
Chromosome-level subgenome-aware de novo assembly provides insight into Saccharomyces bayanus genome divergence after hybridization
- Cory Gardner1,2,5,
- Junhao Chen3,5,
- Christina Hadfield2,
- Zhaolian Lu3,
- David Debruin2,
- Yu Zhan3,
- Maureen J. Donlin2,4,
- Tae-Hyuk Ahn1,2 and
- Zhenguo Lin2,3
- 1Department of Computer Science, Saint Louis University, St. Louis, Missouri 63103, USA;
- 2Program in Bioinformatics and Computational Biology, Saint Louis University, St. Louis, Missouri 63103, USA;
- 3Department of Biology, Saint Louis University, Saint Louis University, St. Louis, Missouri 63103, USA;
- 4Department of Biochemistry and Molecular Biology, Saint Louis University, St. Louis, Missouri 63103, USA
-
↵5 These authors contributed equally to this work.
Abstract
Interspecies hybridization is prevalent in various eukaryotic lineages and plays important roles in phenotypic diversification, adaptation, and speciation. To better understand the changes that occurred in the different subgenomes of a hybrid species and how they facilitate adaptation, we have completed chromosome-level de novo assemblies of all chromosomes for a recently formed hybrid yeast, Saccharomyces bayanus strain CBS380, using Oxford Nanopore Technologies' MinION long-read sequencing. We characterize the S. bayanus genome and compare it with its parent species, Saccharomyces uvarum and Saccharomyces eubayanus, and other S. bayanus genomes to better understand genome evolution after a relatively recent hybridization event. We observe multiple recombination events between the subgenomes in each chromosome, followed by loss of heterozygosity (LOH) in nine chromosome pairs. In addition to maintaining nearly all gene content and synteny from its parental genomes, S. bayanus has acquired many genes from other yeast species, primarily through the introgression of Saccharomyces cerevisiae, such as those involved in the maltose metabolism. Finally, the patterns of recombination and LOH suggest an allotetraploid origin of S. bayanus. The gene acquisition and rapid LOH in the hybrid genome probably facilitated its adaptation to maltose brewing environments and mitigated the maladaptive effect of hybridization. This paper describes the first in-depth study using long-read sequencing technology of an S. bayanus hybrid genome, which may serve as an excellent reference for future studies of this important yeast and other yeast strains.
It has generally been believed that hybridization between closely related species often leads to inviability and sterility, a phenomenon known as hybrid incompatibility. The Dobzhansky–Muller (DM) model proposes that it results from negative epistatic interactions between genes with different evolutionary histories and is a well-regarded explanation for hybrid incompatibility (Dobzhansky 1982; Price et al. 2010). Hybrid incompatibility can act as a reproductive isolating barrier contributing to speciation (Coyne and Orr 2004). Additionally, reduced fertility in hybrids can result from abnormal chromosome segregation during meiosis if the parental genomes are divergent (Coyne and Orr 2004). Nevertheless, recent studies show that interspecies hybridization is prevalent in major eukaryotic lineages, particularly in angiosperms and yeasts, and it is believed to contribute to adaptation to novel environments (Langdon et al. 2019; Taylor and Larson 2019; Gabaldón 2020; Moran et al. 2021; Suvorov et al. 2022). Given that the exchange of genomic content between species is pervasive, it is important to better characterize the impact of hybridization on evolution of hybrid genomes, which will improve our understanding of the genetic basis underlying the adaptation and divergence of species.
Hybrids of different Saccharomyces species have been frequently found in nature and in human-associated environments (Langdon et al. 2019; Gabaldón 2020). New evidence suggests that what was previously interpreted as whole-genome duplication (WGD) in the Saccharomyces lineage was actually the result of an interspecies hybridization event (Marcet-Houben and Gabaldón 2015). Soon after the WGD, there was a period of rapid loss of duplicate genes, and only ∼10% of WGD ohnologs survived. The retained WGD duplicates are enriched in genes related to glucose metabolism or rapid growth, such as glycolysis genes (Conant and Wolfe 2007), hexose transporters (Lin and Li 2011), and ribosomal protein genes (Mullis et al. 2020). These studies suggested that the WGD or hybridization event played a significant role in the adaptation of Saccharomyces species toward aerobic fermentation (Kellis et al. 2004; Thomson et al. 2005; Conant and Wolfe 2007; Lin and Li 2014) and speciation events (Scannell et al. 2006). These studies improved our understanding of the biological significance of interspecies hybridization in speciation and adaptation.
