
An integrative TAD catalog in lymphoblastoid cell lines discloses the functional impact of deletions and insertions in human genomes
- Chong Li1,2,
- Marc Jan Bonder3,4,
- Sabriya Syed5,
- Matthew Jensen6,7,
- Human Genome Structural Variation Consortium (HGSVC),
- HGSVC Functional Analysis Working Group,
- Mark B. Gerstein6,7,
- Michael C. Zody8,
- Mark J.P. Chaisson9,
- Michael E. Talkowski10,11,12,13,
- Tobias Marschall14,15,
- Jan O. Korbel16,
- Evan E. Eichler17,18,
- Charles Lee5,19 and
- Xinghua Shi1,2
- 1Department of Computer and Information Sciences, College of Science and Technology, Temple University, Philadelphia, Pennsylvania 19122, USA;
- 2Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, Pennsylvania 19122, USA;
- 3Department of Genetics, Groningen, University of Groningen, University Medical Center Groningen, Groningen 9713 AV, Netherlands;
- 4Division of Computational Genomics and Systems Genetics, German Cancer Research Center, 69120 Heidelberg, Germany;
- 5The Jackson Laboratory for Genomic Medicine, Farmington, Connecticut 06032, USA;
- 6Department of Molecular Biochemistry and Biophysics, Yale University, New Haven, Connecticut 06510, USA;
- 7Program in Computational Biology and Bioinformatics, Yale University, New Haven, Connecticut 06520, USA;
- 8New York Genome Center, New York, New York 10013, USA;
- 9Department of Quantitative and Computational Biology, University of Southern California, Los Angeles, California 90089, USA;
- 10Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA;
- 11Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts 02114, USA;
- 12Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts 02114, USA;
- 13Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA;
- 14Institute for Medical Biometry and Bioinformatics, Medical Faculty and University Hospital, Heinrich Heine University, 40225 Düsseldorf, Germany;
- 15Center for Digital Medicine, Heinrich Heine University, 40225 Düsseldorf, Germany;
- 16European Molecular Biology Laboratory (EMBL), Genome Biology Unit, 69117 Heidelberg, Germany;
- 17Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195-5065, USA;
- 18Howard Hughes Medical Institute, University of Washington, Seattle, Washington 98195, USA;
- 19Department of Genetics and Genome Sciences, UConn Health, Farmington, Connecticut 06030-6403, USA
Abstract
The human genome is packaged within a three-dimensional (3D) nucleus and organized into structural units known as compartments, topologically associating domains (TADs), and loops. TAD boundaries, separating adjacent TADs, have been found to be well conserved across mammalian species and more evolutionarily constrained than TADs themselves. Recent studies show that structural variants (SVs) can modify 3D genomes through the disruption of TADs, which play an essential role in insulating genes from outside regulatory elements’ aberrant regulation. However, how SV affects the 3D genome structure and their association among different aspects of gene regulation and candidate cis-regulatory elements (cCREs) have rarely been studied systematically. Here, we assess the impact of SVs intersecting with TAD boundaries by developing an integrative Hi-C analysis pipeline, which enables the generation of an in-depth catalog of TADs and TAD boundaries in human lymphoblastoid cell lines (LCLs) to fill the gap of limited resources. Our catalog contains 18,865 TADs, including 4596 sub-TADs, with 185 SVs (TAD–SVs) that alter chromatin architecture. By leveraging the ENCODE registry of cCREs in humans, we determine that 34 of 185 TAD–SVs intersect with cCREs and observe significant enrichment of TAD–SVs within cCREs. This study provides a database of TADs and TAD–SVs in the human genome that will facilitate future investigations of the impact of SVs on chromatin structure and gene regulation in health and disease.
The functional annotation of genomes facilitates the understanding of genotype-to-phenotype relationships in the human genome. One important step in functionally annotating the genome is to determine how the spatial arrangement of DNA impacts genome functionality and gene regulation through the characterization of the three-dimensional (3D) structure of chromatin inside the nucleus (Szabo et al. 2019). Chromosome conformation capture sequencing (Hi-C) is a genome-wide sequencing technique that combines proximity-based DNA ligation with high-throughput sequencing to measure the geographical proximity of any pair of genomic loci. Techniques such as Hi-C are widely employed to characterize the 3D structure of the genome and uncover folding principles of chromatin, such as A/B compartments, topologically associating domains (TADs), and loops (Szabo et al. 2019). TADs are stable genomic regions separated by insulating proteins, e.g., CCCTC-binding factor (CTCF), and provide an encapsulating domain for constraining the chromatin contacts between regulatory elements and genes (Phillips and Corces 2009; Merkenschlager and Nora 2016; Melo et al. 2020; Shrestha et al. 2022). TAD boundaries, which separate adjacent TADs, prove to be well conserved across mammalian species and are more evolutionarily constrained than TADs themselves (McArthur and Capra 2021; Rajderkar et al. 2023). To date, the most comprehensive characterization of 3D genome organization from Hi-C sequencing comes from GM12878, with the Hi-C data of greatest depth so far (Rao et al. 2014), and individuals from the 4D nucleome (4DN) project (Dekker et al. 2017). Despite these efforts and some additional work using deep learning models to enhance Hi-C data (Zhang et al. 2018; Liu and Wang 2019; Liu et al. 2019; Dimmick et al. 2020; Hong et al. 2020; Highsmith and Cheng 2021), the utility of Hi-C data is typically limited due to their relatively low resolution, insufficient read coverage, and the sparse contact matrices, which result in a lack of detailed characterization of chromatin structures in human genomes. Hence, there is a great need to characterize the 3D genome organization of human genomes with a higher resolution to facilitate the understanding of the 3D structural landscape of human genomes.
Recently, there has been a growing interest in uncovering the disruption of gene regulation resulting from pathogenic large genomic rearrangements or structural variants (SVs), including deletions, insertions, inversions, and duplications (Shanta et al. 2020). These genomic rearrangements can disturb the normal 3D structure of the genome and lead to aberrant interactions between chromatin-regulatory elements (Akdemir et al. 2020), with such alterations bringing about the abnormal expression of oncogenic and disease-causing genes (Lupiáñez et al. 2015; Kim et al. 2022). The analysis of gene expression data across multiple tissues in mice and humans reveals that genes residing within TADs display highly conserved expression patterns. When TADs are perturbed by evolutionary rearrangements, they can correlate with alterations in gene expression profiles (Krefting et al. 2018; Szabo et al. 2019). In particular, SVs can affect CTCF-associated border elements, alter gene expression and enhancer–promoter interactions, and consequently result in human diseases (Ibn-Salem et al. 2014; Lupiáñez et al. 2015). Some SVs can cause distinct TADs to fuse, significantly altering chromatin folding maps (Akdemir et al. 2020). Additionally, integrating TAD locations with global gene expression data and genotype variants allows the investigation of how specific variants, including SVs, impact chromatin conformation and subsequently lead to changes in gene regulation (Weischenfeldt et al. 2017; Shanta et al. 2020).
In this study, we were motivated to elucidate the impact of SVs on chromatin organization and gene regulation utilizing our recently released high-quality genotypes of SVs (Ebert et al. 2021), combined with the identified TADs and TAD boundaries among the same individuals. To achieve this, we started with the collection of Hi-C data for 44 of the 1000 Genomes individuals (1000 Genomes Project Consortium 2015) that were sequenced earlier from the Human Genome Structural Variation Consortium (HGSVC) (Gorkin et al. 2019; Ebert et al. 2021), 4DN (Dekker et al. 2017), and Rao et al. (2014). These data allow us to construct a valuable resource of the Hi-C contact map at a significantly improved resolution of 300 bp compared to previous data sets in lymphoblastoid cell lines (LCLs) to assist with a closer observation of these hierarchical genomic regions and their nested derivatives of the identified TADs and TAD boundaries. We identified a set of unique SVs that significantly alter TAD boundaries (TAD–SVs) and demonstrated that TAD–SVs are likely to be enriched for candidate cis-regulatory elements (cCREs) more than is expected by random occurrence. This integrative map of 3D genome TAD structure in humans should serve as an essential resource to help elucidate the impact of SVs on human 3D TAD structure, as well as their disruptive impact on gene regulation. Our study thus provides critical insights into the mechanisms by which SVs influence chromatin dynamics and gene expression, advancing our understanding of the genetic basis of complex traits and diseases.
