
Single-cell spatial transcriptomics reveals a dystrophic trajectory following a developmental bifurcation of myoblast cell fates in facioscapulohumeral muscular dystrophy
- Lujia Chen1,2,8,
- Xiangduo Kong3,8,
- Kevin G. Johnston4,8,
- Ali Mortazavi3,
- Todd C. Holmes2,5,
- Zhiqun Tan2,3,4,6,
- Kyoko Yokomori3 and
- Xiangmin Xu1,2,4,6,7
- 1Department of Biomedical Engineering, University of California, Irvine, California 92697, USA;
- 2Center for Neural Circuit Mapping, University of California, Irvine, California 92697, USA;
- 3Department of Biological Chemistry, School of Medicine, University of California Irvine, Irvine, California 92697, USA;
- 4Department of Anatomy and Neurobiology, School of Medicine, University of California Irvine, Irvine, California 92697, USA;
- 5Department of Physiology and Biophysics, School of Medicine, University of California Irvine, Irvine, California 92697, USA;
- 6Institute for Memory Impairments and Neurological Disorders, University of California, Irvine, California 92697, USA;
- 7Department of Computer Science, University of California, Irvine, California 92697, USA
-
↵8 These authors contributed equally to this work.
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is linked to abnormal derepression of the transcription activator DUX4. This effect is localized to a low percentage of cells, requiring single-cell analysis. However, single-cell/nucleus RNA-seq cannot fully capture the transcriptome of multinucleated large myotubes. To circumvent these issues, we use multiplexed error-robust fluorescent in situ hybridization (MERFISH) spatial transcriptomics that allows profiling of RNA transcripts at a subcellular resolution. We simultaneously examined spatial distributions of 140 genes, including 24 direct DUX4 targets, in in vitro differentiated myotubes and unfused mononuclear cells (MNCs) of control, isogenic D4Z4 contraction mutant and FSHD patient samples, as well as the individual nuclei within them. We find myocyte nuclei segregate into two clusters defined by the expression of DUX4 target genes, which is exclusively found in patient/mutant nuclei, whereas MNCs cluster based on developmental states. Patient/mutant myotubes are found in “FSHD-hi” and “FSHD-lo” states with the former signified by high DUX4 target expression and decreased muscle gene expression. Pseudotime analyses reveal a clear bifurcation of myoblast differentiation into control and FSHD-hi myotube branches, with variable numbers of DUX4 target-expressing nuclei found in multinucleated FSHD-hi myotubes. Gene coexpression modules related to extracellular matrix and stress gene ontologies are significantly altered in patient/mutant myotubes compared with the control. We also identify distinct subpathways within the DUX4 gene network that may differentially contribute to the disease transcriptomic phenotype. Taken together, our MERFISH-based study provides effective gene network profiling of multinucleated cells and identifies FSHD-induced transcriptomic alterations during myoblast differentiation.
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common inherited muscular dystrophies and is characterized by progressive weakening of the facial, shoulder girdle, and upper arm muscles, as well as lower extremity muscles (Tawil and Van Der Maarel 2006; Tihaya et al. 2023). Affected muscles show variability in muscle fiber size and replacement of muscle by fat tissue and fibrosis (Serra and Wagner 2020). Two types of FSHD have been identified: type 1 (FSHD1; MIM 158900) and type 2 (FSHD2; MIM 158901) (Wang and Tawil 2021). FSHD1 represents the majority of FSHD cases (>95%) and is linked to the monoallelic contraction of the D4Z4 macrosatellite repeat (a 3.3-kb repeat containing an open reading frame for the double-homeobox transcription factor DUX4 gene) array on Chromosome 4q to fewer than 10 repeats. FSHD2, which has modest D4Z4 contraction (11–20 units), has been linked to mutations of chromatin modifiers such as SMCHD1 (>95%), DNMT3B, and LRIF1 (Tihaya et al. 2023). The human DUX4 is expressed in early embryos and testes, as well as cancers, and is normally silenced in the somatic cells of healthy humans. Individuals with a 4qA haplotype and a noncanonical polyadenylation signal sequence distal to the last D4Z4 repeat express a full-length DUX4 transcript (DUX4fl) and develop FSHD (Lemmers et al. 2010).
DUX4 target genes have been identified in previous studies (Geng et al. 2012; Yao et al. 2014; Choi et al. 2016; Banerji et al. 2017; Resnick et al. 2019; Jiang et al. 2020), and activation of these DUX4 target genes has been observed in patient muscle cells in multiple studies, supporting the significance of DUX4fl in FSHD to the extent that these target genes are used as markers for the FSHD phenotype (Geng et al. 2012; Jones et al. 2012; Rahimov et al. 2012; Broucqsault et al. 2013; Ferreboeuf et al. 2014; Rickard et al. 2015). However, the DUX4fl transcript is expressed at an extremely low level (not detectable by bulk RNA-seq) in patient muscle cells (Jones et al. 2012). The questions of how limited DUX4 expression impacts disease pathology and how dysregulation of DUX4 target genes contributes to the disease process are not well understood, although recent studies indicate the significant correlation between the clinical phenotype and DUX4 target expression (Wang et al. 2019; Wong et al. 2024). Recent single-cell (sc) and single-nucleus (sn) RNA-seq studies have begun to address these questions (van den Heuvel et al. 2019; Jiang et al. 2020; Zheng et al. 2024). Our previous snRNA-seq analyses indicate heterogeneity of patient myotube nuclei and identify two discrete nuclei clusters, “FSHD-hi” and “FSHD-lo” (Jiang et al. 2020). The former represents nuclei with high expression of DUX4 targets and the latter with low target expression, yet distinct from control non-FSHD nuclei. The prevalence of these gene targets strongly suggests disease-specific changes beyond the rare DUX4-positive nuclei (Jiang et al. 2020).
Although both fusion-inhibited scRNA-seq and snRNA-seq analyses provided important insights, neither of these approaches is suitable for simultaneous analysis of gene profiles of intact myotubes and their incorporated nuclei (Kim et al. 2020; McKellar et al. 2021; Williams et al. 2022). Therefore, to complement our snRNA-seq study, we also performed in situ detection of DUX4 and several representative target gene transcripts using RNAScope (Jiang et al. 2020; Chau et al. 2021). In conjunction with shRNA depletion experiments, these studies provide important evidence for the roles of DUX4 target transcription factors in complex cross-regulation of the DUX4 gene network (Jiang et al. 2020; Chau et al. 2021). Although in situ codetection analyses have yielded new insights into DUX4 target gene expression, only three genes were analyzed in these prior studies (Chau et al. 2021).
The recently developed single-cell spatial transcriptomics technology multiplexed error-robust fluorescence in situ hybridization (MERFISH) can profile spatial locations of hundreds to thousands of RNA species at subcellular resolution (Chen et al. 2015). In this study, we report the first usage of MERFISH for FSHD analysis. We perform MERFISH using a custom-designed 140-gene probe set, including 24 previously identified DUX4 target genes and subclasses of genes shown to be affected in FSHD patient myocytes (Yao et al. 2014; Choi et al. 2016; Banerji et al. 2017; Resnick et al. 2019; Kim et al. 2020). Critically, MERFISH enables combined transcriptomic analysis of both nuclei and the full myotubes containing those nuclei, a feature that cannot be studied using either scRNA-seq or snRNA-seq.
We used an immortalized control, a CRISPR-engineered isogenic D4Z4 contraction mutant (DEL5) (Kong et al. 2024), and an FSHD1 patient myoblast line (Chau et al. 2021), which were allowed to differentiate into myotubes for 3–4 d, capturing both myotubes and unfused mononucleocytes (MNCs). The DEL5 line is an isogenic mutant of the control cell line used in this study, harboring contraction of D4Z4 repeats (Kong et al. 2024). Using these cell lines, we aim to simultaneously characterize DUX4 target gene expression in both the nuclei and cytoplasm of differentiating myotubes and analyze alterations of differentiation trajectory and gene coexpression networks in the FSHD and DEL5 cell lines relative to the control.