There remain unanswered questions about what occurs to the genome after a recent allopolyploidy event, such as the earliest genome rearrangements, the mechanisms of gene loss, recombination between subgenomes, and loss of heterozygosity (LOH) (Morales and Dujon 2012). The ancient hybridization events, such as the WGD in the ancestral Saccharomyces lineage, may not be useful to address these questions as most duplicate genes have been lost. In addition to the ancient hybridization event, recent interspecific hybridization is prevalent in the Saccharomyces lineage as they are used to produce fermented beverages (Langdon et al. 2019). The genomes of these recently generated hybrids may serve as ideal systems to study how genomes evolve after hybridization and contribute to adaptation to specific niches. Saccharomyces pastorianus is an interspecies hybrid between Saccharomyces cerevisiae and Saccharomyces eubayanus that is widely used for brewing lager style beers under low temperatures in Europe (Libkind et al. 2011). Some chromosomes in S. pastorianus strains have up to five copies, suggesting a highly aneuploid genome (van den Broek et al. 2015; Gorter de Vries et al. 2017). The chromosome-level assembly for S. pastorianus strain CBS1483, based on MinION long-read sequencing, enabled the assembly and exploration of the unstable subtelomeric regions, which were found to contain industrially relevant genes such as the maltose metabolism genes (MAL) genes (Salazar et al. 2019).
Saccharomyces bayanus is another interspecies hybrid yeast commonly found in industrial brewing environments but is viewed as a contaminant in some brewing processes owing to the production of undesired byproducts (Rainieri et al. 2003). The taxonomic classification of S. bayanus has been a controversial process (Hittinger 2013). Thanks to the discovery of a wild species S. eubayanus (Libkind et al. 2011), it is now commonly accepted that S. bayanus is a hybrid between S. uvarum and S. eubayanus (Pérez-Través et al. 2014; Peris et al. 2014). S. bayanus isolates have highly heterogeneous genetic and metabolic characteristics, probably resulting from many independent hybridization events between S. eubayanus and S. uvarum (Rainieri et al. 2006; Libkind et al. 2011; Langdon et al. 2019). More than 40 S. bayanus strains, such as CBS380, NCAIM 676, FM1309, and NBRC1948, were analyzed by whole-genome sequencing using the short-read Illumina technology (Libkind et al. 2011; Almeida et al. 2014; Langdon et al. 2019). Mapping the Illumina reads to different Saccharomyces species showed that the contributions of genome content from S. uvarum and S. eubayanus are highly variable among S. bayanus strains. Specifically, the genome content deriving from S. uvarum ranges from 36.6% to 98.8% (Langdon et al. 2019). In addition, small introgressed regions from S. cerevisiae are present in some S. bayanus strains (Nguyen et al. 2011). However, these S. bayanus genome assemblies are fragmented owing to the limitation of Illumina short reads, limiting our understanding of the genome evolution of these hybrid strains.
A chromosomal-level subgenome assembly of S. bayanus will provide much more detail in the genome evolution following a recent allopolyploidy event. In this study, we sequenced the genome of S. bayanus strain CBS380 (BY20106, IFO11022) using the Oxford Nanopore Technologies (ONT) MinION. The strain CBS380 is a representative isolate of S. bayanus, which has been widely used in many studies (Libkind et al. 2011; Nguyen et al. 2011; Caudy et al. 2013; Pérez-Través et al. 2014). We aimed to generate chromosome-level subgenome assemblies based on MinION reads and characterize the evolution of genome structure and gene content. By studying this hybrid genome, we seek to improve our understanding of the genetic basis of hybrid species’ survival and the mechanisms that allow them to overcome hybrid incompatibility.
Results
Inference of the origin and evolutionary relationships of S. bayanus hybrid strains
We first sought to determine the evolutionary relationships between S. bayanus CBS380 and other hybrid strains and to infer their parental strains. It theoretically can be achieved by phylogenetic inferences based on sequences of heterozygous alleles of the same set of genes that are shared by hybrid and parental species strains. However, this is infeasible owing to lack of haplotype-aware assembly in other hybrid strains. In addition, the contributions of the two parental strains vary substantially among hybrid strains (Langdon et al. 2019). Thus, even if haplotype-aware assemblies are available, the chance of having heterozygous genes shared by all hybrid strains is small. To overcome this obstacle, we used the assembly- and alignment-free approach MIKE (Wang et al. 2024) to infer their phylogenetic relationships based on raw sequencing reads of a total of 395 strains of the three species obtained from NCBI SRA database (see Methods) (Supplemental Data Set S1).