Results
Development of a genome-wide TAD catalog using an integrative Hi-C contact map from 44 individuals with genetically diverse genomes
We first sought to construct a 3D chromatin structure TAD map of human genomes from 44 individuals representing five superpopulations (Africa [AFR], n = 19; East Asia [EAS], n = 8; Europe [EUR], n = 6; South Asia [SAS], n = 5; and the Americas [AMR], n = 6) (Table 1; Supplemental Table S1). To generate this integrative genome-wide 3D chromatin structure map, we used Hi-C data that were sequenced earlier on LCLs from 44 individuals and preprocessed using a customized pipeline developed for this project to take into account different restriction enzymes, sequencing technologies, and resolutions of these Hi-C data (Fig. 1A; see Methods).
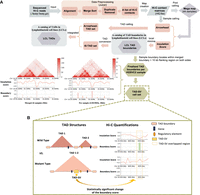
The step-by-step workflow to process raw Hi-C data into TADs/TAD boundaries and TAD–SVs in our Hi-C analysis pipeline. (A) The raw read files from 44 samples were used as input in Juicer for preprocessing and generating Hi-C maps, which were subsequently binned at multiple resolutions. The Insulation Score (IS) algorithm was applied to call an initial TAD boundary for each sample. All 44 Hi-C libraries were merged together to create a “mega” map and used as an input of Arrowhead (Rao et al. 2014) and IS (Crane et al. 2015) algorithms to call TADs, and TAD boundaries for the LCL merged call set. Finalized TAD boundary results for each individual were defined as those sample boundaries located within the merged boundary plus 10 kb flanking regions (the size of the exact TAD boundary called by IS for each individual) on the left side of the boundary start site and the right side of the boundary end site (Yu et al. 2017). The two figures located in the bottom left corner are shown as a comparison between the merged subject level and single subject level, which includes the Hi-C contact maps, the insulation scores, and the boundary strengths for the merged call set (5 kb) and the GM19036 (10 kb) sample over the region Chr 14: 35–35.8 Mb. (B) We examined the impact of SVs on chromatin structure by measuring the boundary score for each TAD boundary with the presence or absence of SVs. The Wilcoxon rank-sum test was employed to identify SVs significantly affecting TAD boundary strength, resulting in a set of TAD–SVs.
The Integrative Catalog in human 3D structure generated from this study aggregates Hi-C data from multiple projects
The combined data represent 27 billion sequenced read pairs and 17 billion contacts that quantify the intensity of interactions between any two genomic regions and result in a high-resolution Hi-C contact map at 300 bp. After processing the data through two famous TAD calling algorithms, namely, Arrowhead and Insulation Score (IS), we developed an Integrative Catalog of TADs at a 5 kb resolution that contains 14,764 TAD boundaries and 18,865 TADs, including 4596 sub-TADs (Supplemental Fig. S1; Supplemental Tables S2, S3). The comparison between our Integrative TAD Catalog and the previously published TAD map for GM12878 human LCLs (Supplemental Table S4; Supplemental Fig. S2; Rao et al. 2014) revealed that our catalog covers all of the TADs of GM12878 (The ENCODE Project Consortium 2004) (1 bp overlap) and 97.76% TADs of GM12878 with a 50% reciprocal overlap (Supplemental Figs. S2, S3), but identified additional 2328 novel TADs (including 356 sub-TADs) (Phillips-Cremins et al. 2013; Rao et al. 2014; Dixon et al. 2016) that were missing from the previously published TAD map for GM12878 (Fig. 2; The ENCODE Project Consortium 2004). The average length of our TADs is 282 kb, while the largest TAD spans 7265 kb, and the smallest TAD measures 20 kb. In contrast, GM12878 TADs exhibit an average length of ∼264 kb, with the smallest TAD at 60 kb and the largest at 3310 kb (Supplemental Fig. S4). Using a 1 bp overlapping criterion, we found that 12.34% of the TADs in our Integrative TAD Catalog call set did not overlap with the ENCODE TAD map for GM12878 and were designated as novel TADs.
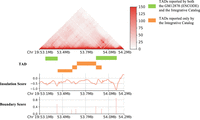
Visualization of one such region containing TADs identified in our Integrative Catalog but missing in the GM12878 released by ENCODE. From top to bottom, these plots show the Hi-C contact maps, the insulation scores, and the corresponding TAD boundaries with the boundary scores over this region. Green regions represent TADs identified by both GM12878 (ENCODE) and our Integrative Catalog, while orange regions highlight TADs identified by our pipeline but not in GM12878 (ENCODE).
Comparison of TADs called using additional algorithms
To obtain increased confidence in the TAD calls made by Arrowhead and IS, we repeated the TAD analysis using two additional TAD callers. One of the TAD callers is a single-level TAD caller, DomainCaller, which is not designed to identify sub-TADs (https://xiaotaowang.github.io/TADLib/domaincaller.html; Wang et al. 2017). The other TAD caller does have a hierarchical TAD calling feature and is named SpectralTAD (Cresswell et al. 2020). Hi-C data from the merged 44 samples were processed through both callers to achieve TAD calls at the same 5 kb resolution. DomainCaller detected 8042 TADs, of which 7963 (99.02%) TADs were already included in 18,611 (98.65%) of our TAD catalog (Supplemental Figs. S5A, S6A). SpectralTAD identified 53,851 TADs, of which 53,637 (99.60%) TADs were already included in 18,171 (96.32%) of our reported Integrative TAD Catalog (Supplemental Figs. S5B, S6B). We also examined the replication of the 301 TADs associated with our identified 185 TAD–SVs by these two methods. We found that DomainCaller was able to replicate 100% of those TADs, and SpectralTAD could detect 92.69% of the associated TADs. These results demonstrate that our Integrative Catalog has highly accurate TAD calls.
Quantifications of the strength of TAD boundaries
The next part of our study aimed to more precisely define the boundaries for the TADs called from the IS method. Given that SVs overlapping with known TAD boundaries have the potential to disrupt these TAD boundaries and subsequently influence gene regulation and phenotypes, it is crucial to define TAD boundaries accurately to examine which SVs actually disrupted chromatin structures. We identified 14,764 boundaries (using boundary scores, BSs ≥ 0.18, see details in Methods) in our Integrative Catalog (Supplemental Table S3; Supplemental Fig. S7). The BS is defined as the difference in the delta vector (the difference between the amount of insulation changes 100 kb to the left and right of the central bin) between the nearest 5′ local maximum and 3′ local minimum relative to the boundary, which can be used to filter out the potential boundary (Crane et al. 2015). Higher BS values represent stronger boundaries, whereas lower BS values indicate weaker boundaries. We asked the question of whether any TAD features were specific for a given human superpopulation (AFR, EAS, EUR, SAS, and AMR); however, we observed that their BSs appeared to be well conserved across different human superpopulations (P-value = 0.5084, Kruskal–Wallis test) (Dixon et al. 2012, 2016).
Disruption of 3D chromatin structure by SVs
To investigate the effect of SVs on disrupting TAD boundaries and thereby changing the landscape of chromatin structure, we examined the HGSVC2 (Ebert et al. 2021) cohort of the data, a subset of the 44 samples consisting of 26 individuals (the GM12329 sample was excluded due to a relatively low Hi-C sequencing quality) with both TAD boundaries and comprehensive SV genotypes available (Fig. 3; Supplemental Fig. S8). The SV call set was compared with the human reference GRCh38 and genotyped by PanGenie (Ebler et al. 2022) (referred to as PanGenie-genotyped SV calls). We started with 12,655 deletions and 17,257 insertions after filtering (Supplemental Fig. S9, see the filtering details in Methods) and observed that 1740 deletions and 2510 insertions overlapped with 1413 and 1750 TAD boundaries, respectively, across these 26 genomes. To assess the impact of SVs on chromatin structure, we measured the boundary strength (quantified as the BS) for each TAD boundary with and without the presence of SVs. Specifically, we compared the changes in BS between individuals with the homozygous genotype (0/0) for this SV and that of individuals who carry genotypes that include at least one alternative allele (1/1, 0/1, or 1/0) of the SV. Although most SVs (95.65% of the PanGenie SV set, 1674 deletions [DELs], and 2391 insertions [INSs]) appear at TAD boundaries do not significantly affect the 3D chromatin structure, we indeed identified 185 SVs (66 deletions and 119 insertions) that significantly disrupted TAD boundaries (referred to as TAD–SVs; False discovery rate [FDR] < 0.05, Wilcoxon rank-sum test, two-sided) (Fig. 1B; Supplemental Table S5).
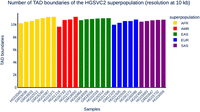
Distribution of TAD boundaries in 27 samples of HGSVC2. The x-axis represents sample IDs, with the superpopulation ordered and represented by a different color, as displayed in the color key in the legend. The y-axis shows the number of TAD boundaries detected using our pipeline in the 27 samples. All of these TAD boundaries were called under a 10 kb resolution for each individual utilizing the IS method, owing to the relatively low sequencing depth and map resolution.
