysregulation (Fig. 4E; Supplemental Fig. S7); more open chromatin in mutants is associated with increased expression level, and more compact chromatin in mutants is associated with decreased expression level. Although we were unable to directly assess gene expression in neurons for technical reasons, we found that the relationship between promoter accessibility disruption and gene expression dysregulation in B/T cells begins to manifest when the magnitude of promoter accessibility changes is below the magnitude shown by the top disrupted peaks in neurons, in both KS1 and KS2 (Supplemental Fig. S7). Collectively, these results provide evidence that (1) chromatin accessibility changes are associated with the expected gene expression changes, and (2) by selecting genes whose promoter regulatory elements are ranked at the top versus the bottom based on their differential accessibility P-value, we enrich and deplete, respectively, for genes whose expression is dysregulated. We therefore asked again whether genes with CpG island promoters and highly coexpressed EM genes are preferentially dysregulated in KS1 and KS2, comparing the top- versus the bottom-ranked promoters. This confirmed our earlier results and further highlighted that the CpG island gene disruption is stronger in KS1 (Fig. 4F), whereas the disruption of highly coexpressed EM genes is stronger in KS2 (Fig. 4F). Finally, in B and T cells, we confirmed the lack of enrichment of top disrupted genes among CpG island genes and highly coexpressed EM genes at the gene expression level as well (odds ratio less than one in all cases).
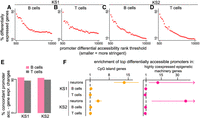
Investigating the relationship between promoter chromatin accessibility disruption and gene expression dysregulation. (A–D) Each point corresponds to a rank threshold for promoter differential accessibility and depicts the proportion (y-axis) of differentially expressed genes among genes whose promoter differential accessibility P-value is ranked below that threshold (see also Methods). For example, the first point from the left in each plot corresponds to the proportion of differentially expressed genes among genes with a promoter differential accessibility P-value among the smallest 500 P-values. Thresholds are calculated sequentially, with a step size of 250; smaller is equivalent to more stringent. (E) The proportion of disrupted promoter–gene pairs with concordant disruption. Concordance is defined as either increased accessibility–increased expression or decreased accessibility–decreased expression. (F) The enrichment (odds ratio) of the top-ranked genes (based on promoter differential accessibility) in the group of genes with CpG island promoters or in highly coexpressed epigenetic machinery genes. The enrichment is calculated by comparing against the bottom-ranked genes (see also Methods). The horizontal dashed line at one corresponds to no enrichment. The 95% confidence intervals are depicted for cases with enrichment exceeding one. Upper CI bound for the enrichment of the top-ranked genes in highly coexpressed epigenetic machinery genes in KS1 and KS2 = 60.01 and 1508.9, respectively.
Aging-related regions show preferentially disrupted chromatin accessibility in KS1 and KS2 neurons but not in B or T cells
Our findings yield insights into (1) the genomic distribution of the chromatin defects in KS1 and KS2 and (2) the cell type selectivity of these defects. However, our findings thus far fall short of providing a link between the chromatin defects and the abnormal development known to be a component of KS. To obtain such a link, we first performed a pathway analysis of the genes with disrupted promoters in neurons (using Reactome; Methods). In both KS1 and KS2, the top pathways indicate disruption of various modes of epigenetic control (consistent with the results of the previous section on EM genes) and disruption of other aspects of transcriptional regulation (Supplemental Tables 5, 6). However, these pathways are rather general. We thus searched for a characterization of the disrupted locations that better captures the abnormal developmental process in KS1 and KS2. To this end, we defined a set of regulatory elements whose chromatin state likely changes as development unfolds by restricting to ATAC peaks containing CpGs whose collective methylation status has been shown to be a highly accurate mouse age estimator, that is, an “epigenetic clock” (582 CpGs in total; Methods) (Thompson et al. 2018). Apart from being biomarkers of biological aging, there is now emerging evidence that such epigenetic clocks track embryonic development as well (Kerepesi et al. 2021; Zhang et al. 2022). Using an approach based on work by Barry et al. (2005), conceptually similar to our approach for between-cell-type comparisons but traditionally used for functional category enrichment analyses, we discovered that clock CpG–containing regulatory elements are preferentially disrupted in KS1 and KS2 neurons. This disruption is present not only at promoter age-associated elements (Fig. 5A,D; lower values of the observed Wilcoxon rank-sum statistic than expected under the null distribution) but also at age-associated elements outside promoters (Fig. 5G,J). In contrast, in B and T cells, there is no preferential chromatin accessibility disruption at clock CpG–containing regulatory elements, in KS1 or KS2 (Fig. 5B,C,E,F,H,I,K,L). In KS2 T cells, there even seems to be a depletion of promoter chromatin accessibility changes at these elements (Fig. 5F), although it is unclear if this has a biological basis, such as an increased dependence of T cells on these elements in the setting of Kdm6a haploinsufficiency.
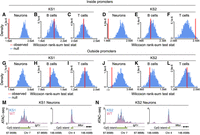
Investigating the chromatin disruption of aging-related regulatory elements in neurons, B cells, and T cells in KS types 1 and 2. (A–L) The observed value (red vertical line) of the Wilcoxon rank-sum test statistic obtained after comparing the differential accessibility P-values of aging-related ATAC peaks (i.e., ATAC peaks that contain epigenetic clock CpGs) to the differential accessibility P-values of other ATAC peaks. Observed values that are lower than expected under the null (blue distributions) indicate the collective disruption of aging-related peaks. Shown separately for neurons, B cells, and T cells, in KS1 and KS2, for ATAC peaks overlapping promoters and ATAC peaks outside promoters. The null distributions were derived by repeatedly (1000 times) sampling random sets of peaks and calculating the Wilcoxon rank-sum test statistic after comparing the P-values of these randomly sampled peaks to the P-values of all other peaks. Each random set contained an equal number of peaks to the number of aging-related peaks. (M,N) The normalized ATAC signal of Igf1r and Mtor, two genes known to regulate lifespan-related pathways, in mutant and wild-type neurons. Shown for KS1 and KS2.