Two phylogenetic trees were generated to illustrate the evolutionary relationship of S. bayanus strains with S. uvarum strains and with S. eubayanus strains, respectively (Fig. 1A; Supplemental Fig. S1). In both phylogenetic trees, hybrid strains form a polyphyletic clade, suggesting that hybrid strains were generated by hybridization from different S. eubayanus and S. uvarum strains. For example, hybrid strains are present in four different clades with S. uvarum strains, suggesting that they were created by hybridization events from at least four different S. uvarum strains. In addition, hybrid strains from the same clade in the S. uvarum tree are found in different clades of the S. eubayanus tree, suggesting that the same S. uvarum strain may have hybridized with different S. eubayanus strains (Fig. 1B). The CBS380 strain is most closely related to S. bayanus FM473 and yHQL566 in both trees.
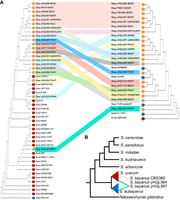
Evolutionary relationships among S. bayanus hybrid strains and their parental species S. uvarum and S. eubayanus. (A) The phylogenetic tree on the left shows the evolutionary relationships among S. bayanus strains with strains of parental species S. uvarum. The right phylogenetic tree shows the evolutionary relationships among S. bayanus strains with representative strains of the other parental species S. eubayanus. Both phylogenetic trees were reconstructed based on shared k-mers of their raw sequencing reads using MIKE. A complete phylogenetic tree that includes all examined S. eubayanus strains was provided as Supplemental Figure S1. (B) Schematic illustration showing a complex origin of S. bayanus strains by independent hybridization events from different strains of S. uvarum and S. eubayanus.
S. bayanus strains were isolated in highly diverse environments, and it was reported that repeated backcrossing with one of the two parental species might have occurred in different strains (Langdon et al. 2019). Together with the observation that many S. bayanus strains were generated by independent hybridization events from different parental strains, it probably explains why genetic and metabolic characteristics are highly diverse among hybrid strains. The high degree of diversity does raise the question of whether we should regard different S. bayanus hybrid strains as the same species.
MinION sequencing, ploidy analysis, and parental inference of S. bayanus CBS380 genome
To confirm the ploidy levels of the S. bayanus CBS380 strain, we assessed its relative genomic DNA content by fluorescence flow cytometry analysis using the haploid yeast strain S. uvarum YJF1450 as a control (Supplemental Fig. S2). Dual peaks of fluorescence were observed in both strains, with the first peak indicating the DNA content of the G1 phase and the second peak showing DNA content after DNA synthesis (G2/M phase). As shown in Supplemental Figure S2, the relative genomic DNA content in the G1 phase of S. bayanus CBS380 is similar to the G2/M phase of the haploid control S. uvarum YJF1450, confirming that two sets of chromosomes are present in S. bayanus CBS380.
Sequencing of S. bayanus CBS380 with the ONT MinION yielded 2.2 Gbp of data (∼170× coverage), with 2.04 Gbp passing quality control (Supplemental Fig. S3). Among these, 100 reads exceeded 100 kbp in length, with the longest extending to 158,255 bp. We hypothesized that most of our reads would map to the suspected parental species, S. eubayanus and S. uvarum, although fewer, if any, reads would map to more distantly related species. Inspired by the sppIDer approach (Langdon et al. 2018), which was designed to investigate hybrid genomes based on short-read sequencing, we developed a modified approach that is better suited for ONT long-read sequencing data (see Methods). We used this method to determine the relative genetic contribution of potential parental genomes. The majority of the reads were assigned as originating from S. uvarum and S. eubayanus, as expected (69.60% from S. uvarum and 27.25% from S. eubayanus). Of the remaining, 2.48% mapped to Saccharomyces mikatae, 0.55% mapped to S. cerevisiae, 0.07% mapped to Saccharomyces kudriavzevii, 0.04% mapped to Saccharomyces paradoxus, and 0.01% mapped to Saccharomyces arboricola (Supplemental Fig. S4). This confirms that the S. bayanus CBS380 strain is a hybrid of S. uvarum and S. eubayanus, with introgressed regions from other Saccharomyces species, such as S. mikatae and S. cerevisiae.