Among these 185 TAD–SVs, which are associated with 301 unique TADs, 53 associated TADs were novel in our TAD catalog, while the remaining TADs were previously reported in GM12878 (Rao et al. 2014). None of these TADs were previously associated with any SV. These findings highlight that numerous SVs in the human genome can alter the insulation strength between nearby TADs, thereby changing the frequency of interactions between sequences within typically isolated domains. Specifically, we observed that deletions (22 out of 66) showed statistical evidence of the TAD boundaries and resulted in the fusion of the adjacent TADs (Fig. 4A,C,E; Ibn-Salem et al. 2014), while the insertion of a sequence (58 out of 119) is capable of splitting a single TAD into two adjacent TADs (i.e., the formation of a neo-TAD) (Fig. 4B,D,F; Supplemental Fig. S10; Willemin et al. 2021).
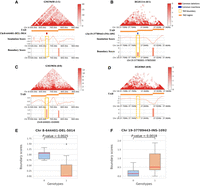
Visualization of two SVs that disrupt TAD boundaries with significant changes in boundary strength. (A,C) A deletion (Chr 8-644401-DEL-5014) that disrupts the TAD boundary and shows differences in Hi-C contact maps, BSs, and insulation scores for individuals with (genotype 1/1) and without (genotype 0/0) the deletion. The orange rectangle shows the TAD location for each sample within the plot region. The dark red rectangle represents the location of this deletion, and the yellow rectangle highlights the TAD boundary location and corresponding boundary strength. The top left figure is the GM19650 sample, whose genotype is 1/1, i.e., it carries this deletion, compared to the sample below, GM19036, whose genotype is 0/0, i.e., it does not have this deletion. The BS panel shows that the GM19650 sample lacks the TAD boundary where it carries that genomic deletion. (B,D) Similar comparisons for an example of an insertion (Chr 19-37789443-INS-1092) between the individual HG01114 (genotype 0/1) and HG03065 (genotype 0/0). The BS panel shows that the HG01114 sample has the TAD boundary where it carries that genomic insertion. (E,F) Boxplots demonstrating the significant differences in BSs for two genotype categories, 0 (genotype 0/0) and 1 (genotypes 0/1, 1/0, or 1/1).
We further explored the different impacts between homozygous and heterozygous SVs. In doing so, we split the genotypes of the 185 TAD–SVs into homozygous (1/1) and heterozygous (0/1 and 1/0) and compared the changes in BS between individuals who do not have the SV (0/0) and who carry these homozygous SV (1/1) and the heterozygous SV (0/1 and 1/0). As a result, we found 33 TAD–SVs (11 deletions and 21 insertions) that show statistically significant changes among the three genotype types (FDR < 0.05, Kruskal–Wallis test) (Supplemental Table S6). One deletion (Chr 7-149878840-DEL-164) and two insertions (Chr 3-84900406-INS-15193 and Chr 6-170399369-INS-349) show a significantly different impact level of the homozygous and heterozygous genotypes on the 3D genome (P-value < 0.05, Conover test) (Supplemental Fig. S11).
Enrichment of TAD–SVs in cCREs
To investigate which TAD–SVs overlap with functional elements, such as ENCODE-defined regulatory elements, we intersected all TAD–SVs with the latest version (V3) of the registry of cCREs produced by the ENCODE Project Consortium (The ENCODE Project Consortium et al. 2020). We observed that 18 deletions and 16 insertions were associated with candidate regulatory elements, corresponding to the disruption of 55 total cCREs (Supplemental Fig. S12; Supplemental Table S7). In aggregate, the TAD–SVs disrupted 42 enhancer elements and six promoter elements. Additionally, given the importance of CTCF-binding sites in the formation and maintenance of TAD boundaries, we found that TAD–SVs disrupted 27 cCREs with evidence of CTCF binding (17 due to deletions and 10 due to insertions). For each cCRE category, we found enrichments of TAD–SVs within cCREs compared with the entire genome (fold-change 3.02–9.85, P-value < 1.76 × 10−10, Fisher's exact test), likely due to the known enrichment of TAD boundaries within cCRE regions (Madani Tonekaboni et al. 2019). A similar hypothesis was supported by permutation test results on 10,000 rounds of putatively generated SVs that shared the same distribution of counts and length per chromosome as the TAD–SVs (Supplemental Fig. S13), which suggested that TAD–SVs are likely to be enriched for cCREs more than is expected by chance.
Enrichment of CTCF at TAD boundaries
TAD boundaries are known to be enriched for CTCF (Dixon et al. 2012). To examine this in our identified TAD boundaries, we evaluated the intersection of the flanking TAD boundaries with the hg38 transcription factor ChIP-seq peaks of CTCF in GM12878 from ENCODE 3 (The ENCODE Project Consortium 2004). Our findings reveal that ∼68.45% of the flanking TAD boundaries overlapped a CTCF ChIP-seq peak, which aligns with previous studies (Dixon et al. 2012; Rao et al. 2014; Long et al. 2022). To further explore, the enrichment of CTCF at TAD boundaries, we randomly shuffled our flanking TAD boundary set and calculated the frequency of overlap between CTCF and the shuffled TAD boundary set, repeating this permutation process 10,000 times and received a P-value < 0.0001 (Supplemental Fig. S14). These results indicate the enrichment of CTCF at our identified TAD boundaries, highlighting that CTCF binding is more common at TAD boundaries than what would be anticipated by random chance.
Depletion of deletion at TAD boundaries
Earlier studies reported that TAD fusion, resulting from a chromosomal deletion, is anticipated to experience negative selection during evolution (Fudenberg and Pollard 2019; Huynh and Hormozdiari 2019). To ascertain whether deletions tend to avoid disrupting TAD boundaries more than is expected by chance, we intersected all the deletions in our recently released high-quality PanGenie genotyped SV call set with our defined TAD boundaries and flanking TAD boundaries, respectively. By permuting deletions while retaining the same number and length distributions of these deletions for each chromosome, we calculated deletions that overlapped with the TAD boundaries and repeated this permutation procedure 10,000 times in total. We observed that the number of deletions intersecting with flanking TAD boundaries was significantly less than that of randomly permuted genomic regions (P-value = 0.0053) (Supplemental Fig. S15). These observations support the hypothesis that deletions are depleted at TAD boundaries, implying that disrupting TADs are indeed under stronger selection pressure compared with random genomic regions (Fudenberg and Pollard 2019; Huynh and Hormozdiari 2019).
Intersection of TAD–SV with GWAS-significant SNPs in high LD
Prior research suggests that some of the single-nucleotide polymorphisms (SNPs) associated with human traits and diseases identified in genome-wide association studies (GWAS) might be linked to diseases through their linkage disequilibrium (LD) with causal SVs (Lappalainen et al. 2013; Liang et al. 2024). Here, we sought to determine which TAD–SVs acting as causal variants could steer SNPs that are in strong LD with them to show strong disease associations. We leveraged a recent database of GWAS-significant SNPs in high LD with the same SVs included in our study on HGSVC2 individuals (Ebert et al. 2021; Liang et al. 2024). Our analysis revealed 19 TAD–SVs (11 deletions and eight insertions) intersecting with 76 entries in the database of GWAS-significant SNPs in high LD, which involved 45 unique SNPs (Supplemental Table S8).
Identification of TAD–SV-QTLs
We hypothesized that SVs in the human population, disrupting TAD boundaries, would also have a functional impact on gene regulation (Kim et al. 2022). To investigate this, we overlapped the TAD–SVs identified in this study with SV expression quantitative trait loci (eQTLs) characterized in Ebert's study (2021), which analyzed 430 individuals whose gene expression profiles were requantified from the RNA sequences generated from the Geuvadis project (Lappalainen et al. 2013) (note that the 26 HGSVC2 samples used in this study form a subset of these 430 individuals) (Supplemental Fig. S16). Specifically, we intersected the 185 TAD–SVs with the 850,482 SV-eQTLs reported by Ebert et al. (2021) and identified 20 TAD–SVs (11 deletions and nine insertions) that are also SV-eQTLs. Although SVs have been previously reported to be associated with gene expression changes as SV-eQTLs (Ebert et al. 2021), here, we disclosed that five such SV-eQTLs (three deletions and two insertions; we termed this kind of SVs as TAD–SV-eQTL) (Supplemental Table S9) may affect the expression of six associated genes through their disruption of TAD boundaries in our 26 HGSVC2 samples (FDR < 0.05, Wilcoxon rank-sum test, two-sided). For example, a 5014 bp deletion that disrupts a TAD on Chr 8 overlaps with an SV-eQTL for the gene ERICH1, whose expression is increased in the presence of the deletion (Fig. 5A,B).