To further explore the link with regions involved in aging, we asked if genes implicated in lifespan-modulating pathways, such as signaling via the IGF1 receptor and mTOR signaling, have disrupted promoters. To do this in an unbiased manner, we examined genes in the mouse “longevity regulating pathway” as defined by KEGG (Methods) (Kanehisa et al. 2017). None of the promoter ATAC peaks of these longevity genes contain any of the 582 clock CpGs, making this an orthogonal assessment of the relationship between chromatin disruption in KS and aging-related regions. We found that in neurons, but not in B or T cells, promoters of longevity genes are collectively disrupted (Supplemental Fig. S8). This is the case both in KS1 and KS2. Notable examples with disrupted promoters are Igf1r and Mtor (Fig. 5M,N). Mutant neurons at the promoters of those two genes show increased accessibility compared with the wild type. If this is accompanied by hyperactivation of signaling via the IGF1 receptor and mTOR signaling, it would suggest that the KS neurons are epigenetically “older” than their wild-type counterparts.
The most significant chromatin accessibility changes in B cells are present, but are very subtle, in neurons
All our investigations reveal substantial differences between the chromatin alterations in neurons and B/T cells. From a methodological standpoint, our results are based on P-values (although, as noted, our approach has less reliance on arbitrary significance thresholds than traditional approaches). In light of this, we returned to our overarching question of whether there is preferential disruption of the same regulatory elements in immune cells and neurons, this time adopting an alternative approach. We asked whether the accessibility of the regions disrupted in B cells is altered in the same direction in neurons, more than expected by chance (e.g., whether ATAC peaks that are more compact in mutant B cells tend to be more compact in mutant neurons, more than expected by chance). In other words, we examined the concordance between the sign of the effect (log-fold change) in the two cell types, without taking the P-value into account (Tukey 1962; Gelman and Tuerlinckx 2000; Stephens 2017). This approach has greater sensitivity and can thus answer whether regulatory elements disrupted in blood are also disrupted in neurons but to a very small degree.
In all comparisons performed, we found statistically significant concordance in the direction of accessibility disruption (Fig. 6A; Supplemental Fig. S9). This includes the neuron versus B cell comparisons that previously showed no overlap based on P-value distributions. As expected, we did see that the concordance between B and T cells is always greater than the concordance between neurons and B cells (Fig. 6A). Closer inspection of the behavior of neuronal peaks overlapping the top disrupted B cell peaks revealed that their accessibility alterations in neurons are of a very small magnitude, which could explain why they do not stand out based on their P-values (Fig. 6B,C, pink curves). However, the distribution of these magnitudes is different—centered away from zero—from the distribution of the magnitude of the changes at elements not disrupted in B cells (Fig. 6B,C, pink vs. blue curves). This suggests that the neuronal disruption of the elements most disrupted in B cells is truly present, albeit very subtle.

Evaluating whether chromatin accessibility changes occur toward the same direction in neurons, B cells, and T cells in KS types 1 and 2. (A) The ratio (y-axis) of the proportion of ATAC peaks that overlap differentially accessible B cell peaks and show accessibility changes in the same direction in neurons (orange dots) or T cells (black dots), over the average proportion of ATAC peaks that overlap randomly sampled B cell ATAC peaks and show accessibility changes in the same direction in neurons or T cells. The dashed horizontal line corresponds to the null expectation when there is no significant concordance in the direction of the accessibility disruption (sign of log 2 (fold change)) between the two cell types tested. (B,C) The distribution of neuronal effect sizes (log2 (fold changes)), shown for the significant disrupted neuronal ATAC peaks (orange), significantly disrupted neuronal peaks overlapping significantly disrupted B cell peaks (pink), and other neuronal peaks (blue). Significance is defined as Q-value < 0.1.
A small subset of regions show changes in chromatin accessibility in all three cell types
As a final analysis, we asked if it would be possible to identify a set of regulatory elements with disrupted accessibility in all three cell types, using a step-wise approach. Our ultimate goal was to derive, in a principled manner, a set of regulatory elements whose accessibility can potentially comprise a blood-based biomarker that also accurately reflects the neuronal chromatin state. We started with regulatory elements disrupted in B cells and then selected the subset that is disrupted in neurons as well (10% FDR level). We then further narrowed these elements down by examining their accessibility in T cells and selecting only those with disrupted accessibility (10% FDR level). This procedure yielded 381 regulatory elements for KS1 and 91 regulatory elements for KS2 (Supplemental Tables 7–9, 11–13). A pathway enrichment analysis of the genes associated with the shared promoter regulatory elements (Supplemental Tables 10, 14) implicated diverse biological processes, including neurotransmission, ion channel activity, metabolism, and various signaling pathways (Methods) (Supplemental Tables 15, 16). This again underscores the existence of a system-level disruption in KS1 and KS2 and argues against a reductionist approach to understanding the molecular basis of KS.
Supporting the validity of the shared regulatory elements, we found that the KS1 shared elements overlap with the KS2 shared elements much more than expected by chance: When restricting to promoter peaks and examining the associated genes, there is a 66.3-fold enrichment of the KS1 genes in the set of KS2 genes (P < 2.2 × 10−16, Fisher's exact test). This overlap between KS1 and KS2 echoes previous results in blood and immune cells (Aref-Eshghi et al. 2021; Luperchio et al. 2021). Moreover, in each KS type, regulatory elements disrupted in both B cells and neurons are substantially more likely to be disrupted in T cells compared with regions only disrupted in B cells (Supplemental Fig. S10). This contrasts with the lack of preferential disruption in the earlier pairwise comparisons of B/T cells versus neurons and suggests that the sequential approach used here can provide additional insights. Taken together, these results reveal the existence of a small set of regulatory elements disrupted both in neurons and immune cells.
