
Ultrasensitive allele inference from immune repertoire sequencing data with MiXCR
- Artem Mikelov1,6,
- George Nefediev2,6,
- Alexander Tashkeev3,
- Oscar L. Rodriguez4,
- Diego Aguilar Ortmans3,
- Valeriia Skatova2,
- Mark Izraelson2,
- Alexey N. Davydov2,5,
- Stanislav Poslavsky2,
- Souad Rahmouni3,
- Corey T. Watson4,
- Dmitriy Chudakov2,5,
- Scott D. Boyd1 and
- Dmitry Bolotin2
- 1Department of Pathology, Stanford University, Stanford, California 94305, USA;
- 2MiLaboratories Incorporated, San Francisco, California 94114, USA;
- 3Unit of Animal Genomics, WELBIO, GIGA-R and Faculty of Veterinary Medicine, University of Liège (B34), 4000 Liège, Belgium;
- 4Department of Biochemistry and Molecular Genetics, University of Louisville School of Medicine, Louisville, Kentucky 40202, USA;
- 5Central European Institute of Technology, Masaryk University, 601 77 Brno, Czech Republic
-
↵6 These authors contributed equally to this work.
Abstract
Allelic variability in the adaptive immune receptor loci, which harbor the gene segments that encode B cell and T cell receptors (BCR/TCR), is of critical importance for immune responses to pathogens and vaccines. Adaptive immune receptor repertoire sequencing (AIRR-seq) has become widespread in immunology research making it the most readily available source of information about allelic diversity in immunoglobulin (IG) and T cell receptor (TR) loci. Here, we present a novel algorithm for extrasensitive and specific variable (V) and joining (J) gene allele inference, allowing the reconstruction of individual high-quality gene segment libraries. The approach can be applied for inferring allelic variants from peripheral blood lymphocyte BCR and TCR repertoire sequencing data, including hypermutated isotype-switched BCR sequences, thus allowing high-throughput novel allele discovery from a wide variety of existing data sets. The developed algorithm is a part of the MiXCR software. We demonstrate the accuracy of this approach using AIRR-seq paired with long-read genomic sequencing data, comparing it to a widely used algorithm, TIgGER. We applied the algorithm to a large set of IG heavy chain (IGH) AIRR-seq data from 450 donors of ancestrally diverse population groups, and to the largest reported full-length TCR alpha and beta chain (TRA and TRB) AIRR-seq data set, representing 134 individuals. This allowed us to assess the genetic diversity within the IGH, TRA, and TRB loci in different populations and to establish a database of alleles of V and J genes inferred from AIRR-seq data and their population frequencies with free public access through VDJ.online database.
Adaptive immune repertoire diversity plays a crucial role in shaping the immune response and forming immunological memory. Most immune repertoire research has focused primarily on somatically derived immune receptor diversity, namely, V(D)J recombination and somatic hypermutation (SHM) diversity. In recent years, however, the extent of population diversity has begun to be appreciated at both the immunoglobulin (IG) (Gidoni et al. 2019; Mikocziova et al. 2021; Corcoran et al. 2023; Gibson et al. 2023; Rodriguez et al. 2023) and T cell receptor (TCR) loci (Omer et al. 2022; Rodriguez et al. 2022; Corcoran et al. 2023). The functional significance of allelic variation in adaptive immune loci has also been recognized in the context of influenza, HIV, and COVID-19 immunity and vaccination (Avnir et al. 2016; Lee et al. 2021; Leggat et al. 2022; Pushparaj et al. 2023).
Sequencing repertoires of adaptive immune receptors encoded by recombined germline variable (V), diversity (D), and joining (J) genes have become a major source of information about adaptive immune functions in health and disease. In recent years, adaptive immune receptor repertoire sequencing (AIRR-seq) has been utilized to discover many novel alleles in TR and IG loci, becoming one of the major sources of information of the allelic diversity of TR and IG genes in different populations. However, the major obstacle for utilizing AIRR-seq data sets for genotyping and allelic discovery is the presence of somatically hypermutated sequences in most available IG AIRR-seq data sets, along with the polymerase chain reaction (PCR) and sequencing errors which affect both B cell receptor (BCR) and TR repertoire data sets. Hot-spot hypermutations and sequence errors have significantly hindered the ability to clearly detect individual polymorphisms. We aimed to overcome these issues with the algorithm described in this paper. However, there are two other challenging obstacles for accurate genotyping and haplotyping of TR and IG loci using AIRR-seq data only. Common structural variants (SVs) in IG loci (Rodriguez et al. 2023), especially gene duplications, in some cases, make it hard to unequivocally map a sequence from AIRR-seq data to a particular germline gene without an additional source of information. Further, some alleles exhibit low usage levels, precluding their detection with AIRR-seq. Despite these limitations, AIRR-seq data remain valuable for applications focused on functional adaptive immune repertoires and their fluctuations in different conditions.