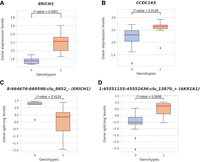
Visualization of two SVs that disrupt TAD boundaries with significant changes in gene expression and splicing levels. (A) Boxplot demonstrates the significant difference in gene expression for two different genotype categories, 0 (genotype 0/0) and 1 (genotypes 0/1, 1/0, or 1/1). The boxplot on the left shows the significant changes between the associated ERICH1 gene expression values (after log transformation) and different genotypes of this deletion Chr 8-644401-DEL-5014. (C) Boxplot shows the significant difference between gene splice ratios (after log transformation and quantile normalization) for the splice junction clusters and genotypes of the same deletion, which is also associated with the ERICH1 gene. (B,D) The same comparisons for an example of insertion (Chr 1-45497763-INS-354) associated with the CCDC163 and AKR1A1 genes between genotype categories 0 and 1.
In some cases, we hypothesized that certain SVs that disrupted TAD boundaries also ultimately impacted gene splicing. In a similar vein, we overlapped our newly identified 185 TAD–SVs with 1,103,872 previously reported SV splicing quantitative trait loci (sQTLs), which were derived from an identical pool of 430 individuals (Ebert et al. 2021). Out of these 185 TAD–SVs, 10 (including six deletions and four insertions) of them intersected with these previously reported SV-sQTLs. In particular, six of these TAD–SVs (four deletions and two insertions; we termed these SVs as TAD–SV-sQTLs) (Supplemental Table S10) were associated with splicing changes of seven genes among our 26 samples, potentially due to the disruption of TAD boundaries (FDR < 0.05, Wilcoxon rank-sum test, two-sided). For instance, we found a 354 bp insertion that disrupts a TAD on chromosome one, overlapping with an SV-sQTL for the AKR1A1 gene, whose splicing increases in the presence of the insertion. These observations reveal that the deletion and insertion of TAD boundaries can result in significant changes in gene splicing within human genomes (Fig. 5C,D).
Three TAD–SVs showed significant alterations in both the patterns of gene expression and splicing (namely Chr 8-644401-DEL-5014 [ERICH1], Chr 8-70671321-DEL-1088 [XKR9], and Chr 1-45497763-INS-354 [CCDC163 and AKR1A1]), where Chr 8-70671321-DEL-1088 was also found to be reported in high LD with disease-associated SNPs in GWAS (Liang et al. 2024). One intriguing TAD–SV, Chr 19-37789443-INS-1092, may serve as a candidate TAD–SV-eQTL. While this insertion has relatively weak significance in terms of affecting the gene expression changes (P-value < 0.0605), it demonstrates statistical support for influencing the gene splicing changes (P-value < 0.0123) and is also documented as exhibiting high LD with disease-associated SNPs in GWAS (Liang et al. 2024).
Mediation analysis of TAD–SVs’ impact on gene regulation
We further investigated TAD–SV-QTLs that demonstrated significant evidence of affecting gene regulation and aimed to determine whether these TAD–SV-QTLs exert their effects on gene regulation directly or indirectly by altering chromatin organization, which acts as a driving mediator. To this end, we performed causal mediation analysis (Supplemental Fig. S17; Imai et al. 2010; see Methods) on each TAD–SV-QTL. Mediation analysis tests a hypothetical causal chain where one variable X affects a second variable M, which in turn affects a third variable Y. As a result, we found one such insertion, Chr 15-76198866-INS-67, associated with the SCAPER gene, whose gene regulatory impact on gene expression is likely to be mediated by the disruption of the corresponding TAD structure (P-value [average causal mediation effect, ACME] = 0.086, P-value [average direct effect, ADE] = 0.226, P-value [Total effect] = 0.024) (Supplemental Tables S11, S12). We observed that the majority of the TAD–SV-QTLs show significant statistical evidence, indicating the SVs themselves directly drive the functional impact on gene regulation.
Evaluation of novel TAD boundaries induced by TAD-INSs
Architectural proteins, such as CTCF and the cohesin complex, play crucial roles in forming boundaries between TADs. The significance of the genomic context in endowing a specific DNA sequence with the capacity to act as a boundary element remains an intriguing subject to be elucidated (Willemin et al. 2021). Here, we specifically focused on the 58 identified TAD-INSs, which exhibit a tendency to add or strengthen TAD boundaries in samples with insertions compared to those without insertions (Supplemental Fig. S18). We argue that the inserted sequence in the TAD-INSs may embrace some necessary components, such as CTCF-binding sites, to strengthen the respective TAD boundaries. To examine this hypothesis, we conducted a motif analysis to identify the CTCF motif among these 58 inserted sequences, with a length between 51 and 54,333 bp (see Methods). In total, we identified 10 insertions that show statistical evidence of containing the CTCF motif (Q-value < 0.2), which we named CTCF-TAD-INSs (Supplemental Tables S13, S14). Additional nine insertions exhibited a small P-value (<0.0001), bound with the evidence of containing a CTCF motif regarding their higher FIMO scores (>10) (Matthews and Waxman 2018). We observed that larger insertions are more capable of being found with a CTCF motif. Among our 58 TAD-INS candidates, 18 insertions have a length larger than 1 kb, and seven demonstrated statistically significant evidence of encompassing the CTCF motif in their sequences. Conversely, only three of the remaining 40 smaller insertions showed the occurrences of CTCF motifs. This suggests that TAD-INSs of different sizes may exhibit variability in their likelihood of introducing CTCF motifs.
Comparison of TAD boundaries with loop anchors and compartment domain borders
With the advance of the improvement of the resolution of Hi-C data and functional genetic research over the years, there are some new observations about the distinct classes of chromatin domains which are commonly referred to as TADs in the literature (and our paper), actually including loop domain and compartment domain, or a combination of both (Rowley and Corces 2018). Specifically, those chromatin domains that align with corner dots at their apexes are known as loop domains (Supplemental Fig. S19), and the chromatin domains whose borders coincide with the inflection points of A or B compartmentalization signals, are defined as compartment domains (Dharanipragada and Parekh 2019), which have recently been shown to significantly overlap (Rowley and Corces 2018). In total, we detected 28,340 loops at 5 and 10 kb merged resolutions with 37,001 unique loop anchors (Supplemental Table S15). Due to the limited computational resources, we only called compartments under 100 kb and received 2261 compartments in total (1139 A compartments and 1122 B compartments) (Supplemental Table S16). We found that 807 (5.47%) TAD boundaries correspond to 792 compartment domain borders and 10,031 (67.94%) TAD boundaries are associated with the 16,199 loop anchors, which are consistent with previous research findings (Rao et al. 2014). The relatively lower portion of overlap between the TAD boundaries and compartment borders could be explained by the different selection of the bin sizes that were used to identify the TADs and compartments in our study.
Recent studies reported evidence that the removal of loop anchors by deletions is under strong purifying natural selection to maintain the integrity of the loop (Radke et al. 2021). We examined this hypothesis in our PanGenie data set to assess whether deletions have a depletion effect on the loop anchor loci. We thus intersected all the deletions from the PanGenie genotyped SV calls with our identified loop anchors (Ebler et al. 2022). We kept the same number and length distributions of these deletions for each chromosome, calculated the deletions that overlapped with the loop anchors, and repeated this permutation procedure 10,000 times. We observed that the number of deletions overlapping with loop anchors was significantly lower in the PanGenie set than in the randomly permuted sets of deletions (P-value = 0.0003) (Supplemental Fig. S20). These results suggest that deletions indeed tend to avoid removing loop anchors (Radke et al. 2021).
Replication in an independent data set
To replicate the findings of the 185 TAD–SVs from our discovery set of 26 individuals, we collected another 17 samples from the 1000 Genomes Project that had both SV genotype and Hi-C data available. We found 3054 SVs (1267 deletions and 1787 insertions) in the replication set, of which 120 SVs (40 deletions and 80 insertions) overlapped with the 185 TAD–SVs from the discovery set. We established a Q-value for each of these 3054 SVs based on each of their genotype associations with the BS at their overlapping TAD boundaries. We then ranked these SVs based on their Q-values and surmised that the SVs that had the most significant impact on the strength of TAD boundaries in the replication set would also overlap with the TAD–SVs found in the discovery set. This analysis reveals that 31.67%, 61.67%, and 84.17% of the 120 replicated TAD–SVs were ranked in the top 25%, 50%, and 75%, respectively, of the TAD–SVs in the independent replication cohort of 17 individuals (Supplemental Table S17). Through this analysis, we thus identified that most of the TAD–SVs from the discovery set were indeed ranked in the top 50% of the SVs in the replication set.