Discussion
We have characterized, compared, and contrasted the genome-wide chromatin alterations ensuing from genetic disruption of the histone methyltransferase Kmt2d and the histone demethylase Kdm6a in three different cell types: hippocampal neurons and peripheral blood B and T cells. Our results paint a nuanced picture. On one hand, the most statistically significant chromatin alterations occur, mostly, at different genomic locations in neurons versus B/T cells (as expected, the alterations in the two immune cell types overlap substantially). On the other hand, the locations harboring the most significant chromatin disruption in B/T cells do in fact show evidence of disruption in neurons, but the degree of this disruption is very small when present.
Broadly speaking, the differences between the cell types are compatible with three different scenarios. In the first scenario, when KMT2D and KDM6A exert their catalytic effects, the relative abundances of their interacting partners (Shilatifard 2012; Li et al. 2016a; Wang et al. 2017, 2022) are different in neurons versus immune cells. KMT2D and KDM6A are recruited to different locations as dictated by these abundances, and as a result, when Kmt2d and Kdm6a are genetically disrupted, the chromatin alterations also occur at different locations. In the second scenario, KMT2D and KDM6A exert their function at the same locations in both neurons and immune cells. However, other epigenetic regulators act after KMT2D and KDM6A to further modify the chromatin landscape in a different fashion in neurons versus immune cells. The chromatin alterations in this case may initially occur at the same locations but are subsequently molded differently. In the third scenario, the chromatin alterations occur at a common progenitor cell type early during development. Then, subsequent chromatin changes occurring as part of the neuronal and immune cell developmental programs (Stergachis et al. 2013; Gorkin et al. 2020) give rise to what we ultimately observe in the mature cell types, which reflects the interplay between the transcription factors driving developmental cell fate transitions in the immune and neuronal lineages and the initially misregulated chromatin accessibility. The distinct chromatin alterations in this case are a reflection of distinct developmental paths. In contrast, the shared, subtle chromatin changes are remnants of the initial defects.
Notwithstanding the underlying explanation, we consider it surprising that a major difference between neurons and B/T cells is the preferential disruption of CpG islands at the promoters of housekeeping genes, regulatory elements that have broad, non–cell type–specific activity. Our RNA-seq versus ATAC-seq comparisons in B and T cells strongly suggest that promoter chromatin accessibility changes cause gene expression changes in neurons as well. This, together with the fact that housekeeping genes control fundamental cellular processes, implies that the neuronal cellular phenotype is more severe; future work should, however, confirm these gene expression changes with direct expression measurements in neurons. Of note, the disruption of EZH2-bound CpG islands (PRC2 targets) is in line with transcriptional changes recently observed in human iPSC-derived neurons and neural crest stem cells (Gabriele et al. 2021), although that study emphasized the down-regulation of some of these genes.
Also of potential mechanistic relevance is our finding that the chromatin disruption in neurons occurs preferentially at regulatory elements containing aging clock CpGs and at promoters of lifespan-associated genes: in other words, at locations that somehow track—and in some cases potentially regulate—the passage of biological time. This may represent a new link between the chromatin disruption in KS and the disruption of normal neural development/maturation. Our observation that, based on some of the accessibility changes, KS neurons may be epigenetically “older” than wild-type neurons fits with a previous report of precocious neuronal differentiation in KS1 (Carosso et al. 2019) and warrants further functional exploration. The hyperactivation of the mTOR pathway that we infer has also recently been seen in cancer contexts upon Kmt2d as well as Kdm6a loss (Revia et al. 2022; Xu et al. 2023). We note, however, that a previous study reported a lower epigenetic age in patients with KS, using the original Horvath epigenetic clock on blood samples (Jeffries et al. 2019). The reasons for the discrepancy between these results are unclear but may include mouse–human differences, differences between cell types, as well as the fact that different variants have different effects on epigenetic age (Jeffries et al. 2019). In addition, it should be pointed out that abnormal immune cell maturation is also known to be a component of the KS phenotype (Lindsley et al. 2016; Pilarowski et al. 2020; Potter et al. 2024). Yet, the aging-associated locations do not show preferential disruption in B/T cells in our study, even though this clock was shown to function as an age predictor in multiple different mouse tissues (including blood) (Thompson et al. 2018). In this respect, the chromatin basis of abnormal immune cell maturation appears distinct from the basis of abnormal neurodevelopment.
Future work should also address the following two puzzles. First, with respect to the very subtle neuronal chromatin alterations at the locations where the immune cells are most saliently disrupted, what our study does not answer is whether these subtle alterations are biologically significant in neurons. In other words, do alterations of such small magnitude have any effect on neuronal cellular function? It is also worth noting here that even the top significant neuronal alterations are of relatively small magnitude, with the majority of changes being much smaller than twofold. However, we anticipate that, collectively, these individually small changes can lead to the emergence of disease phenotypes (Muto et al. 2011; Luperchio et al. 2021). How small such chromatin changes can be and yet still have a biologically significant collective effect is, in our view, an important open question. Second, why do mutant neurons show an increase in promoter accessibility, when haploinsufficiency of either Kmt2d or Kdm6a would be expected to lead to more compact chromatin (Bjornsson 2015) based on the activities of these histone modifiers (H3K4 methyltransferase and H3K27 demethylase, respectively)? Although counterintuitive, this result is consistent with prior results in mouse B cells when examining promoter accessibility (Luperchio et al. 2021), as well as in human peripheral mononuclear blood cells when examining the signal at H3K4me2 peaks (Jung et al. 2023). Three potential explanations to be considered are (1) KMT2D and KDM6A acting in fact as transcriptional repressors at promoter regulatory elements via currently unappreciated mechanisms, (2) the promoter chromatin accessibility changes representing indirect consequences downstream from the initial direct consequences of KMT2D and KDM6A loss, and (3) other epigenetic regulators overcompensating for KMT2D and KDM6A.