The ability to precisely call known allelic variants and infer novel ones from the same AIRR-seq data could enable new analyses of germline variation contribution to immune responses, and also improve the accuracy of many existing downstream approaches. There are several published methods for genotyping and allelic inference of V and J genes from AIRR-seq data (Table 1, numbers 2–5); however, each has important limitations. TIgGER (Gadala-Maria et al. 2015, 2019) and Partis (Ralph and Matsen 2019) are based on the idea that allelic sequence variants show a distinctive pattern over the background of SHMs. On the other hand, IgDiscover (Corcoran et al. 2016), a very robust and reliable tool for novel allele inference, requires data without hypermutations, thus excluding much-published IG repertoire data. The recently developed PIgLET software (Peres et al. 2023) has enhanced genotype inference capabilities through the use of IGHV allele similarity clustering, although it is not designed to infer novel alleles.
Tools for novel allele variant inference from AIRR-seq data and their characteristics
Existing tools also require considerable depth of AIRR-seq data for reliable allele inference (e.g., IgDiscover recommends at least 750,000 sequencing reads per individual library). Such sequencing depth is costly and not available for most publicly available AIRR-seq data sets. Here, we present an algorithm for allelic inference and genotyping from both hypermutated and nonhypermutated repertoires, with low sequencing depth requirements. The algorithm performs well starting with a minimalistic gene reference library of only one allele for each gene, and even with some genes missing. These features make the tool especially useful for studying allelic diversity in nonmodel species where reference gene libraries are sparse and incomplete. The developed approach is integrated into MiXCR software and is available as the findAlleles command. Starting with version 4.0, MiXCR can process immune repertoire data directly from raw sequencing reads in FASTQ format. The MiXCR upstream pipeline supports all commercially available library preparation kits, as well as any custom protocols. It handles preprocessing, sequence alignment, and clonal assembly based on customizable region of interest, such as the CDR3, the whole VDJ region, or any user-defined region. The output of the upstream pipeline can be generated in several formats: a highly efficient binary format, a tabular format with customizable fields, or the AIRR format. The findAlleles command can be executed on clonesets in binary format. For each individual findAlleles outputs a personalized reference allele library in either FASTA, tabular, or json format.
The International ImMunoGeneTics Information System (IMGT), established in 1989, is the oldest widely available source of information about immune receptors, including alleles. Recent advancements in high-throughput AIRR-seq methods enabled a broader view of alleles, and many tools were developed to infer allelic variants from such data. In 2017, the AIRR-Community (a network of over 300 practitioners in the field of AIRR-seq, www.airr-community.org) and IMGT agreed on a process for adding new alleles inferred from AIRR-seq data to the IMGT database (Ohlin et al. 2019). The AIRR-Community also introduced the Open Germline Receptor Database (OGRDB) (https://ogrdb.airr-community.org/, Lees et al. 2020), to track the addition of new alleles. Although being the most recognized source of germline IG sequence data, IMGT lacks information on population allele frequencies and harbors sequences “mapped” to the identified genes at the specific genomic locations. However, structural variation is quite common (Rodriguez et al. 2023) in the IG and TR loci, while most new IG and TR sequence data come from AIRR-seq experiments, and can be hard to map to a particular germline locus position. Other databases of immune receptor gene alleles have been introduced, such as pmTR (Dekker et al. 2022, https://pmtrig.lumc.nl/), IgPdb (https://cgi.cse.unsw.edu.au/~ihmmune/IgPdb) and Karolinska Institutet human T-cell receptor database (Corcoran et al. 2023, https://gkhlab.gitlab.io/tcr/). A comprehensive and well-maintained database of immune receptor gene alleles, including allelic variants inferred from AIRR-seq, is VDJbase (Omer et al. 2020, https://vdjbase.org/). However, VDJbase is not seamlessly integrated with any of the analysis tools, and using it for AIRR-seq data analysis requires conversion of sequence data formats. To accompany the MiXCR software, we have developed VDJ.online (https://vdj.online/library), a free and open database of immune receptor allelic sequences that enable the examination, comparison, and downloading of sequences. The VDJ.online reference library is supplied with the MiXCR, allowing seamless AIRR-seq data processing with accurate V and J gene annotation, genotyping, and novel allele inference.
Results
Novel approach to V and J gene allele variant inference and genotyping
The main challenge of allelic inference from AIRR-seq data is the presence of hypermutations, PCR, and sequencing errors, with a large fraction of them being hot-spot mutations occurring simultaneously in unrelated clones. We have overcome this challenge by consecutively applying several filters based on two major measures. The first one is the lower diversity bound, estimated as the number of unique combinations of J and V genes and CDR3-lengths of clonotypes. The second measure is based on the number of clonotypes with unmutated J and V genes. Filters are applied both at individual mutation and at mutation set levels (see Methods for the detailed description). The mutations at germline-encoded positions in CDR3 are recovered, when possible, by using nonmutated clonotypes matching the inferred variants in the rest of the sequence. This approach allows both to infer novel (undocumented) V and J gene alleles and to perform genotyping with high sensitivity and precision.