Discussion
The 3D genome has revolutionized the field of genomics by demonstrating how 3D organization can impact gene expression. The availability of a thorough 3D genome map would provide a valuable resource to assist with the understanding of human genome structure and function. Although several projects have produced Hi-C data at various resolutions, including 4DN (n = 19, average resolution = 11,571 bp), as well as investigations into GM12878 (resolution = 950 bp), little effort has been made to aggregate these data to generate an integrative genome-wide Hi-C map. In this study, we have integrated these publicly available Hi-C data, together with Hi-C data recently generated on 27 individuals in the HGSVC2 project, and produced a genome-wide Hi-C map at a 300 bp resolution across 44 individuals. Our work not only resulted in the generation of an open-access integrative TAD catalog for future research but also defined a set of TAD–SVs with functional annotations through their enrichment/depletion analysis and overlapping with particularly noncoding regulatory regions and functional data.
Our study develops a strategy for reanalyzing and harmonizing existing data to generate a unified resource to characterize 3D structures across human genomes. Several systematic evaluation studies have revealed a notable lack of concordance among existing methods for calling TADs or sub-TADs, especially regarding the determination of the number and size of TADs. However, TAD boundaries are reported to be more consistently defined across different callers (Forcato et al. 2017; Zufferey et al. 2018; Zhang et al. 2024). Hence, we primarily concentrated on the detection of TAD boundaries in this study. The rationale for choosing the IS to detect TAD boundaries is that the IS is the most recognized method capable of quantifying boundary strength alongside the identified TAD boundary locus, which also provides quantifications of TAD boundaries that make it possible to conduct statistical tests between SV genotypes and TAD boundaries to identify TAD–SVs. Other methods for quantifying TAD boundaries and other TAD callers can be included in future studies.
In the present investigation, we have evaluated how SVs affect the 3D genome using the new TAD catalog we have assembled. We have identified 185 SVs that disrupt TAD boundaries, among which five TAD–SVs are associated with gene expression changes and six TAD–SVs are associated with transcript splicing changes. Previous research has revealed that alterations in the genome's primary DNA sequence can result in chromatin structural modifications, which in turn cause dysregulation of gene expression, leading to pathogenic phenotypes, such as aging-related diseases (Chen et al. 2018b), Alzheimer's disease (Won et al. 2016, 2019; de la Torre-Ubieta et al. 2018), cancer (Dixon et al. 2018; Ibrahim and Mundlos 2020), and developmental disorders (Lupiáñez et al. 2015; Spielmann et al. 2018; Luo et al. 2021). In recent studies, cancer development and progression have also been shown to be influenced by allele-specific expression (ASE) (de Souza et al. 2020; Robles-Espinoza et al. 2021). Among the genes that have expression differences associated with TAD–SVs, we found a few genes, such as ERICH1 (associated with Chr 8-644401-DEL-5014) and XKR9 (associated with Chr 8-70671321-DEL-1088, also reported in the database of GWAS SNP–SV pairs) (Liang et al. 2024), that have been previously reported to be associated with cancer (e.g., such as pancreatic cancer and gastric cancer) as well as tumor progression (Brown et al. 2018; Dharanipragada and Parekh 2019; Li et al. 2020). ERICH1 was also reported to be genomically imprinted and, therefore, linked to ASE (Knight 2004; Morley et al. 2004; Baran et al. 2015; Shao et al. 2019). For XKR9, one recent study reported that its expression is significantly associated with the overall survival rate for all types of cancers and various individual cancer types, which implies that XKR9 might be a novel potential therapeutic target for cancer immunotherapy (Li et al. 2020). We envision that our Integrative Catalog of TADs and TAD boundaries will provide a vital reference map for future studies that want to investigate the disruption of TADs causing phenotypic changes. Genomic variation, including all types of SVs that overlap or intersect with TAD boundaries in human genomes, should be further investigated to better understand the molecular basis of human genetic diseases (Spielmann et al. 2018; Ibrahim and Mundlos 2020; Boltsis et al. 2021; Rajderkar et al. 2023).
However, there is room for improvement in our current strategy for developing a more detailed 3D map of the human genome from multiple studies and data sources. For example, a recent study generated a Hi-C map with 33 billion contacts for 10 individuals (Harris et al. 2023). Although this new Hi-C map offers a higher resolution compared to the one presented here, its primary focus differs from ours as it does not extensively analyze the impact of SVs at the TAD level. We recognize the potential of incorporating these new Hi-C data (Harris et al. 2023) into our next phase of data integration, as well as emerging Hi-C data to be generated from more 1000GP samples in the HGSVC (https://www.hgsvc.org/) and related projects. This addition of future data sets will increase the power of both our discovery study and replication experiment. Additionally, using human pangenomes (Liao et al. 2023) or even personal genomes (Rozowsky et al. 2023) instead of the reference genome for read alignment would allow for more accurate mapping of Hi-C reads and the discovery of individual-specific TADs. Future enhancements of this catalog could also include direct TAD characterization using high-resolution Hi-C data for each individual, should such data become available. This approach would eliminate the need for merging large sample sets, thereby providing a fine characterization of 3D human genome organization and mitigating issues arising from the current merging strategy.
Moreover, our presented Integrative TAD Catalog is limited to LCL cell lines, as we use samples from the 1000 Genomes Project. Due to resource constraints, we are not in a position to explore the TAD variation across different tissue types within individuals, which remains an area for future investigation. Although it is commonly believed that TADs are highly conserved across cell types and possibly different species (Kentepozidou et al. 2020; Dang et al. 2023), some recent studies argue that the extent of this conservation remains uncertain and has been examined in only a limited number of samples. It was reported in Sauerwald et al.’s study (2020) that only 40% of TAD boundaries are shared between different tissue types, consistent with the findings of Schmitt et al.’s study (2016). A similar conclusion is made in McArthur and Capra's study (2021), where they demonstrated significant variability in the landscape of TAD boundaries across different cell types. In Okhovat et al.’s recent study (2023), they found that only 13.6% of the TAD boundaries in the human genome in LCLs were ultraconserved, and only 15% were human-specific. Thus, improved identification of TADs and TAD boundaries using high-resolution Hi-C sequencing and enhanced SV calling and genotyping not only in the LCL cells but also in other tissues and cell types will greatly benefit a more detailed understanding of TADs and TAD–SVs in future exploration (Eres and Gilad 2021).
Furthermore, our analysis utilized the latest characterized SV call set from PanGenie (Ebler et al. 2022) that only contained genotyped deletions and insertions (Ebert et al. 2021). In the future, we will extend our investigation to characterize the impact of other types of SVs (e.g., duplications and inversions) and ancestral alleles on human-specific chromatin structure and gene regulation. Furthermore, the primary focus of our current pipeline centers around TAD calling, given the scope of the current study. It has been shown that chromatin loops are formed through strong interactions between specific genomic regions, particularly involving the CTCF and enhancer–promoter loops (Pal et al. 2019; Tena and Santos-Pereira 2021). Moving forward, we will broaden our evaluation to encompass the impact of genetic variants (including SVs) on loops and gene regulation, which will become feasible as an elevated resolution of Hi-C data becomes available on individuals with high-quality genotypes in human genomes. Additionally, rather than focusing solely on the direct overlap between cCREs and TAD–SVs, as presented in the current study, a closer investigation into whether the disruption of cCREs is caused by SVs or alterations in 3D genome structure will be reserved for future research. This kind of analysis will become feasible once we obtain data on more samples and collect summary statistics data on cCREs, enabling us to conduct a causal mediation analysis. Future work will also include experimental studies to dissect and interpret the overlap between TAD–SVs and cCREs directly.
Methods
Hi-C data collection
We have collected Hi-C data on 44 samples from HGSVC2, HGSVC1, 4DN, and GM12878 (Supplemental Table S1). The first data set (HGSVC2) is the Hi-C data for the 27 HGSVC2 samples generated from the Human Genome Structural Variation Consortium (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/working/20200512_Hi-C/). Hi-C libraries were generated with 1.5 M human cells as input using Proximo Hi-C Kits v3.0 (Phase Genomics) according to the manufacturer's protocol with the following changes: cells were cross-linked, quenched, and lysed sequentially with Lysis buffers 1 and 2, and liberated chromatin immobilized on magnetic recovery beads. During the fragmentation process, a cocktail of 4-cutter restriction enzymes (DpnII [GATC], DdeI [CTNAG], HinfI [GANTC], and MseI [TTAA] was utilized to enhance coverage and facilitate haplotype phasing. Fragmented chromatin was proximity ligated for 4 h at 25°C after fragmentation and filled with biotinylated nucleotides. The cross-links were then reversed. Magnetic streptavidin beads were used to purify DNA and retrieve biotinylated junctions. The dual-unique indexed library was created using bead-bound proximity ligated fragments and Illumina sequencing chemistry. Fluorescence-based assays, such as qPCR with the Universal KAPA Library Quantification Kit and Tapestation, were used to assess the Hi-C libraries (Agilent). The libraries were sequenced at the New York Genome Center (NYGC) in a paired-end 150 bp format on an Illumina NovaSeq 6000 platform.