A limitation of our study is that it is heavily focused on promoters. Although this has the advantage that the unambiguous association between promoters and genes offers a window into the underlying biology, it will be important in future work to integrate analyses of enhancer chromatin accessibility as well, in order to arrive at a more complete understanding of how the genome-wide regulatory landscape is disrupted in KS. Another limitation pertinent to all our results and interpretations is that the sorted cell populations we analyzed were defined by the expression of specific surface markers. However, it is now well appreciated that cells expressing these markers represent a mixed population of cell subtypes (Papalexi and Satija 2018; Zeisel et al. 2018; La Manno et al. 2021). Hence, it is possible that some of our conclusions would be different had we analyzed such subtypes. For instance, we presently cannot exclude the existence of rare B or T cell subtypes whose chromatin disruption more resembles that of neurons in terms of promoter CpG islands and aging-associated regulatory elements. Future single-cell analyses promise to provide an answer to this question, provided that issues related to single-cell differential accessibility analyses are appropriately dealt with.
Finally, we emphasize that our goal when defining the sets of regulatory elements disrupted in all three cell types is not for these regulatory elements to replace those currently comprising existing KS episignatures; the latter have shown their diagnostic utility (Aref-Eshghi et al. 2017, 2021; Levy et al. 2022a). However, if the objective is to somehow leverage the easily accessible epigenomic information from blood in order to draw conclusions pertinent to the chromatin basis of neurodevelopment in KS, we hypothesize that the regulatory elements we discovered in our current study may prove particularly useful. Future work is needed to test this and evaluate if similar results hold for other Mendelian disorders of the EM.
Methods
Mice
All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments received approval by the animal care and use committee of the Johns Hopkins University (protocol number MO18M112). Both the KS1 (Kmt2d+/βGeo) and KS2 (Kdm6a+/−) mouse models and genotyping protocols have been previously described (Luperchio et al. 2021). The Kmt2d+/βGeo mice model haploinsufficiency for the catalytic activity of KMT2D (as the βGeo allele produces a truncated protein that lacks the SET domain). Mice for T cells are the same as reported by Luperchio et al. (2021). Neurons were obtained from 10- to 13-wk-old KS1, KS2, and wild-type mice.
ATAC-seq: cell isolation and processing
B and T cells
B cells (CD19+) from peripheral blood were described by Luperchio et al. (2021). T cells were isolated at exactly the same time as B cells. Specifically, flow through from binding and washing of CD19+ cells on Miltenyi separation columns was collected on ice. These CD19+ depleted samples were then spun, and the supernatant was aspirated, resuspended, and incubated with 2× (20 μL) CD90.2+ microbeads (Miltenyi Biotec 130-121-278). CD90.2+ T cells were bound, washed, and eluted per the manufacturer's protocol and then counted and aliquoted on ice for subsequent processing. Further processing of the T cell samples and ATAC sequencing was performed in parallel with B cells as described by Luperchio et al. (2021). A principal component analysis based on the ATAC signal profiles (after variance stabilization with the vst() DESeq2 function) shows essentially perfect separation between B and T cells (Supplemental Fig. S11), as expected under efficient depletion of B (CD19+) cells before T cell isolation.
Neurons
Hippocampal neurons were isolated using a protocol adapted from the INTACT (Mo et al. 2015) and Omni-ATAC (Corces et al. 2017) protocols. Dentate gyrus was microdissected as previously described (Carosso et al. 2019) and placed into ice-cold 1 × HBS (1× HBS-B, 1 M sucrose, 500 mM EDTA, 10% IGEPAL; 1× HBS-B was initially prepared as 6 × HBS with 1 M CaCl2, 1 M MgAc, 1 M Tris at pH7.8, 1 mM BME). Samples were homogenized in a 2-mL glass dounce, pestle A until resistance went away (approximately 15 strokes) and pestle B until the tissue looked homogenized (approximately eight strokes). Samples were then spun through a discontinuous iodixanol gradient. Gradients were made by diluting homogenized sample 1:1 with 50% iodixanol solution for a final concentration of 25% iodixanol (60% iodixanol solution + HBS) and were overlayed on a 30%–35% iodixanol gradient in 15-mL conical tubes (30% iodixanol solution: 1× HBS, 30% iodixanol solution, 160 mM sucrose; 35% iodixanol solution: 1× HBS, 35% iodixanol solution, 160 mM sucrose). Gradients were spun at 5000g for 20 min at 4°C with no break in swinging bucket centrifuge. Nuclei were collected at the 30%–35% interface, transferred to a new tube, and diluted with 1 × HBS. Samples were spun for 10 min at 4°C 1000g, aspirated, then resuspended in 200 μL 1 × HBS. Nuclei were then stained for RBFOX3 (also known as NeuN; NeuN-488, EMD Millipore MAB377X) and incubated 30 min in the dark. DNA was counterstained with Hoechst 33342 and sorted on RBFOX3+ to collect neurons (to avoid glia). Five thousand nuclei were collected for most samples, although some variation was present. Additional processing, library preparation, and ATAC sequencing was performed as for B/T cells following the protocol described by Luperchio et al. (2021), adjusted for amplification cycles for ATAC.
ATAC-seq: mapping and peak calling
Raw ATAC-seq data were processed as follows. Sequencing reads from the FASTQ files were mapped to the mouse genome (assembly mm10) using Bowtie 2 (Langmead and Salzberg 2012); duplicate reads were removed with Picard (via the “MarkDuplicates” function); and mitochondrial reads were removed with SAMtools (Li et al. 2009). Then, separately for each cell type, we identified genotype-specific peaks as follows. First, we merged the BAM files corresponding to samples of each genotype (KS1, KS2, wild type; one sample from each mouse) using the “merge” function from SAMtools. This created three “metasamples,” each one corresponding to a single genotype (the wild-type mice from both the KS1 cohorts and the KS2 cohort were merged into the same “metasample”). In this merging step, we also included the KS1 and wild-type samples from the replication cohort (see “ATAC-seq: differential accessibility analysis” section). We subsequently called peaks from the three metasamples with MACS2 (Zhang et al. 2008), with the “keep-dup” parameter set to “all.”