Results
MERFISH spatial transcriptomics data are validated by previous bulk RNA-seq data, refined within cellular and structural boundaries
We applied MERFISH to in vitro differentiated immortalized skeletal muscle cell samples of three genotypes: control (four samples), isogenic D4Z4 contraction mutant (DEL5; three samples), and FSHD1 patient-derived (FSHD; four samples) (Fig. 1A). Expression of DUX4 target genes is known to be highly variable in FSHD patient cells (Cowley et al. 2023), so we screened and chose an FSHD1 (the most representative FSHD genotype) cell line showing high expression of DUX4 target genes as a disease prototype for this study. The DEL5 mutant cell line was derived from the control cell line used in this study by deleting D4Z4 repeats down to one copy at the 4qA allele using CRISPR-Cas9, thus serving as an isogenic D4Z4 mutant for comparison (Kong et al. 2024). DEL5 mutant cells upregulate DUX4 target genes in a DUX4-dependent fashion, although the level of DUX4 target gene upregulation is not as robust as that of the FSHD patient cells used in the current study. Our MERFISH gene panel (see Supplemental Table S1) includes differentially expressed marker genes for FSHD identified by previous studies, including 24 DUX4 target genes (Geng et al. 2012; Yao et al. 2014; Jiang et al. 2020; Tihaya et al. 2023).
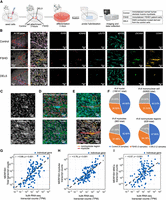
Spatial transcriptomic profiling of differentiated control and FSHD patient/mutant myoblasts by MERFISH. (A) Schematic diagram of the sample preparation process for MERFISH experiments and the genotypes used in the study. (B) Spatial distribution of detected genes by MERFISH. (Column 1) Illustration of the original sample image (cropped). All 140 genes used in the study are illustrated. (Column 2) The magnified example region corresponds to the red square area on the left. (Columns 3–8) Distribution of six example genes in the magnified area. (C) Staining results of the magnified region in B. (Top) DAPI staining of the nuclei in the region; (bottom) cell boundary staining of the region that illustrates the cytoplasm. (D) Automatically segmented mononuclear cells (MNCs) and nuclei inside the magnified region of B. The background is the overlay of cell boundary staining and DAPI staining. (Top) Detected MNCs inside the area. MNCs are circled with red lines. (Bottom) Detected nuclei within the region. Nuclei are circled with red lines. (E) Manually labeled myotube and nonmyotube regions (top), and the corresponding spatial transcriptome illustration with all 140 genes (bottom) in the magnified region in B. (F) Composition of nuclei, MNCs, myotube, and nonmyotube of the different genotypes in this study. (G–I) Comparison of MERFISH results with bulk RNA-seq of control myoblasts at day 3 of differentiation. Comparisons between the log-transformed total transcript counts from MERFISH and total transcript counts of all myocytes from bulk RNA-seq (G), intra-myotube transcript counts from MERFISH and total counts from bulk RNA-seq (H), and intra-MNC transcript counts from MERFISH and total counts from bulk RNA-seq (I). Overall MERFISH detections show a high level of correlation with the detection results from bulk RNA-seq and scRNA-seq methods (G) Pearson's r = 0.66, P < 0.001; (H) Pearson's r = 0.78, P < 0.001; (I) Pearson's r = 0.57, P < 0.001; N = 140 genes.
We first visualized the spatial distribution of all 140 detected genes, including six selected genes of interest, primarily DUX4 target genes and the myogenesis marker gene MYOG (Fig. 1B). MYOG shows high enrichment inside the cellular structure across all three genotypes, whereas DUX4 target genes such as LEUTX, ZSCAN4, and CCNA1 show high expression in the FSHD and DEL5 samples but not the control samples. This indicates that detected transcripts illustrate the condition specificity of FSHD-related genes (Fig. 1B). Analysis of cell boundary staining enables visual identification and annotation of myotube cell structures (Fig. 1C; see also Supplemental Fig. 1).
We focus on studying three classes of biological structures in these samples: nuclei, mononuclear cells (MNCs; consisting of myoblasts and single nucleated myocytes), and myotubes (large multinucleated myocytes). We use the term “myocytes” for muscle cells, which include myoblasts, MNCs and myotubes, in general in this paper. We first use Cellpose, a deep-learning-based cell identification software (Stringer et al. 2021), to extract MNCs and nuclei from DAPI staining images directly (for accuracy and sensitivity measurements, see Supplemental Fig. 1K,L). After subsequent quality control (see Methods), 92,805 MNCs and 156,141 nuclei are extracted for the following analyses (Fig. 1D,F). Myotubes are manually annotated based on cell boundary staining and the MERFISH 140 gene ensemble expression profile. To provide a proper control toward myotube structures, we also define nonmyotube regions, which are contiguous areas containing only MNCs with approximately the same area as a typical myotube for comparison purposes (Fig. 1E). Overall, 983 myotubes and 629 nonmyotube areas are defined for the following analyses (Fig. 1F).
We then compute myotube fusion levels in each sample (Supplemental Fig. 2), defined as the proportion of nuclei contained within myotubes as opposed to MNCs (0.447 ± 0.092, mean ± SD). We also compare pairwise gene expression profiles within the same genotype to ensure consistency between batches (all pairwise gene expression correlates exceed 0.8) (Supplemental Fig. 3).
We next compare the transcript counts of individual genes generated from our MERFISH experiments with corresponding counts generated from bulk RNA-seq samples (Kong et al. 2024) to validate the quality and consistency of the MERFISH experiments. After normalization and log-transformation of the MERFISH transcript counts, we make three types of comparisons: (1) the total transcript counts of all myocytes in MERFISH versus bulk RNA-seq (Fig. 1G), (2) the total intra-myotube transcript counts from MERFISH versus bulk RNA-seq (Fig. 1H), and (3) the total intra-MNC transcript counts from MERFISH versus the total transcript count of all myocytes from bulk RNA-seq (Fig. 1I). Overall, transcript counts returned by MERFISH and bulk RNA-seq show significant positive correlation (Fig. 1G: total transcripts, Pearson's r = 0.66, P < 0.001; Fig. 1H: myotube transcripts, Pearson's r = 0.78, P < 0.001; Fig. 1I: MNC transcripts, Pearson's r = 0.57, P < 0.001; see also Supplemental Fig. 3N) comparable or greater than previous concordances (Liu et al. 2023), indicating that transcriptomic profiles generated by MERFISH in this study are comparable to those generated by RNA-seq. We note that MERFISH myotube transcripts correlate highest with the bulk RNA seq data set, which reflects the higher fusion efficiency of the myocytes used for bulk RNA-seq. As no consensus has been made on the correct normalization technique for bulk RNA comparison (Liu et al. 2023), we also verify that the MERFISH myotube transcripts show high correlation with bulk RNA FPKM (Pearson's r = 0.77).
MERFISH identifies transcriptionally distinct nuclei clusters defined by differential upregulation of DUX4 target genes
We next analyze the expression profiles of individual nuclei across genotypes. The UMAP (McInnes et al. 2020) embedding of the nuclei expression patterns is generally contiguous, and no obvious segregated nuclei subpopulations are noted (Fig. 2A). To obtain biologically meaningful nuclei clusters, we use the scrattch.hicat package (Tasic et al. 2018), an R (R Core Team 2021) package that performs clustering based on differential expression profiles. Clustering analysis reveals two nuclei subpopulations, designated as cluster 0 and cluster 1. Cluster 1 nuclei exist only in FSHD and DEL5 samples and are more common in FSHD than in DEL5 samples, consistent with the milder phenotype of DEL5 (Fig. 2A; Supplemental Figs. 4, 5). Differential expression analysis reveals that 21 of the 24 selected DUX4 target genes are upregulated in cluster 1 nuclei (Fig. 2B,C; Supplemental Fig. 5G,H).