Benchmarking of the V and J gene allele variant inference and genotyping
To assess the performance of the developed algorithm, we utilized publicly available data sets (Rodriguez et al. 2023) containing both AIRR-seq data and highly reliable genotyping data of the IG heavy chain (IGH) locus reconstructed using Pacific Biosciences (PacBio) High Fidelity (HiFi) long-read sequencing from the same individuals. For the sake of comparison, we utilized 33 AIRR-seq data sets of sufficient sequencing depth (>500,000 sequencing reads) and at least 3000 unique full-length clonotypes. Targeted long-read sequencing of genomic DNA (as described in Rodriguez et al. [2023]) allowed us to observe the nonrearranged IGH locus, containing germline, unmutated IGHV, and IGHJ genes. Since these genes were not rearranged, and did not contain SHM, they provide a reliable ground truth for allele identification in our comparison. However, in some individuals, not all genes of interest were captured by the long-read sequencing. Consequently, we excluded the allele calls for these genes from our comparison. AIRR-seq data, which was derived from peripheral blood mononuclear cell samples containing both naive and antigen-experienced B cells, expressing either unmutated or somatically hypermutated BCR sequences, was used to infer the allele variants using our approach, with the PacBio germline DNA sequences as the gold standard for the true alleles present in each individual (Rodriguez et al. 2020b). We also compared the performance of our algorithm to TIgGER, the most widely cited tool for this task.
Upstream analysis, including sequence alignment to reference V and J gene libraries and defining the full-length clonotypes, was performed using the tools’ recommended pipeline, MiXCR's analyze module (Bolotin et al. 2015, https://mixcr.com/mixcr/reference/mixcr-analyze/), and pRESTO (Vander Heiden et al. 2014) and Change-O (Gupta et al. 2015) from Immcantation framework (https://immcantation.readthedocs.io). For further details, please see the Methods section. For the alignment step and V and J gene annotation, we used a custom minimalistic gene set library with only one allelic variant per V and J gene, derived from a custom public genome reference to match the one used for the long-read-based genotyping (Rodriguez et al. 2020b). Then we performed allele variant inference and genotyping with both tools for all data sets containing more than 3000 unique full-length clonotypes and compared the resulting individualized V and J gene libraries with the accurate genotype inferred with the next generation long-read sequencing (Rodriguez et al. 2023), comparing nucleotide sequences of the genes. We also excluded poorly expressed allelic variants as determined by aligning the reads to the individualized gene reference libraries. Thus, in our benchmarking, we focused on the question of detection of particular V or J gene allele sequences in the participants’ AIRR-seq IGH data for the subsequent accurate clonotype annotation, which is crucial for many downstream applications of such data (e.g., lineage trees analysis). Importantly, we compared the abilities of both approaches using the sparse reference libraries and AIRR-seq data, containing varying amounts of errors and sequencing noise, including data sets incorporating unique molecular identifiers and not. Therefore, we consider our benchmarking relevant to real-world applications where the data quality is typically far from ideal in many aspects.
MiXCR on average detected 98% of the allelic variants of the V genes supported by the long-read-based genotyping, while TIgGER detected 81% of the V gene alleles (Fig. 1A). MiXCR produced on average one allele call not supported by long-read-based genotyping, while TIgGER yielded two potential false-positive calls (Fig. 1B). The recall of TIgGER improved up to 94% on average when the upstream analysis and allele inference was performed utilizing the full built-in reference library containing all of the known alleles (Supplemental Fig. S1A). However, the number of the allele calls not supported by the long-read sequencing also increased, up to five potential false-positive calls on average (Supplemental Fig. S1B). We assume that the TIgGER algorithm may exhibit improved performance when utilizing the full reference library, due to the algorithm's inherent design. Even with the most recent implementation utilizing the dynamic window as described by Gadala-Maria et al. (2019), there may be instances where a novel allele could be obfuscated by another novel allele that is more similar to the one in the minimal reference library. For MiXCR, the transition to full reference library resulted in only minor changes in performance (Supplemental Fig. S1A,B).
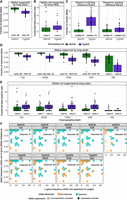
Detection of allelic variants of V genes by inference tools. (A) Fraction of allele calls supported by long-read-based genotyping. (B) Number of allele calls not supported by long-read-based genotyping. (C) Fraction of alleles, missed by MiXCR or TIgGER, by the presence in the initial reference library. (D,E) Sensitivity and specificity testing by downsampling each sample in the benchmarking data set by 500,000, 100,000, 50,000, or 10,000 reads. (D) Fraction of identified allele calls supported by long-read-based genotyping. (E) Number of identified allele calls not supported by long-read-based genotyping. (F) Detection of the allele variants of V genes depending on V usage and allelic imbalance. Each dot represents a V gene allele present in the donor's genotype confirmed by long-read sequencing. The upper row represents detection by the developed algorithm; the lower, allele detection by the comparison tool TIgGER. Columns represent different depths of downsampling by number of aligned reads, from right to left: full set of reads, 500,000, 100,000, 50,000, and 10,000. V gene, and allele frequencies for each facet were calculated using the full set of reads and allele-resolved V and J gene reference library. Alleles excluded due to low expression (<10 clonotypes), are represented as empty crossed points. N = 33 for A–E.