To orthogonally validate the peak locations and ATAC signal intensities we detected in neurons, we first examined ATAC-seq data from the postnatal (day P0) mouse forebrain (because the hippocampus is part of the forebrain). These data were generated by Gorkin et al. (2020) as part of the ENCODE Project and can be obtained at https://hgdownload.soe.ucsc.edu/gbdb/mm10/encode3/atac/. They consist of a set of genomic regions, with each region having a corresponding ATAC signal score. We found that 98% of KS1, 98.5% of KS2, and 97.8% of the wild-type neuronal peaks overlap ENCODE regions that have scores greater than zero. This supports the validity of our peak locations. To then assess the validity of the ATAC signal intensity at these locations, we restricted to ENCODE regions that have scores greater than zero and stratified them according to whether they (1) overlap at least one of our ATAC peaks at promoters, (2) overlap at least one of our ATAC peaks outside promoters, or (3) do not overlap any of our peaks. Examining the scores of the stratified regions, we found that, in each of the genotypes, promoter-overlapping regions have a strong signal, and non-promoter-overlapping regions have a weaker (as expected) but still pronounced signal (Supplemental Fig. S12A). In contrast, ENCODE regions that do not overlap any of our peaks show a very weak signal (Supplemental Fig. S12A).
As a final validation, we compared the ATAC peaks we detected in our samples to the peaks detected by Su et al. (2017) (obtained from the NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] under accession number GSE82010). This study is well suited as a comparison, as it also examines the dentate gyrus of the mouse hippocampus. To ensure a robust comparison, we downloaded the raw FASTQ files corresponding to the wild-type mice, and we processed them in an identical manner as our own FASTQ files. We then called peaks after creating a single wild-type “metasample” by merging all BAM files together. We found that the vast majority of the ATAC peaks identified by Su et al. (2017) are detected in our samples as well (96.4% overlap wild-type peaks, 97.4% overlap KS1 peaks, and 90.4% overlap KS2 peaks) (Supplemental Fig. S12B).
Together, these results provide strong evidence that our neuronal ATAC-seq peaks represent true neuronal regulatory elements in all three of the genotypes.
ATAC-seq: differential accessibility analysis
For neurons and T cells, the differential accessibility analyses were performed separately, as follows. First, we unionized the three sets of peaks called from the three metasamples (in which each metasample corresponded to one genotype; see previous section) and excluded peaks which had at least 1-bp overlap with ENCODE blacklisted regions (obtained from GitHub [https://github.com/Boyle-Lab/Blacklist/archive/v2.0.zip] [Amemiya et al. 2019] for neurons, whereas an earlier version [Carosso et al. 2019] was used for T cells). This yielded a common (across genotypes) set of peaks for each cell type: 172,558 peaks in neurons and 93,446 peaks in T cells (Luperchio et al. 2021). Using this common set of peaks, for each differential analysis (e.g., KS1 vs. wild type in neurons), we created a peak-by-sample matrix by counting the number of reads from each sample that map to each peak with the featureCounts() function from the Rsubread R package (requiring an overlap of at least 3 bp between a read and the corresponding peak location) (Liao et al. 2019). We specified the parameters of featureCounts() so as to exclude reads that map to multiple locations and reads belonging to chimeric fragments. The resulting counts served as the entries of the peak-by-sample matrix. Using the peak-by-sample matrix, we excluded peaks with median count (across samples) of fewer than 10. Finally, we performed the differential analysis with DESeq2 (Love et al. 2014), after adjusting for unknown confounding variables estimated via surrogate variable analysis (Leek and Storey 2007). For B cells, we obtained the differential accessibility results from Luperchio et al. (2021) but also included peaks that had been excluded from the analysis therein owing to adjusted P-values set to NA after independent filtering by DESeq2.
To validate our differential accessibility results in KS1 neurons, we first examined a replication cohort, consisting of three wild-type and three KS1 mice. Even though the smaller sample size meant we had lower power compared with the original differential analysis, we estimated that 73% of the top disrupted promoter regulatory elements in the original analysis (smallest 10% of P-values) are differential in the replication cohort based on their P-value distribution (1 − π0). Furthermore, in 95% of these promoter elements the direction in which accessibility changes is consistent between the original and the replication analysis. In stark contrast, only 11% of the promoter elements with the highest 10% of P-values in the original differential analysis are estimated to be differential in the replication analysis, with only 54% showing accessibility change in the same direction (essentially equivalent to random chance). This shows the robustness of the differential accessibility signal at promoters. We also found clear, albeit weaker, evidence for replication at regulatory elements outside promoters, with 76% of nonpromoter elements with a Q-value less than 0.1 changing accessibility in the same direction in the replication cohort, compared with 52.2% of nonpromoter elements with a Q-value greater than 0.8 (again essentially reflecting random chance).
As an additional validation, we stratified our ATAC peaks based on whether they overlap regions where KMT2D has been previously shown to bind in ht22 cells (via ChIP-seq) (Carosso et al. 2019). This is a mouse hippocampal neuronal cell line and thus provides a reasonable comparison. We found that ATAC peaks overlapping KMT2D ChIP-seq peaks have a higher probability of showing disrupted accessibility compared with other ATAC peaks, as evidenced by the corresponding P-value distributions (see also next section). This is true both for ATAC peaks at promoters and for ATAC peaks outside promoters (Supplemental Fig. S13). It provides orthogonal support for our differential results and indicates the enrichment of direct KMT2D targets among regulatory elements showing disruption.
Finally, we found a strong signal of preferential disruption of the same regulatory elements in KS1 and KS2 neurons (Supplemental Fig. S14).
Comparing the chromatin disruption between different cell types
Using the results of the differential accessibility analyses, we compared the chromatin disruption between cell types using the approach developed by Luperchio et al. (2021) (where it was applied for comparisons between genotypes). This approach involves the following steps:
-
Obtain ATAC peaks differentially accessible in B cells as well as ATAC peaks not differentially accessible in B cells (i.e., the rest of the B cell peaks).
-
Obtain neuronal (or T cell) peaks that have an overlap of at least 1 bp with the differential B cell peaks from step 1.