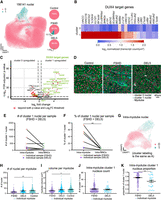
Nuclei clusters show differential expression of DUX4 target genes detected by MERFISH. (A) Two-dimensional projections (UMAP) of gene expression profiles of all identified nuclei. (Left) UMAPs overlaid with the corresponding cluster identities identified by scrattch.hicat algorithm; (right) proportion of each cluster in each genotype. (B) Heatmap illustrating DUX4 target gene expression level of the nuclei clusters. The expression profile for each nucleus cluster is the averaged, area-normalized, log-transformed expression across different samples and genotypes. (C) Volcano plot illustrates the differential expression between the two identified nuclei clusters across all samples. About 88% of DUX4 target genes show upregulation in cluster 1 nuclei, whereas one DUX target gene (ZNF596) shows upregulation in cluster 0 nuclei (Log2FC threshold: 0.5; P-value threshold: 0.001). (D) Examples of DAPI and cell boundary staining overlaid with nuclei, colored with their corresponding cluster identities. Annotated myotubes are circled in cyan. (E) Total number of intra-myotube and intra-MNC cluster 1 nuclei for each FSHD and DEL5 sample. No significant difference was noted in counts between intra-myotube and intra-MNC cluster 1 nuclei (intra-myotube: 551.0 ± 240.7; intra-MNC: 206.0 ± 66.1; mean ± SE; P = 0.3939, Mann–Whitney U test; N = 6 samples). (F) Proportion of intra-myotube and intra-MNC nuclei to be cluster 1 for each FSHD and DEL5 sample. Intra-myotube nuclei contain a higher percentage of cluster 1 nuclei compared with intra-MNC nuclei for all samples (intra-myotube: 0.3570 ± 0.1130; intra-MNC: 0.0316 ± 0.0098; mean ± SE; P = 0.0087, Mann–Whitney U test; N = 6 samples). (G) UMAP of gene expression profiles of intra-myotube nuclei. UMAPs are overlaid with the clusters identified by scrattch.hicat algorithm. (H) Distribution of intra-myotube nuclei counts for different genotypes. Overall, control myotubes show a significantly higher nuclei count compared with FSHD, whereas DEL5 myotubes do not show significantly different nuclei counts compared with FSHD (370 FSHD myotubes: 15.47 ± 0.60; 213 DEL5 myotubes: 24.60 ± 1.29; 404 control myotubes: 27.42 ± 1.07; mean ± SE; control vs. FSHD: P = 0.0092; DEL5 vs. FSHD: P = 0.0996; linear mixed effects model). (I) Distribution of myotube volume by genotype. Overall, myotubes from control and DEL5 samples show higher volume than those in FSHD (370 FSHD myotubes: 11499 ± 381.9 µm2; 213 DEL5 myotubes: 14652 ± 686 µm2; 404 control myotubes: 15768.0 ± 483.9 µm2; mean ± SE; DEL5 vs. FSHD: P = 0. 0174; control vs. FSHD: P = 2.4655 × 10−6; linear mixed effects model). (J) Comparison of intra-myotube cluster 1 nuclei between FSHD and DEL5 genotype myotubes. Distribution of counts of intra-myotube cluster 1 nuclei per myotube. Only myotubes with cluster 1 nuclei are included. No significant differences are noted between the two genotypes (234 FSHD myotubes: 12.54 ± 0.61; 29 DEL5 myotubes: 11.31 ± 1.38; mean ± SE; FSHD vs. DEL5: P = 0.7145, linear mixed effects model). (K) Proportion of cluster 1 nuclei per myotube. The percentage is calculated as the ratio of cluster 1 nuclei counts to total nuclei counts. Only myotubes with cluster 1 nuclei are included. FSHD myotubes show a significantly higher proportion of cluster 1 intra-myotube nuclei compared with DEL5 (234 FSHD myotubes: 0.7470 ± 0.0150; 29 DEL5 myotubes: 0.5705 ± 0.0535; mean ± SE; FSHD vs. DEL5: P = 0.0011, linear mixed effects model).
Motivated by exploratory visualization of the FSHD myotubes, which contain large numbers of cluster 1 nuclei (Fig. 2D), we examine how nuclei are distributed across different types of muscle cells and genotypes. We first compare the total counts and percentage of intra-myotube and intra-MNC cluster 1 nuclei. Among all FSHD and DEL5 samples, four out of six samples show higher counts of intra-myotube cluster 1 nuclei compared with intra-MNC cluster 1 nuclei, although the trend did not reach significance thresholds (intra-myotube: 551.0 ± 240.7; intra-MNC: 206.0 ± 66.1; mean ± SE; P = 0.3939, Mann–Whitney U test) (Fig. 2E). We next compare the percentage of cluster 1 nuclei among the whole intra-myotube or intra-MNC nuclei population. A proportion comparison shows that the intra-myotube nuclei population is significantly more enriched with cluster 1 nuclei (intra-myotube: 0.3570 ± 0.1130; intra-MNC: 0.0316 ± 0.0098; mean ± SE; P = 0.0087, Mann–Whitney U test) (Fig. 2F), which indicates that compared with MNCs, nuclei in myotubes are more enriched in cluster 1 and show higher DUX4 target gene expression.
We then examine the profiles of intra-myotube nuclei across genotypes. UMAP projection of the intra-myotube nuclei's expression profile identifies two segregated regions (Fig. 2G). FSHD samples show a significant trend of having a lower number of nuclei per myotube compared with the control (FSHD: 15.47 ± 0.60; DEL5: 24.60 ± 1.29; control: 27.42 ± 1.07; mean ± SE; control vs. FSHD: P = 0.0092; DEL5 vs. FSHD: P = 0.0996; linear mixed effect modeling) (Fig. 2H). This may be explained by the significantly smaller size of FSHD myotubes compared with the control (FSHD: 11,499 ± 381.9 µm2; DEL5: 14,652 ± 686 µm2; control: 15,768.0 ± 483.9 µm2; mean ± SE; DEL5 vs. FSHD: P = 0.0174; control vs. FSHD: P = 2.4655 × 10−6; linear mixed effects model) (Fig. 2I). Next, we investigate the concentration of cluster 1 nuclei in FSHD and DEL5 myotubes. Overall, FSHD myotubes and DEL5 myotubes are heterogeneous regarding inclusion of cluster 1 nuclei, with no significant difference noted in term of cluster 1 nuclei counts (FSHD: 12.54 ± 0.61; DEL5: 11.31 ± 1.38; mean ± SE; FSHD vs. DEL5: P = 0.7145, linear mixed effects model), but the proportion of cluster 1 nuclei is significantly higher in FSHD myotubes (FSHD: 0.7470 ± 0.0150; DEL5: 0.5705 ± 0.0535; mean ± SE; FSHD vs. DEL5: P = 0.0011, linear mixed effects model) (Fig. 2K). These results reveal the enrichment of genetically characterized nuclei subpopulations inside of multinucleated myotubes, which cannot be discerned by snRNA-seq studies owing to the lack of concomitant nuclei and myotube information (Jiang et al. 2020; Williams et al. 2022).
MERFISH reveals distinct differentiation states of MNC subpopulations
Next, we examine the characteristics of the detected MNCs and their expression profiles as revealed by MERFISH. As observed for nuclei, UMAP embedding of MNC expression patterns is largely contiguous (Fig. 3A). However, clustering reveals two distinct MNC subpopulations (type A and type B MNCs) that contain similar proportions of each genotype (Fig. 3A, pie charts), in contrast to the genotype-specific distributions of nuclear clusters (Fig. 2A). Differential expression analysis illustrates that although some DUX4 target genes are enriched in type B, they do not separate the two MNC types. Instead, four myotube marker genes (NEB, TTN, MYH8, MYH3) are upregulated in type B MNCs across all three genotypes (Fig. 3B,C; Supplemental Fig. 5M–O). We hypothesize based on this gene profile that the MNC clusters are not indicative of FSHD but, instead, reflect differentiation states toward multinucleated myotubes. To test this hypothesis, we pool all the MNCs together with the myotubes and examine their position in the UMAP embedding of their expression profiles. The majority of myotubes overlap with type B MNCs on the UMAP plot regardless of genotype (Fig. 3D). This is consistent with higher expression of myotube marker genes (Fig. 3E, upper panels), suggesting that type B MNCs represent a late-stage prefusion myocyte. The localized positive correlation between differentiation marker and DUX4 target gene expression in mutant/patient MNCs (Fig. 3E, lower panels), combined with the previous nuclei data, indicates that pathological nuclei and myocyte alterations occur relatively late in muscle development.