The difference in the number of called alleles between the two algorithms was also apparent when we compared rates of detection of the de novo inferred alleles. TIgGER did not detect on average 14% of alleles absent in the starting reference gene library, while MiXCR missed none of the alleles (Fig. 1C).
To test the sensitivity of the approaches, we also downsampled the data set to 500,000, 100,000, 50,000, and 10,000 raw sequencing reads. MiXCR allele detection rates decreased by 9 percentage points down to 89% on average when downsampled to 50,000 reads, which is more than 10× downsampling for all of the data sets. TIgGER detection rates also deteriorated by 23 percentage points, detecting on average 58% of alleles with 50,000 reads. At the extreme level of downsampling by 10,000 sequencing reads MiXCR was able to detect 70% of alleles on average, while TIgGER yielded an error for 21 of the samples due to the low number of clones assigned to any of the V genes (Fig. 1D). The number of potential false-positive calls did not increase with lower downsampling depth. For MiXCR, there was a slight decrease in false positives at each downsampling step. Expectedly, both tools produced no potential false-positive calls at extreme downsampling depths (Fig. 1E). For MiXCR, the detection of the alleles clearly depended on the two variables—the frequency of the V gene in a particular repertoire and the imbalance in usage between different alleles for a particular V gene. For TIgGER, these parameters appeared to have little influence on detection rates (Fig. 1F). Sequencing quality influenced inference for both tools during upstream processing. TIgGER filters sequences based on average Phred quality scores, while MiXCR uses an adjustable threshold for each position. MiXCR's stringent default criteria resulted in no alleles being recovered for one sample at a 50,000 reads downsampling depth and for several more samples at 10,000 reads due to extensive read filtering. For the task of detecting J gene allelic variants, for which could not be performed with TIgGER, MiXCR yielded 100% sensitivity and specificity even with the data sets downsampled to 50,000 reads (Supplemental Fig. S2).
Furthermore, we compared the runtime of both tools and found that, on average, there was no significant difference. However, the runtime variability for TIgGER was considerably greater (Supplemental Fig. S3).
To assess the influence of sequencing and PCR errors on MiXCR inference performance, we compared results from data generated with and without unique molecular identifiers (Supplemental Fig. S4A,B), which are known to eliminate these errors (Shugay et al. 2014). Additionally, we evaluated the impact of SHM load on allele inference performance (Supplemental Fig. S4C,D). The number of potential false-positive calls was unaffected by SHM frequency or by the presence of sequencing errors in data generated without using unique molecular identifiers (Supplemental Fig. S4B,D). However, the fraction of false negative calls was influenced by both parameters (Supplemental Fig. S4A,C).
Numerous IGH, TRA, and TRB novel alleles detected using MiXCR allele inference
To investigate allelic diversity in human populations, we applied the developed algorithm to a large collection of IGH (450 individuals) and full-length TCR alpha and beta chain (TRA and TRB) (134 individuals) AIRR-seq data sets. The MiXCR allele inference and genotyping pipeline resulted in the identification of both known and previously undocumented alleles, 384 IGHV, 128 TRAV, 144 TRBV, 14 IGHJ, 64 TRAJ, and 14 TRBJ in total.
Numerous previously undocumented alleles, absent from major databases mentioned above (OGRDB or IMGT), were detected: 183 IGHV, 33 TRAV, 7 TRAJ, and 41 TRBV (Fig. 2A–D,F). Of note, we did not detect any novel variant for any of the IGHJ and TRBJ genes (Fig. 2E). All of the novel allele sequences were contributed to the public database of allelic variants and are available for download at https://vdj.online/library.

Number of observed novel and known alleles. (A) TRAV, (B) TRBV, (C) IGHV, (D) TRAJ, (E) TRBJ, and (F) IGHJ.
Divergent allele frequency distribution in IGHV genes in the African population
The considered IGH AIRR-seq data sets included repertoires from African, Asian, European, and Hispanic/Latino individuals (Fig. 3A). We did not observe significant differences in the number of detected novel IGHV alleles per donor between these groups (Fig. 3B). The sufficient sample sizes in the European and African populations allowed us to investigate differences in the number of IGHV and IGHJ alleles and allelic distributions between these two groups. The number of detected alleles was similar for all of the J genes (Fig. 3C) and most of the V genes with the exception of IGHV1-3, IGHV1-69, IGHV3-53, and IGHV4-30-2 (Fig. 3D).
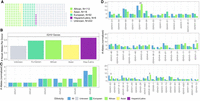
IGHV and IGHJ allelic diversity by major ethnic groups. (A) Cohort composition. (B) Number of detected novel alleles, normalized per number of individuals. (C) Total number of detected alleles by IGHJ gene in European, African, and general population, normalized by downsampling to a fixed number of individuals (N = 92). (D) Total number of detected alleles by IGHV gene in European, African, and general population, normalized by downsampling to a fixed number of individuals (N = 92). Comparison between ethnicities in each (B–D) was performed using a permutation test (1000 permutations, [*] P ≤ 0.05, [**] P ≤ 0.01, nonsignificant not shown).