The second (HGSVC1, https://www.ebi.ac.uk/ena/browser/view/PRJEB11418) and third (4DN, https://data.4dnucleome.org/publications/b8c7c5f5-c76f-457f-9a0d-6c567924b816/#expats-table) pilot data sets are the Hi-C sequencing data, which were generated for the 1000 Genomes Project SV group by the laboratory of Bing Ren using the Illumina HiSeq (2000, 2500, or 3000) paired-end sequencing (Dekker et al. 2017; Chaisson et al. 2019; Gorkin et al. 2019) and were performed using a “dilution” 6-cutter HindIII (AAGCTT) protocol. The second pilot data set was obtained from Hi-C sequencing of three trios: Yoruba (NA19238, NA19239, and NA19240), Han Chinese (HG00512, HG00513, and HG00514), and Puerto Rican (HG00731, HG00732, and HG00733). Three individuals, namely, HG00514, HG00733, and NA19240, were excluded from this data set since these samples were re-sequenced in the HGSVC2 study. For the same reason, GM19238 from the 4DN pilot data set was also excluded.
We included the Hi-C data from GM12878 B-lymphoblastoid cells in our analysis from Rao et al. (2014), which has almost 5 billion mapped paired-end read pairs and is considered the largest coverage for the accurate identification of 3D chromatin structures. The sequence data were produced as in Rao et al. (2014) and can be downloaded from ENCODE (https://www.encodeproject.org/experiments/ENCSR410MDC/). Note that 26 HGSVC2 samples were used in the functional analysis in this study due to the low sequencing quality of GM12329. We still include this sample in the 44 samples used to generate the Integrative TAD Catalog in this paper.
Hi-C data processing
We required a minimum alignment quality for each read included in our Hi-C maps. We used the mapping quality score (MAPQ), which quantifies the probability that a read is misplaced, to filter out the read pairs where the alignment of one or both reads failed to meet these two thresholds: MAPQ > 0 and MAPQ ≥ 30 (Rao et al. 2014). In our study, we used the MAPQ ≥ 30 filtered data to be stringent about avoiding false positives caused by poor alignment. The result of filtering low-quality alignments is a list of Hi-C contacts.
For each sample, the raw reads were mapped to the GRCh38 reference genome and processed using Juicer software tools (version 1.6) with default aligner BWA-MEM (Li and Durbin 2009; Durand et al. 2016b). Unmapped reads such as abnormal split reads and duplicate reads were removed, and low mapping quality read pairs were filtered out if their mapping quality value MAPQ was less than 30. Those filtered read pairs (Hi-C contacts) were subsequently used to construct chromatin contact maps for each sample by Juicer. To construct a Hi-C contact map on an Integrative TAD Catalog of LCLs basis, contacts were pooled across all 44 individuals using the mega.sh script provided by Juicer. Previous studies report that Arrowhead consistently outperforms numerous other TAD callers in tests for the enrichment of TAD-associated biological features and the detection of hierarchical TAD architecture (Zufferey et al. 2018), which are central to our analysis in this study. Arrowhead demonstrates superior performance, particularly at high-resolution matrices (5–10 kb), as evidenced by the limited number of TADs identified by Arrowhead for nearly all individuals in our data sets. Thus, we aggregated individual contacts together to achieve a higher resolution and leverage the capabilities of Arrowhead to generate an Integrative Catalog of TADs and TAD boundaries in this study. The outputs of the previous processes are two sets of HIC files, a particular format of the highly compressed binary file used to store contact matrices with various resolutions (i.e., bin sizes). They can be supported mainly by Juicer, JuicerTools, and Juicebox command line tools for downstream analysis and visualization (Durand et al. 2016a). All analyses and results reported in our study employ the contact frequency matrices normalized with SCALE matrix balancing (Durand et al. 2016b), which remedies the issue that Knight and Ruiz (KR) normalization sometimes does not provide coverage for a particular region or chromosome (Knight and Ruiz 2012).
We used verifyBamID to verify whether the Hi-C sequencing reads in our aligned files match the PanGenie genotyped SV calls (https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/release/v1.0/PanGenie_results/) of the 44 individuals used in this study (Jun et al. 2012; Ebler et al. 2022). It can also identify whether the reads have been contaminated or swapped by a mixture of two samples. We did this for each of our samples (Supplemental Table S18), and we observed that one of the samples (GM19204) has 12% or more nonreference based observed on the reference cites, which is much greater than the normal 2% standard criteria and indicated that this sample is very likely to have contamination. For this reason, we excluded this sample from any of our genotype-related downstream analyses.
Calculation of Hi-C map resolution
To determine the applicable TAD map resolution to be called for each sample, we first applied the script from Juicer to calculate the map resolution of each sample (Durand et al. 2016b). The map resolution is intended to reflect the finest scale at which local features can be reliably detected. The lowest resolution is 17,800 bp (GM12329), while the highest is 4650 bp (GM19650). Juicer created a specific HIC file format to describe the input reads under nine different bin sizes (base pair-delimited resolutions: 2,500,000, 1,000,000, 500,000, 250,000, 100,000, 50,000, 25,000, 10,000, and 5000), and usually based on the sequencing depth of the Hi-C file, 5 kb or 10 kb resolution is used to call Arrowhead (Durand et al. 2016b). Considering that GM12878, with the largest contact map and deepest depth of sequencing in its Hi-C file to date (map resolution of 950 bp), was called under 5 kb resolution in the Arrowhead (Rao et al. 2014), we chose the same 5 kb as the practical resolution of TAD calling on all of our 44 merged samples (kilobase map resolution of 300 bp).
We applied the method used by Rao et al. (2014) to calculate the Hi-C map resolution of our 27 samples. The map resolution is defined as the smallest bin size for which 80% of bins have at least 1000 contacts, intended to reflect the finest scale at which local features can be detected reliably. The scripts for calculating the Hi-C resolution of each sample were directly downloaded from Rao's study (2014).
Identification of TADs and TAD boundaries
Two TAD callers, JuicerTools (version 1.22.01) Arrowhead and Insulation Score (IS), were used and compared to call TADs for 43 samples (excluding GM12878), respectively, at 10 kb resolution from the human LCLs (Crane et al. 2015; Durand et al. 2016b). Arrowhead is more accurate and sensitive for ultra-high-resolution data and focused on detecting the corners of the domains to locate the boundaries of TADs, while the IS algorithm is initially created to find TAD boundaries and quantify the boundary strength of Hi-C data with a relatively low resolution (Chen et al. 2018a; Zufferey et al. 2018; Yardımcı et al. 2019). For pool calling, a SCALE normalized merged contact matrix (44 samples) at 5 kb resolution was used to calculate the insulation scores and corresponding BSs using the “fanc boundaries” function FAN-C toolkit version 0.9.26b2 with default parameters to detect the TAD boundaries at a 100 kb window size (which was referenced from the 4DN domain calling protocol) (Dekker et al. 2017; Kruse et al. 2020). The same method was applied for sample calling, and the SCALE normalized contact matrix at 10 kb of each sample was used as input. Specifically, the IS defines a sliding window and sums contacts in this window to align the Hi-C matrix diagonal. The BS (also known as boundary strength) is defined as the difference in the delta value between the local maximum to the left and the local minimum to the right of the boundary bin. The delta value can be calculated as the difference in insulation between 100 kb to the left and 100 kb to the right of each central bin along the diagonal of the genome contact map. The insulation valleys or minima are considered TAD boundaries, whereas deeper insulation valleys indicate stronger TAD boundaries (Crane et al. 2015). Regions with low insulation scores (high BSs) are insulating and are referred to as TAD boundaries, while regions with high insulation scores (low BSs) are most typically found inside domains and are referred to as TAD regions, which are also considered as the regions between the two neighboring TAD boundaries (Kruse et al. 2020).