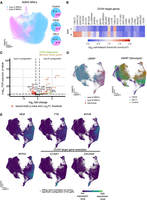
MNC clusters with differential expression of myogenesis and muscle function genes identified by MERFISH. (A) Two-dimensional UMAP projection of gene expression profiles of all MNCs. (Left) UMAP overlaid with the corresponding cluster identities; (right) Composition of each cluster in each genotype. (B) Heatmap illustrating DUX4 target gene expression level of the MNC clusters. The expression profile for each MNC cluster is the averaged, normalized, log-transformed expression across different samples and genotypes. (C) Volcano plot visualizes differential expression between the two identified MNC clusters across all samples. Myoblast and myotube marker genes (TTN, MYH3, MYH8, and NEB) show upregulation in cluster B MNCs (Log2FC threshold: 0.5; P-value threshold: 0.001). (D) Comparison of expression profiles of the two MNC clusters and myotube populations. (Top) UMAP calculated from the expressions of both MNCs and myotubes from all samples. The myotube population mostly overlaps with cluster 1 MNCs in UMAP space. Each dot represents a single MNC or a myotube. (Bottom) UMAP colored by genotype. Each dot represents a single MNC or myotube. (E) UMAP as in D, but overlaid with the normalized expression level of the myotube target genes, as well as two example DUX4 target genes (CCNA1, ZSCAN4). Cells with higher expression levels of TTN, MYH3, MYH8, and NEB mainly colocalize with cluster B MNCs, further indicating that cluster B MNCs are at a more mature differentiation state. CCNA1 and ZSCAN4 are highly expressed in the area occupied by FSHD and DEL5 myotubes.
MERFISH reveals a cell differential bifurcation modulated by FSHD
We next examine the manually annotated myotubes compared with nonmyotube regions (Supplemental Figs. 6, 7). Differential expression analysis of myotubes and nonmyotube regions indicate that myotubes of all three genotypes show upregulation of myotube marker genes (e.g., NEB, MYH3) relative to nonmyotube regions (Supplemental Fig. 7G–I).
After batch correction (Supplemental Fig. 8A), clustering analysis identified 11 myotube and nonmyotube region clusters (Fig. 4A). Although nonmyotubes of all three genotypes cluster relatively closely (clusters b and g for control, c for FSHD, and i and part of h for DEL5), control myotubes (clusters a and k) are clearly separated from mutant/patient myotubes (Fig. 4B,C). FSHD myotubes localize to clusters d, e, and j, whereas DEL5 myotubes are primarily localized to cluster f, with subpopulations overlapping with FSHD myotubes (clusters d and e) and with control and DEL5 nonmyotube regions (cluster h) (Fig. 4B,C). Although the expression levels of individual DUX4 target genes vary, their significant upregulation is found exclusively in clusters d and e (Fig. 4D). Thus, we term the myotubes in clusters d and e “FSHD-hi” and the remaining FSHD/DEL5 myotubes “FSHD-lo” (Fig. 4F,G).
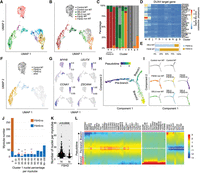
Clustering and pseudotime analysis identify a developmental bifurcation in the myoblast population, associated with a distinct reduction in myotube volume. (A) UMAP plots of MERFISH data from 987 myotubes and 629 nonmyotube regions. The plots are colored according to the clusters determined by the shared nearest neighbor (SNN) algorithm after batch correction using the Harmony algorithm. (B) UMAP from A colored by cell types, including the myotubes and nonmyotube regions of the differentiated control, FSHD and DEL5 cells. (C) Bar plot of the proportion of cell types in each cluster from the UMAP, colored by annotated cell types as in B. The myotubes for FSHD-hi and FSHD-lo are determined by DUX4 target gene expression levels. (D) Average expression profiles of the DUX4 target genes in each cluster. (E) The percentage of FSHD-hi and FSHD-lo cells in FSHD and DEL5 myotubes. (F) UMAP from A colored by designation of FSHD-hi or FSHD-lo versus control myotubes and remaining cells. (G) UMAP from A colored by normalized and scaled expression of example marker genes. (H) Trajectory analysis of batch-corrected MERFISH data using Monocle. The top 18 genes with the highest variability in expression within the myotube and nonmyotube regions are used to construct the pseudotime tree. Myotubes or nonmyotube regions on the tree are colored by pseudotime. (I) The pseudotime tree from H is colored by cell types and displayed individually. (J) Bar plot of the distribution of two types of myotubes, FSHD-hi and FSHD-lo, based on the percentage of cluster 1 nuclei in each myotube. The x-axis represents the range of cluster 1 nuclei percentages. The majority of FSHD-lo myotubes have no cluster 1 nuclei, with a few exceptions. To highlight these rare occurrences, the FSHD-lo myotube numbers (1 or 2) are indicated on the bars with red arrows. (K) The number of nuclei per myotube quantified in the FSHD-hi and FSHD-lo groups of FSHD myotubes. (L) The pseudotime heatmap displays the expression level of significantly changed genes (P < 0.01) in the two branches for comparison. Color key from blue to red indicates relative expression levels from low to high.
Approximately 80% of FSHD myotubes are FSHD-hi, whereas <20% of DEL5 myotubes are FSHD-hi. (Fig. 4E,F). As expected, MYH8 is expressed in control myotubes but not in nonmyotubes (Fig. 4G). However, MYH8 is higher in FSHD-lo myotubes and is significantly suppressed in FSHD-hi myotubes despite their clear myotube morphology (Figs. 1E, 4G).
We next performed pseudotime differential trajectory analysis using Monocle (Fig. 4H; Trapnell et al. 2014; Qiu et al. 2017; Cao et al. 2019; Yoshihara et al. 2022). This identifies a bifurcated developmental trajectory consisting of a prebranch component and two branches. The prebranch component consists exclusively of nonmyotube regions, whereas branch 1 is enriched for control and FSHD-lo myotubes, and branch 2 is enriched for FSHD-hi myotubes (Fig. 4H,I). Projection of MNCs to the differential trajectory indicates that type A MNCs appear on the prebranch, whereas type B MNCs are split between prebranch and both branch 1 and branch 2, depending on the genotype (Supplemental Fig. 8B). This suggests that the pathway bifurcation likely occurs during or shortly before myotube fusion.
To investigate the relationship of cluster 0 and 1 nuclei (Fig. 2A) with FSHD-hi and FSHD-lo myotubes, we examine their distributions in myotubes along the pseudotime trajectory. Notably, the majority of cluster 1 nuclei are observed in FSHD-hi samples, whereas FSHD-lo myotubes contain almost exclusively cluster 0 nuclei, with only 2% showing a mixture of cluster 0 and 1 nuclei (Fig. 4J; Supplemental Fig. 8C). Although the average total number of nuclei per myotubes is significantly lower in FSHD patient myotubes, with no significant change in DEL5 myotubes compared with the control (Fig. 2H), total numbers of nuclei are higher in FSHD-hi myotubes than in FSHD-lo myotubes in patient samples. A possible explanation is that myotubes with high numbers of nuclei are more likely to include cluster 1 nuclei and that even low proportions of cluster 1 nuclei are sufficient to propel myotubes to the branch 2 trajectory (Fig. 4K). FSHD-hi myotubes show variable percentages of cluster 1 nuclei within each myotube, with only ∼20% of FSHD-hi myotubes containing >90% cluster 1 nuclei (Fig. 4J; Supplemental Fig. 8C). Notably, the higher the percentage of cluster 1 nuclei is, the farther down the myotubes are in the branch 2 in the pseudotime trajectory, with myotubes with >90% cluster 1 nuclei composition distributed almost exclusively at the tip of branch 2 (Supplemental Fig. 8D). These results indicate that even a small number of cluster 1 nuclei is sufficient to drive the FSHD-hi phenotype, and strongly suggest that cluster 0 nuclei can be converted to cluster 1 in FSHD-hi myotubes via DUX4 and target transcription factor proteins cross-migration through the cytoplasm post-fusion.