On the other hand, the allele frequency distribution in the African population was significantly different than that of other populations for 38 IGHV genes (Supplemental Figs. S5, S6). Some V genes, even those with similar frequencies in a typical human repertoire, showed very distinct allele distributions. For example, several alleles of IGHV1-69 and IGHV3-48 appear at intermediate frequencies in the populations studied. In contrast, IGHV3-23 had only one predominantly represented allele, while IGHV3-7 had several alleles at the level of nucleotide sequence differences, but encoding the same amino acid sequence (Fig. 4). For these V genes, where the alleles were more evenly distributed, the allele distributions also showed greater differences between ethnic groups (Fig. 4; Supplemental Figs. S5, S6).
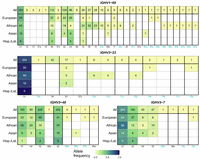
IGHV gene allele frequencies in major ethnic groups for selected IGHV genes. Each column in heatmaps represents a particular allele; numbers for novel alleles, first reported in this study, are colored in green; the letter “x” designates alleles inferred from AIRR-seq data, either in this study or previously, with the same sequences already present in OGRDB; bold lines separate groups of alleles with different amino acid sequences; groups of alleles with the same amino acid sequence are ordered by the aggregated frequencies of alleles; alleles within groups are order by allele frequency in the general population. Color represents the allele frequency within the ethnic group; numbers in cells represent the number of occurrences of the corresponding allele.
The same difference was also observed for two of the IGHJ genes: IGHJ3 and IGHJ6 (Supplemental Fig. S7). In TRBV and TRAV loci for most of the gene frequencies, distributions were heavily skewed toward particular single allele variants (Supplemental Figs. S8, S9), which may be attributed to a more homogeneous cohort composition by ethnicity, with the predominant majority of participants being of European descent.
Discussion
Immune receptor repertoire sequencing data sets have become a valuable source of information for studying immune responses across different health conditions, tissues, and cell subsets. Recently developed specialized algorithms (Corcoran et al. 2016; Zhang et al. 2016; Gadala-Maria et al. 2019; Ralph and Matsen 2019) allow inference of allelic variants of V and J genes of adaptive immune receptors from AIRR-seq data, scaling up the process of novel allele discovery and allowing AIRR-seq data analysis using individualized gene reference libraries, which significantly increases the accuracy and quality of many types of downstream repertoire analyses. However, all of the current approaches demand high sequencing depth and a significant number of unique receptor sequences for the analysis. Moreover, many prior approaches do not allow allelic inference from both hypermutated and nonhypermutated repertoires. The most comprehensive approach for precise genotyping and allelic inference, utilizing long-read sequencing of the immune receptor gene loci (Rodriguez et al. 2020b, 2023; Gibson et al. 2023), has the greatest accuracy, and also allows for the investigation of SVs. Although being the most desirable and accurate way to obtain donor-specific V and J gene genotypes, this methodology is costly and requires special experimental procedures. Here, we addressed this unmet need by developing an alternative approach for inferring allelic variants of V and J genes directly from AIRR-seq data, offering improved sensitivity and accuracy compared to existing tools.
Our method allows for successful allelic inference from data sets downsampled to as few as 50,000 sequencing reads. Moreover, the algorithm applicability is not restricted to a particular type of AIRR-seq data; it can be applied to both repertoires containing hypermutated sequences (e.g., IGH repertoires generated from any isotype) as well as data sets containing only nonhypermutated sequences (e.g., TCR repertoires). Multiple filtering steps integrated into our pipeline prevent false-positive polymorphism calling which typically arises due to the presence of hot-spot hypermutations and PCR and sequencing errors. Furthermore, we demonstrate the high sensitivity and specificity of the approach utilizing a very sparse starting reference gene library, containing only one allelic variant per gene, which makes it even more useful for studying allelic diversity in nonmodel species for which V and J gene reference libraries are incomplete and lack allelic variants. We assume that the improved sensitivity compared to the comparison tool is due to our algorithm bypassing the regression modeling component, which requires a certain number of sequences for a specific V gene to reliably infer alleles. The developed approach is integrated within the MiXCR (Bolotin et al. 2015, https://mixcr.com) pipeline for immune repertoire analysis, and allows seamless allelic inference and realigning repertoires to a personalized reference library.
Applying the developed approach to large collections of IGH, TRA, and TRB repertoire data sets, we were able to identify a large number of previously undocumented V and J gene alleles. The number of novel IGHV alleles, normalized per donor, did not significantly differ among the different population groups. This finding suggests that the genetic diversity of IGHV genes even in the relatively better-studied European populations is still not fully characterized. Each additional sampling in the different population groups we studied continues to reveal novel alleles at similar rates. To facilitate sharing and usage of the discovered allele sequences we have established a database of allelic variants integrated with MiXCR and publicly available at https://vdj.online/library.