-
From the differential accessibility analysis in neurons (or T cells), obtain the P-values corresponding to the peaks from step 2.
-
Apply Storey's method (Storey 2003; Storey and Tibshirani 2003) to the P-values from step 3 to estimate the proportion (1 − π0) of peaks from step 2 that are differential in neurons (or T cells). π0 is calculated using the pi0est() function from the “qvalue” R package, with the “pi0.method” argument set either to “bootstrap” or to the default with specified λ (see below).
-
Test whether the proportion estimated in step 4 is statistically significant: Derive a null distribution by repeatedly (10,000 times) sampling random sets of B cell peaks and estimating the neuronal (or T cell) 1 − π0 from these peaks in an identical manner as for the B cell differential peaks in steps 2–4. The number of peaks contained in each randomly sampled set is chosen to be equal to the number of differential B cell peaks.
-
For visualization purposes (Fig. 1, 2; Supplemental Fig. S2), repeat steps 2 and 3 to also obtain P-values corresponding to neuronal (or T cell) peaks that have an overlap of at least 1 bp with peaks nondifferential in B cells. Then, compare the distribution of the neuronal (or T cell) P-values corresponding to differential B cell peaks to the distribution of the neuronal (or T cell) P-values corresponding to nondifferential B cell peaks. These distributions are plotted using the density() function in R.
Intuitively, in any given cell type, the greater the difference in the height of the peak close to zero when comparing the P-value distribution corresponding to peaks differential in another cell type to the P-value distribution of peaks nondifferential in another cell type, the greater the signal of preferential disruption of the same regulatory elements in the two cell types.
Step 1 in the above procedure requires us to choose a threshold in order to define differentially accessible ATAC peaks in B cells. For Figure 2, we used a Q-value threshold of 0.1. To verify that our findings are not dependent on this specific B cell threshold, we performed the same analysis, but this time in step 1, we started with the differential accessibility results in neurons and T cells instead of B cells. That is, we obtained differentially accessible regulatory elements in neurons (Q-value < 0.1) and T cells (Q-value < 0.1). We subsequently examined the P-value distributions of these peaks in B cells and compared them to the P-value distributions corresponding to the rest of the neuronal and T cell peaks. Our results unambiguously recapitulated our original findings: There is preferential chromatin accessibility disruption of the same regulatory elements in B and T cells but not in B cells and neurons (Supplemental Fig. S2). Similarly, there is no preferential disruption of the same regulatory elements when starting with neurons and comparing them to T cells (P > 0.9 in all cases).
With regards to the null distributions corresponding to Figure 2, as stated in step 5 above, in each case we estimated both the observed 1 − π0 and the null distributions in an identical manner to ensure a fair comparison. We used either the bootstrap method as implemented in the pi0est() function or, when the bootstrap failed to yield an estimate for the null π0 during the permutations (likely owing to the paucity of large P-values), the default method with a specified value for the “lambda” parameter. We used λ = 0.5 for the KS2 comparisons in Figure 2C,D,G,H, and for the KS1 promoter comparisons when taking directionality into account (Fig. 2I). We have empirically observed that this value for λ generally yields estimates consistent with these of the bootstrap method, with smaller λ values being more conservative. The only exception was the KS2 B cell versus neuron comparison when starting with ATAC peaks more compact in mutant B cells, where we used λ = 0.4 as values 0.5 and 0.45 yield negative π0’s during the permutations. In all cases, the same λ was used to calculate both the observed value and the permutations.
Finally, for our analysis of the concordance in the direction of accessibility disruption between cell types (section “The most significant chromatin accessibility changes in B cells are present, but are very subtle, in neurons”), the null distributions were derived as follows. We repeatedly (1000 times) sampled random sets of B cell peaks. The sampling was performed so that, within each random set, (1) the balance of peaks with increased versus decreased accessibility in the mutant versus wild-type mice was the same as in the set of B cell differential peaks (see above for defining differential peaks), and (2) the total number of peaks sampled was equal to the number of B cell differential peaks. For each random set of B cell peaks, we obtained overlapping neuronal or T cell peaks (overlap of at least 1 bp) and examined the proportion of these peaks whose accessibility changes in the same direction as in the B cell differential peaks. These random proportions formed the null distribution.
Mouse promoter and enhancer coordinates
We defined promoters as regions ±2 kb from the TSS. We obtained coordinates of such promoter regions in mm10 using the promoters() function from the EnsDb.Mmusculus.v79 R package, with the “upstream” and “downstream” parameters set to 2000. In cases of genes with multiple alternative promoters, all of these were included.
We defined enhancers as ATAC peaks within 100 kb from the TSS, except these within the promoter sequence (defined as above). Although this definition excludes some enhancers that act at longer distances from their target genes, we reasoned that it enriches for true enhancer–gene pairs because distance from TSS is a strong predictor of whether an enhancer regulates a given gene. In cases in which multiple TSSs were within the 100-kb sequence downstream from or upstream of an enhancer, we considered all of the corresponding genes as target genes for that enhancer. To examine the direction of accessibility disruption at enhancers near disrupted promoters (Supplemental Fig. S1), we first obtained a single promoter P-value for each gene by selecting the peak with the smallest P-value out of the peaks that overlap the gene's promoter (or the gene's multiple promoters, in cases in which there are more than one annotated TSSs). We then obtained the genes with the smallest and highest 10% of promoter P-values, and examined enhancers within 100 kb from the TSS of these genes as described above (Supplemental Fig. S1, orange vs. pink densities). Promoters were split into those more compact and those more open in mutants based on the log2 (fold change) of the peak with the smallest P-value. Enhancers near promoters that are not disrupted (highest 10% of P-values) served as controls.
RNA-seq: cell and RNA isolation
Each sample whose RNA was extracted and sequenced was an aliquot of the same cell harvest, from the same mouse, as the sample used for ATAC-seq. For B cells, RNA isolation and sequencing are as previously described (Luperchio et al. 2021). For T cells, RNA was isolated and sequenced in exactly the same way.