Corresponding gene expression analyses reveal progressive gene expression changes along the pseudotime trajectory, reflecting cell state alteration (Fig. 4L). Although there are some common changes (Fig. 4L, orange cluster), distinct gene expression patterns are observed in branches 1 and 2. As expected, DUX4 target genes are uniquely upregulated in branch 2 (Fig. 4L, pink cluster). The expression of genes related to muscle structure development and function, such as MYH8, TNNI2, KLHL41, and ACTA1, is low at the beginning of the prebranch and gradually increases toward the branch point (Fig. 4L, blue cluster). Although their expression levels continue to increase toward the end of branch 1, they appear to reverse and be suppressed at the end of branch 2 with the exception of TTN, which appears to continue to be upregulated toward the end of branch 2. This impaired muscle gene expression may be linked to the observed characteristics of FSHD myotubes (i.e., fewer nuclei in smaller size myotubes compared with the control) (Fig. 2H,I). This difference is not apparent in DEL5, consistent with the fact that only a subset of DEL5 myotubes show the FSHD-hi phenotype (<20%), whereas the majority of FSHD myotubes are FSHD-hi (∼80%) (Fig. 4E).
Gene coexpression network analysis reveals significant dysregulation of non-DUX4 networks in DEL5 and FSHD samples
We next performed gene coexpression network analysis for control samples, with the intention of measuring module preservation across genotypes, to identify any DEL5 or FSHD correlated dysregulation. We first removed genes with low variation across samples (P > 0.05, monocle differential gene test) and eliminated genes with high FDR (>5%, based on unmapped gene barcodes). This resulted in 20 DUX4 target genes and 77 non-DUX4-target genes for downstream coexpression analysis. Weighted gene coexpression network analysis (WGCNA) of the control myotubes divides the 77 non-DUX4-target genes into three modules (Fig. 5A, blue [18 genes], brown [12 genes], and turquoise [35 genes]; Supplemental Table S2). An additional 12 genes in the data set are unassigned (Fig. 5A, gray). We then performed functional enrichment analysis of the three modules. In the control group, the turquoise module shows enrichment of genes related to the extracellular matrix (ECM), the blue module contains genes involved in endoplasmic reticulum (ER) stress response processes, and the brown module is enriched for muscle contraction–associated genes (Fig. 5B). Differential expression of some of these genes was also captured in the comparison of branches 1 and 2 in the pseudotime analysis described above (Fig. 4L).
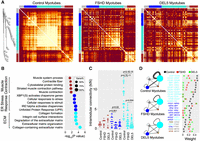
Weighted gene coexpression network analysis (WGCNA) of the non-DUX4-targets reveals gene network disruptions in FSHD and DEL5 myotubes. (A) Heatmaps depict the topological overlap matrix (TOM) among the 77 non-DUX4-target genes in the analysis. This matrix indicates similarity between genes by analyzing the extent to which individual genes coexpress with the same subset of genes (including each other). This results in a more robust similarity measure than pure correlation matrices. Lighter colors represent higher overlap, and darker colors represent lower overlap. The three heatmaps are plotted with the same order of genes based on the gene dendrogram of control myotubes. The modules of control are shown along the left side and the top. (B) The bubble plot shows Gene Ontology (GO) enrichment analysis of genes in the modules of control using the online tool g:Profiler. The bubble colors correspond to the module colors in A. The x-axis displays log10-adjusted P-values used Bonferroni correction and a threshold of 0.05. Bubble size indicates, in each term, the percentage of total genes in the given module. (C) The intramodular k-values of the genes in the blue and turquoise modules, identified in control myotubes, are decreased significantly in the FSHD and DEL5 myotubes. (D) The intramodular and intermodular connections of genes change significantly in FSHD and DEL5 myotubes compared with the control. The width of the edges and self-loops is proportional to sum of the weights (topological overlap values) of the edges, thresholded at 0.1. The module size indicates the number of genes within it. Some gene pairs are shown on the right panel as examples.
We computed the intramodular connectivity for each module (intramodular connectivity kIN), defined as the sum of a gene's connection strengths with all other genes in its module (Oldham et al. 2006). The gene–gene connectivity patterns in the blue and turquoise modules are significantly decreased in FSHD and DEL5 myotubes, compared with the control (Fig. 5A–C). Although there is a difference between FSHD and DEL5 gene coexpression in the turquoise module (P = 0.004), they are more similar to each other than to the control (Fig. 5A,C). Because the most significant connectivity loss is observed in the turquoise module (ECM) in FSHD/DEL5 compared with the control (Fig. 5C,D), we further analyze the genes in this module. The results show a significant decrease in the coexpression of collagen family members and other ECM-related factors such as SOX9, SERPINE2, and ADAM19 (Fig. 5D; Supplemental Fig. 9C; Buchholz et al. 2003; Athwal et al. 2018; Peixoto et al. 2019). ATF6B, a gene primarily involved in ER stress (Benedetti et al. 2022), shows relatively high connectivity with certain collagen genes in the control group, but not in FSHD and DEL5 (Fig. 5D).
Although the three modules defined in control myotubes are highly unpreserved in FSHD and DEL5 cells, four modules are detected in both FSHD and DEL5 myotubes (Supplemental Fig. 9A; Supplemental Table S2). We next quantify and compare the preservation of these modules (Jardim-Perassi et al. 2019; Yu et al. 2021) by projecting modules from DEL5, serving as a reference, to control and FSHD. Notably, all the preservation Zsummary values (statistical measures of preservation) of DEL5 modules in FSHD are higher than those in the control (Supplemental Fig. 9B). Some of the differences in the gene module composition are caused by the disconnection of genes in the same module for the control, such as COL4A2 and SERPINE2/SOX9 (Fig. 5D, right panel; Supplemental Fig. 9C). Some of the differences are caused by the reconnection of genes or their attribution to another module. For example, PTAR1, which belongs to the gray module in the control, is connected to CCNL1 (Fig. 5D, right panel) and joins its module in the FSHD and DEL5 (Supplemental Fig. 9C). These results indicate that not only the gene expression level but also the gene coexpression networks are altered in the FSHD and DEL5 myotubes.
Here we investigate whether the correlation with DUX4 target genes influences the shift of gene modules in FSHD and DEL5 cells compared with the control. We include DUX4 target genes in the gene expression correlation analysis and superimpose the correlations with corresponding DUX4 target genes on the coexpression gene modules of 77 non-DUX4-target genes identified above in FSHD and DEL5 (Supplemental Fig. 9A,D). We find that new modules formed in FSHD and DEL5 are predominantly driven by their correlation with DUX4 target genes (Supplemental Fig. 9D). In contrast, four collagen genes whose link is maintained in FSHD and DEL5 as the control do not show any strong correlation with DUX4 target genes (Supplemental Fig. 9C,D). Taken together, our results depict that global gene network alterations correlate with DUX4 target gene expression.
Gene coexpression analysis of DUX4 target genes identifies two distinct gene groups that show differential coexpression patterns with other DUX4 and nonmuscle genes
To dissect the DUX4 gene network, we perform both direct pairwise correlation and WGCNA analyses of the 20 DUX4 target genes in both FSHD and DEL5 myotubes (control is omitted owing to low expression of DUX4 target genes). Although there is a general positive pairwise correlation in expression between most of the DUX4 target genes, the strengths of the correlations vary across gene pairs and are relatively consistent between FSHD and DEL5 (Fig. 6A; Supplemental Fig. 10A). WGCNA identifies two distinct subgroups common in both DEL5 and FSHD: LEUTX-CCNA1-KHDC1L (the LEUTX group) and ZSCAN4-KDM4E-RFPL4B (the ZSCAN4 group), with Pearson pairwise correlations within each group exceeding 0.8 (Fig. 6A; Supplemental Fig. 10B). Other DUX4 target genes show differential association with the two groups (Fig. 6A–C). VMO1, DUXB, PRAMEF20, and ZNF705G show stronger correlation with the LEUTX group relative to the ZSCAN4 group. In contrast, TAF11L11, PRAMEF12, and ZNF296 show stronger correlation with the ZSCAN4 group relative to the LEUTX group. H3.Y, RFPL2, and SLC34A2 show similar correlation with both groups, whereas KFL17, DUXA, and PRAMEF19 do not show any correlations passing the threshold (weights > 0.2 for both cell lines) with either group. SLC38A1 does not show a significant correlation with any known DUX4 target genes in this study.