Differences in allele frequency distributions may have major implications for the susceptibility of different populations to diseases and vaccination outcomes (Avnir et al. 2016). Large sample sizes (450 individuals for IGH and 134 for TRA/TRB) allowed us to estimate allele frequencies for most of the studied genes in the population. For IGHV and J gene allelic variants, we identify striking differences in allele frequency distributions between African donors and other major population groups. We also contributed the information on V and J gene allele frequencies to VDJ.online, making it a valuable public resource of such information. Having incorporated this database of allelic variants into the MiXCR platform, we hope that it will facilitate further advancement in the immune repertoire analysis field, adding the dimension of allele analysis with little additional effort and cost to many further studies.
Methods
Allele variant detection algorithm
The algorithm utilizes alignment and clonotype assembly information from the upstream AIRR-seq data processing, specifically, mutation calls from reference V and J gene reference library for BCR or TCR clonotypes and V and J gene annotations, readily available after running the “analyze” command in the MiXCR software (Bolotin et al. 2015). The clonotype definition for the purpose of allele inference may vary depending on the region covered by sequencing.
Using these defined sets of mutations which differentiate the particular clonotype sequences from the corresponding reference V or J gene, the algorithm then separately infers alleles for V and J genes. For simplicity, we describe the algorithm steps for V genes only, the J gene inference follows the same logic:
-
Clonotypes are grouped by the V genes. For the data without unique molecular barcodes, only clonotypes with a read count greater than one are utilized for subsequent analysis.
-
For each mutation within the group, including insertions and deletions, we define a set of clonotypes which contain this mutation.
-
The mutations are filtered based on the lower diversity bound, estimated as the number of unique combinations of J genes and CDR3-lengths of clonotypes containing that mutation. The mutations that do not exceed a predefined threshold for the value are removed from each of the clonotype's mutation sets.
-
Clonotypes are grouped by filtered mutation sets, including “empty” mutation sets, containing no mutations. The lower diversity bound is calculated for each of the groups as described above. Additionally, the number of clonotypes containing no mutations in J gene after filtering as described in step 3 is calculated. Mutation sets are then filtered by thresholds of these two parameters, resulting in a list of allele candidates.
-
Clonotypes are then assigned to the closest allele candidates. Clonotypes which cannot be unambiguously assigned are filtered out. The lower bound of naive diversity is calculated as the number of unique combinations of J genes and CDR3-lengths for clonotypes with unmutated J gene sequences. Candidates are sorted by the score which represents the weighted sum of the lower bound of diversity and lower bound of naive diversity, calculated as described above. The formula for the score:
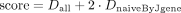 where Dall is the lower bound of diversity for all clonotypes; DnaiveByJgene—lower bound diversity, calculated only for clonotypes with no mutations in J gene.
where Dall is the lower bound of diversity for all clonotypes; DnaiveByJgene—lower bound diversity, calculated only for clonotypes with no mutations in J gene.
-
Candidates with the score not lower than 0.35 of the maximum score are then selected for the subject-specific gene set library.
-
Mutations at germline-encoded positions in CDR3 are recovered using the nonmutated clonotypes, which totally match the inferred variants by the rest of the sequence excluding CDR3. Each position is considered if it has at least five clonotypes covering it and 70% nucleotide concordance. The right-most position in CDR3, which meets these criteria, is reported by MiXCR (reliableRegion field in tabular output). The rest of CDR3 is picked from the closest allele in the database.
The process for inferring J gene alleles is the same; however, the initial grouping is performed by J genes and V genes are used for all of the filtering steps.
This stepwise approach-based sequential filtering first on the level of individual mutation and then on the level of mutation sets reduces noise introduced by SHM and sequencing and PCR errors. The threshold of 0.35 for the final allele filtering was initially chosen from a theoretical consideration of possible distributions of expressed alleles for a V gene allowing the presence of three allelic variants due to possible V gene duplications. This was then corroborated by examining empirical score distributions for alleles in the sequencing of the IGH repertoire of a healthy donor with known genotype; in this case, the donor was different from the one in the benchmarking of the algorithm.
In case of a significant difference between the reference library and a particular individual's genotype, the algorithm repeats the steps described above twice, with two different sets of parameters. The first step generates preliminary allele calls, which allows more precise estimation of the numbers of clonotypes with unmutated gene sequences.
The algorithm always utilizes only one allele variant per gene as starting reference, preventing potential biases toward particular known sequences. In case there is a weak signal in a particular gene (usually represented by less than 20 clonotypes), the algorithm falls back to assigning one of the known alleles.
Finally, for all of the allele calls, the allele names are looked up in a reference database (the same as available at https://vdj.online/library) by an exact match of the nucleotide sequence. If there is no match the new name is derived from concatenation of the closest allele and sequence hash.
The described algorithm is integrated into MiXCR as the findAlleles command.