The sex Chromosomes X and Y were eliminated from all analyses because of sex disparities in our samples. ENCODE has processed the GM12878 files for hg38 and released the TADs called by Arrowhead on the SCALE normalized Hi-C matrices (The ENCODE Project Consortium 2004). We considered it a benchmark TAD map; thus, we compared their TAD results with the GM12878 results processed by our pipeline to determine a minimum boundary strength cutoff value (0.17) for GM12878, which yielded the most repetitive results. The value was then transformed to a percentile (∼51.95%) to find the corresponding cutoff score in our LCL call set. After the removal of the missing and duplicated BSs, a minimum BS cutoff value of 0.18 was chosen. The insulation scores, TAD boundaries, and corresponding BSs were visualized using Juicebox software and the FAN-C toolkit in Python 3.7 (Durand et al. 2016a; Kruse et al. 2020).
We next sought to generate a detailed TAD call set aligned with our TAD boundary locations for further downstream analysis (Fig. 1). Specifically, (1) we first called the TADs directly from Arrowhead under 5 kb resolution as our Arrowhead-TAD set; (2) the boundaries we previously received from the IS were first converted to TADs and then filtered by (a) excluding those TADs that contain more than 10% of the length of those TADs that have missing insulation scores and (b) removing TADs that do not have any maxima of insulation scores to be our IS-TAD set; (3) we then used BEDTools “intersect” (or “intersectBed”) (Quinlan and Hall 2010) to find the overlapping regions that are both detected by Arrowhead (Arrowhead-TAD set) and IS (IS-TAD set) when the reciprocal overlap was above 50%. For those overlapping regions that contain more than one TAD detected from Arrowhead, we kept the TAD locations identified from Arrowhead instead of from IS since Arrowhead is able to find the sub-TADs nested or overlapping within the larger TADs, which are demonstrated to have more cell or species-specific gene regulation activities within it (Dixon et al. 2012; Rao et al. 2014; Beagan and Phillips-Cremins 2020). We subtracted those regions both from the Arrowhead-TAD set and IS-TAD set to avoid duplicates; (4) we then merged the remaining overlapping regions from step 3 by the smallest start position and largest end position for each TAD; (5) we finally added the TADs that were distinctly detected from Arrowhead and IS, the sub-TAD regions from step 3, and the merged regions from step 4 to generate our final Integrative TAD Catalog.
TAD calling using DomainCaller and SpectralTAD
We selected two other popular TAD callers, DomainCaller and SpectralTAD. The DomainCaller is a TAD caller that implements the original directionality index (DI) proposed by Dixon et al. (2012). The original HIC format merged contact matrix was converted to .cool format using the tool hic2cool (https://github.com/4dn-dcic/hic2cool). Then “cooler balance” was added to the converted .cool file to perform the built-in filtering and balancing using iterative correction (Abdennur and Mirny 2020). The new balancing normalization and the same window size of 100 kb were specified in the calling process of DomainCaller. The SpectralTAD was run on the original HIC contact map without normalization as suggested (Cresswell et al. 2020). Specifically, we detected TADs with two levels of hierarchical structures and turned on the “quality filtering” option.
Identification of loops and compartments
Chromatin loops of the LCLs merged set (44 samples) were identified by HiCCUPS GPU (Rao et al. 2014) after filtering contacts with MAPQ below 30, with a merged resolution at 5 kb and 10 kb, respectively, on the SCALE normalized Hi-C matrix with -m 2048 -r 5000,10000 -k SCALE -f .1,.1 -p 4,2 -i 7,5 -t 0.02,1.5,1.75,2 -d 20000,20000 ‐‐ignore-sparsity parameters.
The A/B compartments were identified at the same filtered and normalized Hi-C matrix at 100 kb bin size using the “fanc compartments” function from the FAN-C toolkit version 0.9.26b2 with a hg38 genome FASTA file provided by the -g command and all other default parameters. Positive entries are correlated with a sign of “A” (high GC content, active compartment), and negative entries are correlated with a sign of “B” (low GC content, inactive compartment) according to the orientation of the eigenvector.
Identification of TAD–SVs
SVs used in our discovery analysis include deletions and insertions that met the following criteria: (1) genotyped in our PanGenie SVs calls; (2) present in at least 5/26 samples; (3) <50% of samples have missing genotypes, and (4) with homozygous reference alleles (0/0) present at least once in our 26 HGSVC2 individuals. We further excluded those SVs for which the BSs were missing among all 26 samples. A Wilcoxon rank-sum test (Mann–Whitney U test) was performed on the BS calculated by the IS for each of those SVs located within the flanking TAD boundaries to compare subsets of the 26 samples with homozygous reference (0/0) and heterozygous/homozygous deletions (1/1, 0/1, and 1/0). If multiple TAD boundaries of one sample were located inside one single TAD boundary from the merged call set, we calculated the median value of the BS of TAD boundaries for that sample as its final boundary strength quantification. For those SVs intersected with more than one TAD boundary, the multitest correction was conducted locally, and an FDR < 0.05 was considered significant for deciding whether or not these SVs were considered TAD–SVs. For the analysis of homozygous SVs and heterozygous SVs, the Kruskal–Wallis test was performed, followed by a post-hoc test (Conover test). The same manner of multiple test correction was conducted, and FDR < 0.05 was considered significant. All test statistics were estimated using the SciPy library, and box plots were generated using Matplotlib in Python 3.7.3. The FDR correction was conducted in the qvalue 2.28.0 package in R v.4.2.1.
Associations of TAD–SVs with SV-eQTLs and SV-sQTLs
We overlapped our 185 TAD–SVs with the SV-eQTL (https://github.com/shilab/Hi-C-integrative-catalog/raw/main/data/qtl_results_all_v4_fdr0.05.txt.gz) and SV-sQTL (https://github.com/shilab/Hi-C-integrative-catalog/raw/main/data/qtl_results_all_v2_fdr0.05.txt.gz) mapping results from Ebert's study (2021). For each intersecting SV-eQTL and SV-sQTL, we extracted their corresponding quantifications of gene expression and transcript splicing in the 26 HGSVC2 samples used in this study. We performed the Wilcoxon rank-sum test (Mann–Whitney U test) on the gene expression and transcript splicing for each of those overlapped SV-eQTLs and SV-sQTLs, respectively, to compare subsets of the 26 samples with the homozygous reference (0/0) and heterozygous/homozygous genotypes (1/1, 0/1, and 1/0). For those SVs intersected with more than one gene, multitest correction was conducted locally, and an FDR < 0.05 was considered as being significant.
Identification of CTCF motif
The CTCF Motif Scanning was implemented through the FIMO tool in MEME Suite 5.5.5 (Grant et al. 2011; Bailey et al. 2015) to scan a set of inserted sequences for individual matches canonical-binding motif (M1) downloaded from the JASPAR database (JASPAR motif MA0139.1) (Sandelin et al. 2004) and CTCFBSDB 2.0 database (https://insulatordb.uthsc.edu/download/CTCFBSDB_PWM.mat) (Ziebarth et al. 2013). Default parameters were used for the search, except that the set of insertion sequences was used to compute a zeroth-order Markov background model for the search.
Mediation analysis between the TAD–SV, gene regulation, and chromatin structure
The mediation analysis was conducted using the statsmodels package (v.0.13.5) in Python 3.7. We formulated two models: the mediator model aimed to examine how the independent variable (SV) affects the mediator variable (boundary strength), and the outcome model assessed how the independent variable (SV) and the mediator variable (boundary strength) together influence the dependent variable (gene expression and splicing level) (Supplemental Fig. S17). The P-values for the ACME, ADE, and Total effect were extracted from the model output, with a P-value < 0.05 considered significant.
Replication studies in an additional data set
The GM19193 individual was excluded from the analysis because of the missing genotype from the PanGenie genotype calling set. SVs used in our replication analysis include deletions and insertions that met the following criteria: (1) genotyped in our PanGenie SVs calls; (2) present in at least 5/17 samples; (3) <50% of samples have missing genotypes; and (4) with homozygous reference alleles (0/0) presented at least once in our 17 HGSVC2 individuals. The same statistical test for BS per genotype category was conducted to examine the significance of each SV, as we did for the discovery set.
Overlap with cCREs
ENCODE v3 cCRE regions (The ENCODE Project Consortium et al. 2020) were downloaded from the SCREEN regulatory element database (https://screen.encodeproject.org/). TAD–SVs were overlapped with each cCRE data set using the BEDTools “intersect” command (Quinlan and Hall 2010). The enrichment of TAD–SVs in cCRE regions was calculated from Fisher's exact tests on 2 × 2 bp-count contingency tables (TAD–SVs overlapping cCREs, TAD–SVs without cCREs overlap, cCREs without TAD–SVs overlap, and the rest of the genome binned into windows of average cCRE length) using R v.4.1.2 (R Core Team 2024).