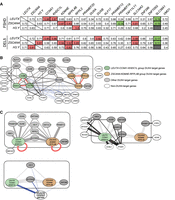
Differential correlation between DUX4 target genes in FSHD and DEL5 myotubes reveals network hubs showing differential association with non-DUX4 targets. (A) Pearson correlation analysis of DUX4 target genes in FSHD and DEL5 myotubes. Correlation between genes is determined by normalized and batch-corrected expression values of three DUX4 target genes versus all 20 DUX4 target genes. The top panel shows data for FSHD myotubes, and the bottom panel shows data for DEL5 myotubes. Some specific results are highlighted, with red indicating a very strong positive relationship, green indicating a very weak relationship, and gray indicating a P-value > 0.05. (B) The expression values of 20 DUX4 target genes (enclosed in the block box) and 14 non-DUX4-target genes in FSHD and DEL5 myotubes are analyzed by WGCNA. Red edges indicate gene pairs that show strong coexpression in both cell lines, with weights greater than 0.4 and mean values exceeding 0.5. Dark gray and light gray edges represent positive correlations between genes that do not meet the threshold of the red edges. Dark gray edges indicate that the correlation values between FSHD and DEL5 are not significantly different. Blue edges indicate negative correlations between genes. The thickness of each edge represents the average absolute weight of the corresponding gene pair in both cell lines. For the correlations between DUX4 target genes, only weights greater than 0.2 are displayed, except for DUXA, ZNF296, and PRAMEF19. These three DUX4 target genes have a lower threshold of 0.06 and are presented with their largest mean weight. For the correlations between DUX4 target genes and non-DUX4-target genes, the network represents positive correlations with weights above 0.06 or negative correlations above 0.025 in both cell lines. The correlations between non-DUX4-target genes are not shown. (C) Three subfigures extracted from B. The LEUTX-CCNA1-KHDC1L groups, ZSCAN4-KDM4E-RFPL4B groups, and six genes related to muscle function or development are classified into three groups, respectively, with the width of the edges proportional to the sum of the weights of the edges between two individual genes in B. To highlight the differences in edge weights, the scale used in these subfigures differs from that of B. The red self-loop is not scaled in the same manner as the other edges. In the top left panel, the two DUX4 target gene groups and other DUX4 target genes connected to them are displayed. SMCHD1 and DUXA are also included in this panel. The top right panel presents seven non-DUX4-target genes along with the DUX4 target genes/groups they are connected to by dark edges. The bottom left panel displays all three gene groups and shows the DUX4 target genes that are negatively correlated with the muscle genes group. The connections between DUX4 target individual genes and the groups and in top right and bottom left panels are represented by very light edges. For all three panels, the edges between individual DUX4 target genes are not shown, except for DUXA in the top left panel.
In addition to DUX4 target genes, we identify 14 non-DUX4 target genes (out of 77 genes) that are positively (eight genes with weight > 0.06) or negatively (six genes, weight > 0.025) correlated with DUX4 target genes in both FSHD and DEL5. Although the correlation between genes is typically directionless, seven out of eight positively correlated genes are upregulated in FSHD/DEL5 (Supplemental Fig. 10C). SERPINE2, MYDGF, NCOA3, SMCHD1, CCNL1, and PTAR1 are highly correlated with the LEUTX group rather than the ZSCAN4 group (Fig. 6C, top right), whereas LOLX2 and SOX9 are positively correlated with the ZSCAN4 group. SERPINE2 is identified as a LEUTX target gene (Gawriyski et al. 2023), and the NCOA3 protein was found to interact with LEUTX protein (Gawriyski et al. 2023), supporting the connection. SOX9 has been shown to be upregulated by DUX4, but the mechanism is unclear (Resnick et al. 2019). SMCHD1, a FSHD modifier gene also linked to FSHD2, binds to D4Z4 in a H3K9me3-dependent manner, and decreased SMCHD1 binding to D4Z4 results in DUX4 expression (Lemmers et al. 2012; Zeng et al. 2014). A positive correlation and upregulation of SMCHD1, together with the LEUTX group, suggest a possible compensatory feedback mechanism in DUX4-activated cells. In FSHD/DEL5 cells, all the six LEUTX group correlated genes join the same module, whereas LOLX2 and SOX9, which show higher correlation with the ZSCAN4 group, are in gray (unassigned) module or fail to separate from the original group of control cells (Fig. 9C; Supplemental Table S2). Six muscle-related genes (Bolcato-Bellemin et al. 2003; Potthoff et al. 2007; Schiaffino 2018; Woo et al. 2020; Murgia et al. 2021) downregulated in FSHD-hi show negative correlation specifically with the LEUTX group as well as with SLC34A2, rather than the ZSCAN4 group (Fig. 6C, bottom left). This raises the possibility that different subgroups of DUX4 target genes may make distinct contributions to the FSHD transcriptomic phenotypes.
Discussion
In this study, we applied MERFISH to spatial transcriptomic profiling of FSHD in individual cells, analyzing a rare DUX4 expression profile that is present in a low percentage of patient myocytes that show a strong disease-specific phenotype. Because of the rarity of the phenotype, this analysis cannot be precisely addressed by bulk RNA-seq methods, and single-cell and single-nuclei techniques cannot resolve the nuclei and cellular structures. MERFISH analyses allow us to capture differential transcriptional states of MNCs and myotubes (as well as their integrated nuclei) of control, isogenic D4Z4 deletion mutant, and FSHD1 patient samples, highlighting the deviation of FSHD myotube differentiation accompanied by downregulation of muscle-related genes and global gene network alteration compared with the healthy control. Our results also provide evidence for multiple DUX4 target coexpression modules, suggesting more complex regulation of DUX4 targets than previously appreciated.
FSHD patient cell phenotypes are highly variable. In our current study, we chose a specific FSHD1 patient cell line that shows high DUX4 target gene expression as a disease prototype, and compared it with a healthy control cell line and an isogenic mutant with D4Z4 repeat contraction, which shows moderate DUX4 target gene expression compared with patient cells. We find that nuclei from all three genotypes can be separated into two clusters, cluster 0 and cluster 1, with cluster 1 only present in FSHD and mutant samples, strongly associating with DUX4 target expression. Our results show the presence of two states in patient and mutant myotubes, “FSHD-hi” and “FSHD-lo,” clearly separated from control myotubes, characterized by expression of DUX4 targets and suppression of certain muscle-related genes. Comparison of myocytes with different prevalences of DUX4 target gene expression enabled us to observe different ratios of FSHD-hi and FSHD-lo myotubes as well as DUX4 target upregulated cluster 1 nuclei in the two cell types. We observe similar characteristics of cluster 1 nuclei as well as FSHD-hi and FSHD-lo myotubes in FSHD and mutant cells distinct from the control, strongly supporting that the observed phenotype can be attributed to D4Z4 contraction. On the other hand, myotubes in FSHD samples show both decreased volume and decreased nuclei counts, a feature largely not found in the D4Z4 contraction mutant. Comparable non-DUX4 target gene network disruption and coexpression of similar subsets of DUX4 targets are observed in both FSHD and mutant cell types, arguing that these are important features of the disease. Our analysis of DUX4 targets identifies two highly connected modules, showing differential connectivity with the remaining genes.