Data collection and repertoire sequencing
For the benchmarking purposes, we utilized IGH repertoire sequencing data, accompanied by a targeted long-read IGH locus sequencing from Rodriguez et al. (2023), selecting samples which had at least 500,000 sequencing (N = 40), which was necessary for compatibility in downsampling experiments. IGH locus assembly and variant detection characterizing novel alleles were performed using iGenotyper (https://github.com/oscarlr/IGenotyper, Rodriguez et al. 2020b) as previously described (Rodriguez et al. 2023). Briefly, IGenotyper utilizes BLASR (Chaisson and Tesler 2012), WhatsHap (Martin et al. 2023), MsPAC (Rodriguez et al. 2020a), and Canu (Koren et al. 2017) for read alignment, calling and phasing single nucleotide variants, phasing reads, and assembling phased reads, respectively. Using the genotypes generated with IGenotyper, we constructed reference allele libraries in FASTA format for each participant. These libraries were then used to match with AIRR-seq-derived allele sequences in the benchmarking.
For calculating population allele frequencies, we used publicly available IGH AIRR-seq data from six published studies (total N = 450) (Roskin et al. 2015; Davis et al. 2019; Gidoni et al. 2019; Nielsen et al. 2019, 2020; Rodriguez et al. 2022).
For generating high-quality full-length TCR repertoires, peripheral blood was collected from 134 individuals without major chronic immunological conditions at CHU of Liège, including COVID-19 patients and individuals after vaccination; 2.5 mL of blood was collected on PAXGene RNA tubes from each participant and stored at −80°C until use, RNA was extracted using the PAXgene Blood RNA Kit (Qiagen). cDNA libraries were generated using SMARTer Human TCR a/b Profiling Kit v2 (Takara Bio). Briefly, a rapid amplification of cDNA ends (RACE) approach with a template-switch effect was used to introduce 5′ adaptors during cDNA synthesis. cDNA corresponding to TRA and TRB transcripts was further amplified and prepared for sequencing, which was performed on a MiSeq instrument with paired-end 2 × 300 bp reads using the NovaSeq 6000 SP Reagent Kit v1.5 (500 cycles) (Illumina). The protocol was approved by the ethics committee of Liège University Hospital (approval numbers 2021-54 and 2020/107).
Benchmarking of allele variant detection and genotyping
Processing of the AIRR-seq data was performed using MiXCR v4.4.0 (https://mixcr.com, Bolotin et al. 2015) upstream pipeline “analyze” command, parallelized using GNU Parallel (Tange 2018). Importantly, for the alignment step and V and J gene annotation, we used a custom minimalistic gene set library with only one allelic variant per V and J gene, derived from a custom public genome reference to match the one used for the long-read assembly (Rodriguez et al. 2023). After processing we excluded samples with the resulting number of full-length IGH clonotypes less than 3000 (N = 7), which probably related to samples either with low cell counts or with low RNA yield. Then the allelic variants were inferred and individual genotypes were reconstructed for each individual sample (N = 33) with the algorithm described above integrated into the MiXCR pipeline as findAlleles command.
To infer the alleles with the comparison tool, TIgGER (Gadala-Maria et al. 2019), we used the same set of samples (N = 33). For initial AIRR-seq data processing, we utilized tools pRESTO (Vander Heiden et al. 2014) and Change-O (Gupta et al. 2015), which are the part of the Immcantation framework along with TIgGER (https://immcantation.readthedocs.io), using commands and settings, recommended by the documentation. TIgGER v1.0.1 functions findNovelAlleles and inferGenotype were used for inferring novel alleles and reconstructing genotypes.
To test the sensitivity of the approaches, we downsampled the data set to 500,000, 100,000, 50,000, and 10,000 raw sequencing reads using seqtk (https://github.com/lh3/seqtk) v1.3 and applied the same upstream processing, allele inference, and genotyping pipelines as for the full data sets.
The resulting sets of allele sequences were exported from both tools in FASTA format and matched with the sequences of the alleles present in the genotype of the donor, previously recovered with iGenotyper (Rodriguez et al. 2023), and the number of matches was determined. The comparison was performed on the sequences remaining after removing primers, 5′ untranslated regions, and leader sequences. Importantly, due to the fact that IGH repertoire sequencing data utilized for comparison was derived using RNA-based technology, inference could be performed only for expressed V and J gene alleles. Thus, we excluded nonfunctional alleles and also those alleles from comparison which had less than 10 total clonotypes or less than 3 “naive” clonotypes with no mutation calls assigned to these alleles, when utilizing the same MiXCR v4.4.0 upstream pipeline, but with the individual allele-resolved V and J gene reference libraries constructed from long-read-based genotypes. Also, we have excluded from comparison genes which were not captured by the long-read sequencing. In particular, IGHJ genes were covered only for 9 of 33 considered individuals. For the benchmarking purposes, we excluded alleles for low abundance genes with too low abundance, as defined by each of the tools. TIgGER could not infer novel alleles for genes with less than 50 clonotypes assigned to it, so we excluded such alleles from comparison for TIgGER. MiXCR reports the low abundance genes for which the analysis is impossible with the parameters described above. The average number of clonotypes assigned to those genes was less than 10, we excluded such alleles from comparison for MiXCR too. Finally, we have not taken into account false negative and false-positive polymorphism calls in the whole CDR3 region for TIgGER; for MiXCR we applied stricter criteria and have not considered false negative and false-positive polymorphism calls outside reliableRegion, defined by the tool as described above. The runtime for each of the samples was benchmarked with the R package “bench” v. 1.1.3 (Hester and Vaughan 2023).