RNA-seq: mapping and differential analysis
For both B and T cells, we first used Salmon to build an index and pseudoalign the raw RNA-seq reads (FASTQ files) to a FASTA file (http://uswest.ensembl.org/Mus_musculus/Info/Index, version 91, downloaded January 2018) containing all mouse cDNA sequences from Ensembl. Following pseudoalignment and transcript quantification, we used the tximport R package to import the quantifications into R and obtain gene-level counts. Subsequently, using the resulting gene-by-sample matrix, we performed the differential expression analysis in an identical manner as the differential accessibility analysis for ATAC-seq. This is also the same procedure we used for the differential expression analysis in B cells in the work by Luperchio et al. (2021). However, in downstream analyses here we kept genes whose adjusted P-values were set to NA by independent filtering in DESeq2.
For our analysis examining the percentage of differentially expressed genes when varying the threshold for promoter differential accessibility (Fig. 4A), we associated each promoter with the smallest P-value out of all the P-values corresponding to peaks in that promoter, as described earlier. At each threshold applied to the rank of these promoter P-values (Fig. 4A–D, x-axis), we estimated the proportion of differentially expressed genes from their P-value distribution (1 − π0). For our analysis of the concordance between promoter accessibility changes and gene expression changes (Fig. 4B; Supplemental Figs. S6, S7), for each promoter we used the log2 (fold change) corresponding to the ATAC peak with the smallest P-value.
For Figure 4F, we calculated the enrichment as the odds ratio. We applied a P-value rank threshold of 500 for CpG island genes (i.e., we compared the genes whose promoter accessibility P-value rank was in the lowest 500 to genes whose promoter accessibility P-value rank was in the highest 500) and a rank threshold of 3000 for EM genes. These rank thresholds were chosen after starting from 500 and proceeding in steps of 250 until the first threshold that yielded an odds ratio not equal to infinity. The enrichment analysis for the top versus bottom differentially expressed genes in B and T cells was performed with a rank threshold of 500.
Mouse orthologs of human episignature genes
We obtained the coordinates of differentially methylated CpG positions (DMPs) in KS1 patients versus controls from Aref-Eshghi et al. (2017). We then used the EnsDb.Hsapiens.v75 R package to obtain the coordinates of human promoters in hg19, and restricted to promoters that contain at least one DMP. To minimize false-positive associations between promoters and DMPs, here we defined promoters as regions ±500 bp from the TSS. We mapped the Ensembl IDs of the genes corresponding to these promoters to their mouse ortholog Ensembl IDs using the biomaRt R package (with the “mmusculus_homolog_orthology_confidence” parameter set to one). To perform the analysis, we then obtained gene-level P-values for these genes as described in the “Mouse promoter and enhancer coordinates” section above.
CpG islands
We obtained coordinates of CpG islands in the mouse genome (mm10 assembly) using the ucscTableQuery() function from the “rtracklayer” R package. We defined ATAC peaks that overlap CpG islands as peaks with at least one overlapping base pair.
As expected, peaks overlapping CpG islands have higher read counts compared with peaks that do not. Because the number of read counts can be inversely related to the uncertainty around log fold-change estimates, we sought to verify that the strong quantitative relationship between probability of CpG island overlap and differential accessibility P-value (Fig. 3) is not driven by the higher read counts of CpG island–overlapping peaks in the setting of lower overall counts in neurons (across-peak median average normalized read count = 151.4 and 124.2 in KS1 and KS2, respectively) versus B (across-peak median average normalized read count = 368 and 400.4 in KS1 and KS2, respectively) or T cells (across-peak median average normalized count = 296.2 and 302.8 in KS1 and KS2, respectively). We downsampled the BAM files corresponding to the B and T cell samples to 25% of the initial number of reads, generated new peaks-by-samples matrices using the same procedure as before, and performed a new differential accessibility analysis. We found that, even with overall counts similar to these in neurons, there is still no relationship between probability of CpG island overlap and differential accessibility P-value in B or T cells; this is true both in KS1 (1 − ratio of deviance to null deviance = 1.58 × 10−5 in B cells and 1.57 × 10−5 in T cells before downsampling versus 7 × 10−5 and 10−3 after downsampling) and KS2 (1 − ratio of deviance to null deviance = 0.003 in B and T cells before downsampling versus 8 × 10−4 in B cells and 2 × 10−4 in T cells after downsampling).
To identify mouse CpG islands bound by the Polycomb Repressive Complex 2 (PRC2), we first identified locations bound by EZH2, the catalytic subunit of the PRC2 complex, in the human genome. To do this, we used the ucscTableQuery() function from the “rtracklayer” R package to query the UCSC Table Browser. Specifically, from the “Txn Factor ChIP” track (which belongs to the “Regulation” group), we obtained the “wgEncodeRegTfbsClusteredV3” table. This provided us with a set of genomic locations and their coordinates, with each location having an associated score. The maximum possible score was 14. That score represents the number of EZH2 ChIP-seq peaks detected in that location, after uniform processing of ENCODE ChIP-seq experiments from 14 cell lines (H1-hESC, embryonic stem cells; HeLa-S3, cervical carcinoma; HMEC, mammary epithelial cells; HSMM, skeletal muscle myoblasts; NH-A, astrocytes; NHDF-Ad, dermal fibroblasts; NHEK, epidermal keratinocytes; NHLF, lung fibroblasts; Dnd41, T cell leukemia with Notch mutation; GM12878, lymphoblastoid; HepG2, hepatocellular carcinoma; HSMMtube, skeletal muscle myotubes differentiated from the HSMM cell line; HUVEC, umbilical vein endothelial cells; and K562, lymphoblasts). To minimize the inclusion of potential false positives in our analysis, we restricted to locations having at least five EZH2 peaks (2776 regions). We subsequently identified human genes whose promoters overlap these locations (i.e., have at least 1 bp in common) using the promoters() function from the “EnsDb.Hsapiens.v75” R package. Then, we obtained mouse orthologs of these human genes using the “biomaRt” R package. We only selected high-confidence ortholog pairs by setting the “mmusculus_homolog_orthology_confidence” parameter equal to one. This yielded a total of 1515 mouse orthologs. Finally, we identified mouse CpG islands that overlap the promoters (±2 kb from the TSS) of these mouse orthologs.