Differential trajectory inference via pseudotime analysis identified a bifurcation of the developmental path (branches 1 and 2) between healthy and pathological myotubes, with FSHD-hi myotubes enriched in branch 2. Our analyses reveal that myotubes show the FSHD-hi phenotype with varying numbers of cluster 1 nuclei. An increased cluster 1 nuclei percentage composition correlates with increased pseudotime, which posits a more developed myotube. This strongly suggests that nuclei clusters are metastable and that cluster 0 nuclei are converted to cluster 1 nuclei by nuclear communication through the shared cytoplasm in FSHD and mutant myotubes. We do express some caution in interpreting these results, owing to the relative lack of cells near the pseudotime bifurcation and owing to the lack of genes differentially upregulated at that pseudotime point. Further experiments are required to confirm and analyze the gene regulatory roles associated with this bifurcated path.
Gene coexpression analyses in non-DUX4-target genes further capture global gene network alteration in patient and mutant myotubes, particularly notable with the ECM-related genes in our probe set. In skeletal muscle, the ECM plays an important role in regulation of muscle development, growth, and repair and is essential for effective muscle contraction and force transmission (Csapo et al. 2020). It provides the necessary support and communication between muscle fibers and surrounding tissues and contributes to muscle regeneration after injury. The ECM in skeletal muscle is primarily composed of collagen, which forms a meshwork around individual muscle fibers, holding them in place and transmitting forces generated by muscle contractions (Gillies and Lieber 2011). Significant changes of ECM gene connectivity may represent rewiring of the gene coexpression network in patient/mutant cells related to the FSHD disease process.
Downregulation of muscle genes (MYH8, etc.) has been reported previously in FSHD myocytes (Bosnakovski et al. 2017). Our UMAP and gene network analyses reveal specific downregulation of certain muscle genes only in FSHD-hi myotubes. Our pseudotime trajectory analyses indicate an apparent transient upregulation of muscle genes in FSHD-lo myotubes, which is not sustained as the myotubes transition into FSHD-hi. Although overall DUX4 target gene expression inversely correlates with muscle gene expression, coexpression analyses suggest significant anticorrelation of at least those muscle-related genes tested with the LEUTX subgroup. There are additional non-DUX4-target genes that were not previously linked to FSHD but that appear to show specific correlations with a subset of DUX4 target genes in both patient and mutant myotubes in our study. For example, the expression of PTAR1, CCNL1, and MYDGF specifically correlates with the expression of CCNA1 and KHDC1L in the LEUTX group, which are involved in protein modification, cell cycle, and cell growth and have been verified as cancer-related genes (Muller et al. 2006; Bortnov et al. 2018; Zhao et al. 2020). However, because our probe set and cell sample numbers are limited, additional studies would be necessary to systematically capture affected genes and their relationship with the DUX4 gene network. Further investigation of high-resolution gene expression correlation in FSHD as well as experimental validation of their relationship may provide valuable insights into the muscle cell-intrinsic alteration associated with the disease. Additionally, MERFISH analysis on patient muscle biopsy samples will enable the analysis of muscle fibers that cannot be obtained in vitro, as well as cell–cell communications with other cell types that reside in patient muscles, to understand their possible collective contributions to the dystrophic phenotype. Expansion of analysis beyond the limited number of cell lines used here will help determine whether the phenotypic and transcriptomic alterations identified are representative of the disease state as a whole.
Imaging-based spatial transcriptomics, particularly MERFISH, uniquely enables the simultaneous multiscale transcriptomic analysis of both nuclei and whole multinucleated myotubes containing those same nuclei, which is highly effective in examining the low prevalence of DUX4 pathology in patient myocytes and is also applicable to the integrated analyses of any multinucleated cell types. We optimized the hybridization protocol as well as image sampling and measurement tailored to in vitro cell culture. This technique opens doors for future projects analyzing multinucleated hepatocytes, osteoclasts, and cancer cells (Estradas et al. 2009; Zhang et al. 2011; Kodama and Kaito 2020) in the context of healthy function and disease.
Methods
Human myocyte culture
Control myoblasts (20 D4Z4 units at 4qA; doubling time, 25.2 h) and FSHD myoblasts (FSHD1; two D4Z4 units at 4qA; doubling time, 39 h) are immortalized myoblast cells as previously described (Chau et al. 2021). Control-derived D4Z4 contraction mutant DEL5 myoblasts (one D4Z4 unit at 4qA; doubling time, 34 h) are generated using CRISPR engineering as described in our previous paper (Kong et al. 2024). The cells at passage 10–15 were grown in high-glucose DMEM (Gibco) supplemented with 20% FBS (Omega Scientific), 1% Pen-Strep (Gibco), and 2% Ultraser G (Crescent Chemical). Because the fusion efficiency of the DEL5 myoblasts was lower than that of others, differentiation of DEL5 cells was induced first using differentiation media, the high-glucose DMEM medium supplemented with 2% FBS, and ITS supplement (insulin 0.1%, 0.000067% sodium selenite, 0.055% transferrin; Invitrogen). After 24 h growing at 37°C, the control, FSHD1, and DEL5 cells were harvested with TrypLE express (Thermo Fisher Scientific), washed, and resuspended in fresh differentiation media. The cells were counted, and the volumes were adjusted to produce a cell concentration of 0.5 million cells per milliliter for the control and FSHD1, as well as 1 million cells per milliliter for the DEL5. A drop of 60 μL cell suspension from each line was separately plated without mixing onto the center region of a 4-cm poly-lysine-precoated round glass coverslip in a 60-mm cell culture dish (Fig. 1A). Once cells attached to the glass coverslip in a 37°C tissue culture incubator with 100% humidity and 10% of CO2 for 3 h, cell attachment were confirmed under a microscope, and then floating cells were washed off and attached cells were maintained in the differentiation media, which was refreshed every 24 h. Three days later, cells were fixed with 4% paraformaldehyde and permeabilized in 70% ethanol overnight after a rinse with 1 × PBS.
Generation of D4Z4 contraction mutants with CRISPR-Cas9
We used the Alt-R CRISPR-Cas9 genome editing system (Alt-R S.p. HiFi Cas9 nuclease V3, Alt-R CRISPR-Cas9 tracrRNA, and custom Alt-R CRISPR-Cas9 guide RNA [gRNA]; IDT) to remove D4Z4 repeat units from control myoblast cells. To ensure precise targeting, we designed gRNAs to selectively bind to a specific 1-kb subregion of the D4Z4 sequence located at 4q/10q (Ehrlich et al. 2007) while excluding any homologous regions found on other chromosomes, as well as the DUX4 gene and its promoter region. Following the screening of hundreds of CRISPR-Cas9/gRNA-treated single colonies, we obtained several mutated cell lines with reduced D4Z4 copy numbers, confirmed by nanopore sequencing. These cell lines were then used as a disease model to investigate the mechanisms underlying FSHD pathogenesis. Specifically, we chose the DEL5 cell line, which has only one copy of D4Z4 repeat at its 4qA and shows the highest levels of DUX4 target gene expression among the D4Z4 contraction mutant cells (Kong et al. 2024), to study its gene expression patterns by using MERFISH technology in this paper.
Gene selection for MERFISH
To assess gene expression differences and correlations among control, patient, and mutant cells with MERFISH, we designed a panel of 140 genes. Twenty-four genes are previously identified as “possible DUX4 targets” or “FSHD-induced genes” from myoblasts with inducible DUX4 (Resnick et al. 2019), endogenous DUX4 (Jiang et al. 2020), or FSHD biopsies (Yao et al. 2014). At least 19 of them are found the presence of DUX4-binding sites adjacent to the transcription start site based on previous DUX4 ChIP-seq data (Geng et al. 2012). The remaining 116 genes are referred to as “non-DUX4-target” genes, which were selected based on previous analyses (Cabianca et al. 2012; Hamanaka et al. 2020). We exclude sex-specific genes, as well as genes that are relatively short or showed high expression levels, which are potentially challenging for highly multiplexed FISH imaging experiments (Moffitt et al. 2018). Vizgen provided the corresponding encoding probes, readout probes, barcodes, as well as 40 “blank” barcodes that were not assigned to any specific genes and used for quality control.