Novel allele inference and population frequencies
Processing of the AIRR-seq data for both BCR and TCR repertoires was performed using the MiXCR v4.3.2 analyze command. Repertoires containing less than 3000 unique clonotypes were not used for downstream analysis. The algorithm described above for inferring novel alleles and genotyping was used by invoking findAlleles MiXCR command under default settings. Alleles lacking designated names by the International Union of Immunological Societies were given interim names composed of a number continuing the existing sequence, with “x” letter added after the number. Those not found in OGRDB, the most up-to-date database of alleles inferred from AIRR-seq, were labeled as undocumented.
To compare the total number of alleles per V gene, we selected only European and African populations due to their sufficient size. We downsampled these populations to match the number of participants in the smallest group (N = 92) and performed a permutation test to statistically validate the findings. For both TCR and IG V and J gene alleles, the number of haplotypes with these alleles was estimated using output tables from findAlleles command, utilizing only those alleles for which inference could be reliably performed as mentioned in the generated reports. Each case where the only one allele per gene was indicated was treated as a gene in the homozygous state, thus not taking into account possible deletions of the genes on one of the chromosomes. We also limited our analysis with the genes detected in at least 15% of the donors. Allele frequencies were then calculated by dividing the number of haplotypes for a particular allele by the total number of haplotypes for this gene in the population. For the IGH data, we also were able to calculate allele frequencies for four major ethnic groups—African, Asian, European, Hispanic/Latino (self-reported by participants, where missing assigned to “unknown”).
To evaluate pairwise similarity between IGH allele frequency distributions in different populations, we utilized Hellinger distance (Hellinger 1909), calculated using the following formula: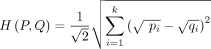 where P and Q represent the distributions of alleles of a particular V or J gene in two populations, and pi and qi represent frequencies of individual member i (one particular allele) of a total number of alleles for the gene k.
where P and Q represent the distributions of alleles of a particular V or J gene in two populations, and pi and qi represent frequencies of individual member i (one particular allele) of a total number of alleles for the gene k.
We utilized a permutation test to statistically validate differences in the number of novel alleles (Fig. 3B), the total number of alleles per gene (Fig. 3C,D), and Hellinger distance between allele distributions (Supplemental Fig. S6), reshuffling ethnicity labels 1000 times, and then calculating the fraction of permutations where we observed the same absolute difference (or Hellinger distance) between groups or greater.
Software and packages
All downstream data analyses and visualizations were conducted using R version 4.3.2 (R Core Team 2023) with the following packages: bench v. 1.1.3 (Hester and Vaughan 2023), Biostrings v. 2.70.3 (Pagès et al. 2024), ComplexHeatmap v. 2.15.4 (Gu et al. 2016; Gu 2022), cowplot v. 1.1.3 (Wilke 2024), fuzzyjoin v. 0.1.6 (Robinson 2020), ggpubr v. 0.6.0 (Kassambara 2023), spgs v. 1.0.4 (Hart and Martínez 2023), tidyverse v. 2.0.0 (Wickham et al. 2019), and waffle v. 1.0.2 (Rudis and Gandy 2023).
Data access
Sequencing data generated in this study have been deposited in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-13593. MiXCR software, including findAlleles module, is free for academic nonprofit research, and is available at GitHub (https://github.com/milaboratory/mixcr) and as Supplemental Code. Code required to reproduce the work is also included as Supplemental Code.
Competing interest statement
A.M. consulted MiLaboratories Inc. G.N., M.I., A.N.D., S.P., D.C., and D.B. are employed by MiLaboratories Inc. S.P., D.C., and D.B. are co-founders and shareholders of MiLaboratories Inc. S.D.B. has consulted for Regeneron, Sanofi, Novartis, Genentech, and Janssen on topics unrelated to this study and owns stock in AbCellera Biologics.
Acknowledgments
We thank the study participants for their contributions to this research. A.M. and S.D.B. were partially supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases (NIH/NIAID) grants U19Al104209, R01Al127877, R01Al125567, R01Al130398, U19AI167903, and U19AI057229.
Author contributions: A.M., G.N., S.P., and D.B. conceived the study. G.N. and A.M. developed and benchmarked the described algorithm. O.L.R. and C.T.W. performed contig assembly and allele calling from long-read sequencing data. A.M., D.A.O., M.I., V.S., and A.N.D. performed data analysis for the large population cohort. A.T. performed experimental work. S.R. supervised data collection for the TCR cohort. D.C., S.D.B., and D.B. supervised the study. A.M. wrote the manuscript with inputs from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278775.123.
-
Freely available online through the Genome Research Open Access option.
- Received November 26, 2023.
- Accepted October 3, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