Normalized ATAC signal plots
The BAM files corresponding to the genotype-specific “metasamples” were indexed (using SAMtools) and converted to bigWig files using deepTools (Ramírez et al. 2016; see https://deeptools.readthedocs.io/en/develop/). Reads were CPM-normalized also using deepTools (bamCoverage –normalizeUsing CPM). Mouse transcript annotations and CpG island annotations (both from mm10) were obtained from UCSC. All plots were then generated using the karyoploteR R package. We plotted regions between 5 kb upstream of and 8 kb downstream from the selected promoters using the plotKaryotype() function. The normalized ATAC signal tracks were plotted with the kpPlotBigWig() function, from the bigWig files. Gene tracks were added by addGeneNames() and mergeTranscripts() and plotted with the kpPlotGenes() function. CpG islands overlapping the promoter regions were added to the plot by using kpPlotRegions(). Labels and text positions were determined by the “r0” and “r1” parameters in karyoploteR.
EM genes
EM genes and their coexpression status (highly coexpressed vs. noncoexpressed) were obtained from Boukas et al. (2019). This catalog of EM genes was compiled based on the presence of specific protein domains known to confer biochemical functions central to epigenetic regulation: writing/erasing/reading epigenetic marks and/or chromatin remodeling (see also http://www.epigeneticmachinery.org). It was also shown that there is a subset of “highly coexpressed” EM genes whose expression levels are correlated (across individuals) with those of many other EM genes in multiple different tissues (Boukas et al. 2019). Noncoexpressed EM genes are these whose expression is not correlated with the expression of other EM genes.
Pathway analysis
For the pathway analysis in KS1 and KS2 neurons, we defined differential genes as those with P-values in the bottom 5%. We obtained gene-level P-values as described above in the “Mouse promoter and enhancer coordinates” section. We subsequently used these differential genes and the Reactome pathway annotations (Fabregat et al. 2018) to identify disrupted pathways with the “goseq” R package (Young et al. 2010), comparing differential genes to all genes that were assigned a P-value in the differential accessibility analysis (this excludes, e.g., genes whose promoter regulatory elements were filtered out because of very low ATAC-seq counts, suggesting inactivity in neurons; see “ATAC-seq: differential accessibility analysis” section).
Reactome pathway annotations and the “goseq” R package were also used for the pathway analysis of genes with shared disruption in all three cell types, separately in KS1 and KS2. These genes were those whose promoters contained regulatory elements with shared disruption in all three cell types, as described in the section “Identification of regulatory elements disrupted in all three cell types” below. They were compared against all genes that were assigned a P-value in all three differential accessibility analyses.
Mouse epigenetic clock CpGs and mouse longevity genes
We obtained mouse epigenetic clock CpGs from Thompson et al. (2018). Specifically, we used the “elastic net clock,” which is composed of 582 CpGs. This clock is a linear model whose predictors are the methylation values of these 582 CpGs. It was shown by Thompson et al. (2018) that the age predicted by this clock correlates strongly with chronological age across a diverse set of tissues, including blood and cerebral cortex.
To examine the chromatin disruption at promoters of genes associated with lifespan regulation in mammals, we used the KEGGREST R package to obtain genes in the “longevity regulating pathway” (pathway: mmu04213). We then analyzed peaks within promoters (±2 kb) of these genes. For Supplemental Fig. S8, the gene-level P-values were obtained as described in the “Pathway analysis” section above.
Identification of regulatory elements disrupted in all three cell types
In both KS1 and KS2, we adopted the following stepwise strategy. We started by obtaining ATAC peaks disrupted in B cells (Q-value < 0.1). Among these genes, we then selected peaks disrupted in neurons as well (at 10% FDR level) using the qvalue() function from the “qvalue” R package (Storey and Tibshirani 2003). Finally, from the resulting peaks, we selected these disrupted in T cells (10% FDR), again using the qvalue() function.
Genome assembly
All our analyses were conducted using the mm10 mouse genome assembly as reference. Our focus is predominantly on promoters throughout, which are well-annotated regions of the genome with well-characterized sequences in mm10. In addition, we have performed several QC checks validating our ATAC peaks in coding and noncoding genomic regions. Thus, use of the latest GRCm39 mouse reference genome would not be expected to significantly impact our conclusions. In terms of human genome assembly, we used the hg19 assembly to obtain promoter coordinates containing differentially methylated CpGs as described in the section “Mouse orthologs of human episignature genes,” because this is the assembly in which the coordinates of the differentially methylated CpGs were provided.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE239339 (superseries containing the neuron and T cell ATAC-seq and RNA-seq data as subseries). Code for all analyses can be found at GitHub (https://github.com/hansenlab/mdem_neuron_paper) and in the Supplemental Materials. Code was executed using various versions of R; the most recent one was 4.3.2 (R Core Team 2023).
Competing interest statement
H.T.B. is a paid consultant for Mahzi Therapeutics. The other authors declare no competing interests.
Acknowledgments
H.T.B. and T.R.L. were supported by a grant from the Louma G. Foundation. H.T.B. was also supported by the Icelandic Research fund (217988, 195835, 206806) and the Icelandic Technology Development fund (2010588). Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of the Health under award number R01GM121459 and CZF2019-002443 from the Chan Zuckerberg Initiative DAF, an advised fund of the Silicon Valley Community Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions: L.B., T.R.L., K.D.H., and H.T.B. conceived of the study and designed it. L.B. performed analyses. T.R.L. performed experiments. L.B. performed data visualization, with the exception of the normalized ATAC signal plots, which were performed by A.R. L.B. wrote the initial draft of the manuscript, which was reviewed and edited by L.B., T.R.L., K.D.H., and H.T.B.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278416.123.
- Received August 17, 2023.
- Accepted April 15, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