MERFISH experiment
MERFISH procedures were performed based on Vizgen's protocol using the materials/buffers included in the MERSCOPE cell boundary stain kit, sample preparation kit, and 140-gene imaging kit. Briefly, 70% ethanol-permeabilized prefixed cells on the glass coverslips were first stained with the Vizgen cell boundary staining kit before staining with gene panel encoding probes. The cells were refixed with 4% paraformaldehyde, incubated in the formamide wash buffer at 37°C, and stained with the custom-designed and synthesized MERFISH probes that contained a panel of 140 genes and blank controls for ∼40 h at 37°C. Then, the stained cells were incubated in the formamide wash buffer at 47°C (30 min × 2), rinsed with the sample preparation buffer, and stained with the DAPI and poly(T) staining reagent, as well as the fiducial fluorescent beads. The glass coverslip was finally assembled into the MERFISH gasket for imaging after incubation in the formamide wash buffer (10 min at RT) and a brief wash with the sample preparation buffer. The MERFISH imaging was conducted on Vizgen merscope with a 140-gene panel imaging kit controlled by the MERSCOPE program (software version 231.220531.1390) with default settings (poly(T) and DAPI and cell boundary channels “on,” scan thickness: 10 μm) as Vizgen recommended. Once the imaging was completed, the raw imaging data were automatically converted by the program into “vzg” meta output files, which are subject to downstream in-depth analysis with both MERFISH Visualizer and our customized bioinformatic pipelines.
Comparison of MERFISH versus bulk RNA-seq
The day 3 differentiated control cell lines have been previously sequenced using bulk RNA-seq. The RNA-seq raw reads were aligned with STAR (Dobin et al. 2013) using human genome reference hg38. Read count was performed using RSEM with gene annotations from GENCODE v28, normalized by TMM in edgeR (Robinson et al. 2010), and then converted to transcripts per million (TPM) (Kong et al. 2024). The raw RNA-seq data are available in The database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) with accession number phs002554.v2. Comparisons of bulk RNA-seq TPM and MERFISH counts are performed with smplot R package (Min and Zhou 2021). In practice, bulk RNA-seq preparations showed greater fusion efficiency compared with merslide preparations, so that a higher percentage of myotubes were present relative to the MERFISH data.
Machine learning–based MNC and nuclei segmentation
We used the deep-learning-based segmentation software (Cellpose) (Stringer et al. 2021) to segment the MNCs and nuclei, using DAPI and cell boundary staining to separately isolate individual nuclei and MNCs from the same image. Across the 10 sample regions, the sensitivity for MNC is 0.858 ± 0.039, the sensitivity for nuclei is 0.989 ± 0.007, the accuracy for MNC is 0.976 ± 0.007, and the accuracy for nuclei is 0.986 ± 0.003 (see also Supplemental Methods).
After training and validation, the segmentation process on the remaining ROIs was automatic, and no manual intervention was included. Final segmentation results were presented as a mask containing individual MNCs or nuclei footprints. Based on the masks, we calculated the boundaries of MNCs and nuclei and then determined the transcripts inside based on their spatial coordinates given by the MERFISH system.
Quality control and preprocessing
Automatically detected MNCs and nuclei were examined with their volumes and transcript counts to identify and remove potential false detections. For MNCs, an empirical volume range of 85–1500 µm2 was used to exclude false positives. For nuclei, the empirical volume range was 15–700 µm2. A transcript count range across 40–5000 was applied to MNCs to further trim false detections. For nuclei, the transcript count range was set as five to 4000. The lower count thresholds were based on previous experience detailing the minimum requirement of cell type classification in previous experiments. We noted some samples had blurry and low-quality imaging results at peripheral regions; hence, we further limited our MNCs and nuclei selection to the center of each sample, which is illustrated in Supplemental Figure 1. The myotube and nonmyotube regions were not subject to the center limit because they are manually picked and do not exist in low-quality regions.
The transcript counts of each MNC, nucleus, and myotube were divided by their corresponding volumes for normalization purpose (Zhang et al. 2021). In practice, volume versus library size normalization did not show major differences. For differential expression analysis, however, the unnormalized raw counts were used as input to the DESeq2 package as it has interior mechanisms for data normalization (Love et al. 2014). For nonmyotube regions, their intra-region transcript counts were calculated as the sum of all the transcripts inside.
Myotube/nonmyotube region identification and segmentation
Because of incompatibility of the current Cellpose model with cell boundary detection of myotubes (likely owing to the large variance in size and morphology for these cells), myotubes were manually segmented based on the combined DAPI-cell boundary stain. Therefore, myotubes were manually segmented based on the labeling of cell membrane antibodies and the empty spaces between the RNA transcripts signals. Combined with DAPI staining, these allowed us to identify differentiated myotubes as cells containing three or more nuclei. On the DAPI transcript overlay image, the area containing an individual myotube was circled manually. This resulted in processed images containing the single-myotube boundaries and the RNA transcripts inside. The gene expression data at single-myotube resolution were then used for downstream bioinformatic analysis.
Nonmyotube regions are manually selected contiguous areas satisfying two criteria: (1) volumes of nonmyotube regions are within the size distribution of myotubes (e.g., approximately the same size as the mean myotube volume), and (2) nonmyotube regions do not intersect with any manually annotated myotubes. We feel that these regions better represent differential comparisons with myotubes than do individual cells, as such regions contain multiple nuclei and are of similar volume to myotubes.
Transcriptomic analysis
MNCs or nuclei across all samples were pooled together for clustering analysis and clustered using the scrattch.hicat package. Transcripts counts of single-myotube and nonmyotube regions were normalized by area and processed using Seurat (Hao et al. 2021), with batch effect correction using Harmony (for clustering) (Korsunsky et al. 2019) and clustering using the Louvain algorithm (Blondel et al. 2008). Differential expression analyses of transcripts were conducted using a pseudobulk approach and statistical analysis with DESeq2 (v1.36.0) (Love et al. 2014). Differential expression analysis between the myotube and nonmyotube regions was restricted to genes exceeding the blank (control barcode) gene expression within the myotube and nonmyotube regions (Supplemental Fig. 7C–E). For pseudotime analysis, the myotube and nonmyotube region data were reintegrated using Seurat's anchor method to create a batch-corrected gene expression matrix. This was then analyzed using Monocle to construct pseudotime trajectories. Gene coexpression analysis was conducted using WGCNA (Langfelder and Horvath 2008). GO and pathway enrichment analysis for the identified modules was conducted using g:Profiler (Reimand et al. 2007). The statistical domain scope was used for the analyzed non-DUX4-target genes as custom background.
Statistical analysis
Data are presented as the mean ± SEM. A two-tailed Wilcoxon rank-sum test was used for testing statistical significance between distributions of individual samples. Linear mixed effect modeling was used for test statistical significance between distributions of individual myotubes pooled from multiple samples. The level of statistical significance was defined as follows: (*) P ≤ 0.05, (**) P < 0.01, and (***) P < 0.001. Linear mixed effects modeling was used to address issues related to repeated measurements.
Data access
Code used to generate results is available at the Mendeley data repository (https://data.mendeley.com/datasets/mkg3yhtmh7/6) and as Supplemental Code. Raw data from the MERFISH experiment, as well as individual histology images of all the cell culture samples, are available at Zenodo (https://doi.org/10.5281/zenodo.11100095).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by grant NS104897 from the National Institute of Neurological Disorders and Stroke (NINDS) and grant R01AR071287 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS). The work was also supported in part by the National Institute on Aging (NIA) grants U01AG076791 and R01AG082127, as well as the National Institute on Deafness and Other Communication Disorders (NIDCD) grant T32 DC010775-14.
Author contributions: X.X., K.Y., and A.M. designed the study, secured the funding, and supervised the operations. Z.T. and X.K. performed the experiment. L.C. and X.K. performed the analysis and worked with K.G.J., T.C.H., X.X., and K.Y. to write the manuscript. All authors have read and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278717.123.
-
Freely available online through the Genome Research Open Access option.
- Received November 9, 2023.
- Accepted May 17, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