Permutation analysis
To conduct the permutation analysis to examine the enrichment between TAD–SVs identified in this study and cCRE regions, TAD–SVs were overlapped with the whole cCRE data set using BEDTools “intersect”. Deletions used in the depletion analysis of the TAD boundaries and loop anchors were retrieved from the PanGenie SV call set filtered by the type (deletion) and length (>49 bp) of each SV. The filtered deletions were overlapped with the identified TAD boundaries using BEDTools “intersect” with flags “-f 0.5 -F 0.5” with the same 50% reciprocal overlap criterion to boost the accuracy of the test. To explore the enrichment of CTCF at TAD boundaries, we randomly shuffled our flanking TAD boundary set while preserving the same number and length distributions across each chromosome and calculated the frequency of at least 1 bp overlap between CTCF and the shuffled TAD boundary set. For any of the permuted sets, we retained the same number and length distributions of the features for each chromosome and repeated this permutation procedure 10,000 times in total.
Data access
All original code and data required for the statistical analysis and pipeline have been deposited on GitHub https://github.com/shilab/Hi-C-integrative-catalog and as Supplemental Code. The integrative TAD catalog (includes the TAD boundaries, compartments, and loops) and the curated collection of TAD–SVs are readily accessible for download at https://github.com/shilab/Hi-C-integrative-catalog/tree/catalog. The TAD boundary call set for each sample (excluded GM12878) is available at https://github.com/shilab/Hi-C-integrative-catalog/tree/main/data/sample_bound. All processed HIC files are available at https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/working/20230515_Shi_hic_files/.
Human Genome Structural Variation Consortium (HGSVC)
The members of the Human Genome Structural Variation Consortium (HGSVC) are Evan E. Eichler (Cochair), Jan O. Korbel (Cochair), Charles Lee (Cochair), Tobias Marschall (Cochair), Hufsah Ashraf, Peter A. Audano, Ola Austine, Anna O. Basile, Christine R. Beck, Marc Jan Bonder, Marta Byrska-Bishop, Mark J.P. Chaisson, Zechen Chong, André Corvelo, Scott E. Devine, Peter Ebert, Jana Ebler, Mark B. Gerstein, Pille Hallast, William T. Harvey, Patrick Hasenfeld, Alex R. Hastie, Mir Henglin, Kendra Hoekzema, Wolfram Höps, PingHsun Hsieh, Sarah Hunt, Matthew Jensen, Miriam K. Konkel, Jennifer Kordosky, Peter M. Lansdorp, Charles Lee, Wan-Ping Lee, Alexandra P. Lewis, Chong Li, Jiadong Lin, Mark Loftus, Glennis A. Logsdon, Ryan E. Mills, Yulia Mostovoy, Katherine M. Munson, Giuseppe Narzisi, Andy Pang, David Porubsky, Timofey Prodanov, Tobias Rausch, Bernardo Rodriguez-Martin, Xinghua Shi, Likhitha Surapaneni, Michael E. Talkowski, Feyza Yilmaz, DongAhn Yoo, Xuefang Zhao, Weichen Zhou, and Michael C. Zody.
HGSVC Functional Analysis Working Group
The members of the Human Genome Structural Variation Consortium (HGSVC) functional analysis working group are Xinghua Shi (colead), Jan O. Korbel (colead), Anna O. Basile, Christine Beck, Marta Byrska-Bishop, Marc Jan Bonder, Mark J.P. Chaisson, Ken Chen, Evan E. Eichler, Mark B. Gerstein, Pille Hallast, Wolfram Höps, Daniel Ben-Isvy, Matthew Jensen, Yunzhe Jiang, Kwondo Kim, Miriam Konkel, Tobias Marschall, Bernardo Rodriguez-Martin, Gianni Martino, Ryan E. Mills, Nicholas Moskwa, Yuia Mostovoy, Lingbin Ni, Charles Lee, Chong Li, Jiaqi Li, Yang I. Li, Qingnan Liang, Mark Loftus, Glennis A. Logsdon, Carolyn Paisie, Oliver Stegle, Sabriya Syed, Michael E. Talkowski, Yukun Tan, Xuefang Zhao, Weichen Zhou, Michael C. Zody, Alex Yenkin, and DongAhn Yoo.
Competing interest statement
E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc. C.Lee is an SAB member of Nabsys and Inocras. M.E.T. is a SAB member of BridgeBio and receives research reagents and/or support from Pacific Biosciences, Illumina, and Microsoft Inc. The other authors declare no competing interests.
Acknowledgments
We thank Weichen Zhou from the HGSVC Functional Analysis Working Group for providing critical and constructive advice and discussion about Hi-C analysis. We thank Mohammad Erfan Mowlaei, Emily Thyrum, and Tom Lu for their feedback on the project and diligent proofreading. We sincerely extend our gratitude to the individuals who contributed samples to the 1000 Genomes Project. This work was supported by the National Institutes of Health (NIH) grants U24HG007497 (to C.Lee, E.E.E., J.O.K., T.M., M.B.G., and X.S.), R01HG002385 and R01HG010169 (to E.E.E.), and MH115957 (to M.E.T.); the German Human Genome-Phenome Archive (DFG [NFDI 1/1] to J.O.K.); the European Research Council (ERC Consolidator grant 773026 to J.O.K.). E.E.E. is an investigator of the Howard Hughes Medical Institute. This research includes calculations carried out on HPC resources supported in part by the National Science Foundation through major research instrumentation grant number 1625061 and by the US Army Research Laboratory under contract number W911NF-16-2-0189.
Author contributions: The study was designed by C.Li and overseen by X.S., HGSVC (cochaired by T.M., J.O.K., E.E.E., and C.Lee), and the HGSVC Functional Analysis Working Group. C.Li conducted data analysis. M.J.B., S.S., M.J., M.C.Z., M.J.P.C., and M.E.T. assisted in the data analysis and provided valuable feedback, and M.J.B., M.J., and M.B.G. provided additional relevant data sets for this work. C.Li, X.S., and C.Lee wrote the manuscript, incorporating contributions from all other coauthors. All authors read and approved the final manuscript.
Footnotes
-
Human Genome Structural Variation Consortium (HGSVC)
-
The members of the Human Genome Structural Variation Consortium (HGSVC) are Evan E. Eichler (Cochair), Jan O. Korbel (Cochair), Charles Lee (Cochair), Tobias Marschall (Cochair), Hufsah Ashraf, Peter A. Audano, Ola Austine, Anna O. Basile, Christine R. Beck, Marc Jan Bonder, Marta Byrska-Bishop, Mark J.P. Chaisson, Zechen Chong, André Corvelo, Scott E. Devine, Peter Ebert, Jana Ebler, Mark B. Gerstein, Pille Hallast, William T. Harvey, Patrick Hasenfeld, Alex R. Hastie, Mir Henglin, Kendra Hoekzema, Wolfram Höps, PingHsun Hsieh, Sarah Hunt, Matthew Jensen, Miriam K. Konkel, Jennifer Kordosky, Peter M. Lansdorp, Charles Lee, Wan-Ping Lee, Alexandra P. Lewis, Chong Li, Jiadong Lin, Mark Loftus, Glennis A. Logsdon, Ryan E. Mills, Yulia Mostovoy, Katherine M. Munson, Giuseppe Narzisi, Andy Pang, David Porubsky, Timofey Prodanov, Tobias Rausch, Bernardo Rodriguez-Martin, Xinghua Shi, Likhitha Surapaneni, Michael E. Talkowski, Feyza Yilmaz, DongAhn Yoo, Xuefang Zhao, Weichen Zhou, and Michael C. Zody.
-
HGSVC Functional Analysis Working Group
-
The members of the Human Genome Structural Variation Consortium (HGSVC) functional analysis working group are Xinghua Shi (colead), Jan O. Korbel (colead), Anna O. Basile, Christine Beck, Marta Byrska-Bishop, Marc Jan Bonder, Mark J.P. Chaisson, Ken Chen, Evan E. Eichler, Mark B. Gerstein, Pille Hallast, Wolfram Höps, Daniel Ben-Isvy, Matthew Jensen, Yunzhe Jiang, Kwondo Kim, Miriam Konkel, Tobias Marschall, Bernardo Rodriguez-Martin, Gianni Martino, Ryan E. Mills, Nicholas Moskwa, Yuia Mostovoy, Lingbin Ni, Charles Lee, Chong Li, Jiaqi Li, Yang I. Li, Qingnan Liang, Glennis A. Logsdon, Carolyn Paisie, Oliver Stegle, Sabriya Syed, Michael E. Talkowski, Yukun Tan, Xuefang Zhao, Weichen Zhou, Michael C. Zody, Alex Yenkin, and DongAhn Yoo.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279419.124.
- Received March 29, 2024.
- Accepted October 4, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