De novo assembly and subgenome phasing
Our genome assembly process examined several tools, detailed in the Methods section and in Supplemental Table S1, to address the challenges posed by the diploid nature of the target organism. Among the various tools tested, Flye stood out by producing a collapsed-consensus assembly with the highest quality, as reflected in a 96.6% completeness score according to BUSCO analysis. This high score indicates a successful capture of the genomic features we aimed to assemble.
We aimed to address the inherent complexity of the diploid genome of S. bayanus CBS380 by assembling the two subgenomes separately. We used the MinION platform's long-read technology to generate the sequence and, of the different approaches tested, found that phasing the Flye collapsed-consensus assembly via the WhatsHap pipeline proved the most successful at constructing a high-fidelity diploid representation of S. bayanus CBS380 (Patterson et al. 2015). Our methodology successfully assembled two distinct subgenomes, randomly named as haplotype-a and haplotype-b, allowing for a sophisticated analysis of the dual genome architecture (Table 1; Supplemental Table S2; Supplemental Data Set S2).
Assembly statistics for the S. bayanus genome and subgenome grouping
To assess the completeness of our genome assembly, we searched for yeast telomere repeat sequences using TelFinder (Sun et al. 2023) and sequence motif T(G)2-3(TG)1-6 as described by Teixeira and Gilson (2005). Telomere repeat sequences were detected in 50 of 64 chromosome ends (Supplemental Table S3). In addition, telomere repeat sequences were at both ends of 20 of all 32 chromosomes, indicating that the assembly of most chromosomes is truly telomere-to-telomere (T2T). Telomere repeats were detected in one end of 10 chromosomes and were not detected in only two chromosomes: Chr VIa and Chr VIIb (Supplemental Table S3). In terms of assembled chromosome length, Chr VIIb is similar to Chr VIIa (1,042,311 vs. 1,055,917), so the assembly of Chr VIIb can be considered as complete (Supplemental Table S2).
Our assembly generated a circular mitochondrial genome with a size of 65 kb. To determine the origin of the CBS380 mitochondrial genome, we first mapped our raw MinION sequencing reads to the combined mitochondrial genomes of S. eubayanus (NW_017264706.1) and S. uvarum (CP113780.1) and inferred their origin using sppIDer. Our results show that 9.31% of all MinION reads were mapped to mitochondrial genomes (Supplemental Fig. S5). Of the reads mapped to mitochondria, 97.7% of reads were mapped to S. uvarum, suggesting that the mitochondrial genome of CBS380 was inherited from S. uvarum, which is consistent with results based on Illumina reads (Langdon et al. 2019). In addition, a nucleotide BLAST search, using the assembled mitochondrion as the query sequence, showed a hit for the S. uvarum mitochondrion, with 100% query coverage and 99.89% identity.
Genome annotation
We used the GALBA pipeline to predict and annotate the protein-coding genes for each subgenome/haplotype of S. bayanus CBS380 (Brůna et al. 2023). The pipeline is well suited to our use case, given its capability to leverage high-quality protein sequences from closely related species. The output revealed a total of 11,545 protein-coding genes identified across both haplotypes, with 5737 genes in haplotype-a and 5808 genes in haplotype-b (Table 1; Fig. 2; Supplemental Data Set S3). The variation in gene count between the two haplotypes is in direct proportion to their chromosomal lengths.
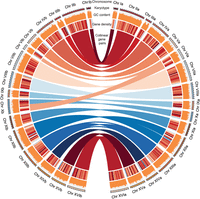
Circos circular visualization of the genome assembly for the 16 pairs of chromosomes of the S. bayanus genome. Different genomic features across four concentric circles. The outermost circle represents the karyotype of the S. bayanus genome for two haplotypes, with the right part representing haplotype-a and the left part representing haplotype-b. The second outermost circle represents the GC content on each chromosome of the genome. The third circle provides information on gene density within the chromosomes, and a darker color indicates a higher gene density. The innermost circle highlights synteny blocks between haplotypes, illustrating collinear gene pairs between haplotype-a and haplotype-b.
We assessed the completeness of the genome annotation with BUSCO (Manni et al. 2021), using the saccharomycetes_odb10 database as the reference, and observed a 98% degree of completeness (2095 of 2137). The BUSCO analysis is based on both haplotype assemblies, but most genes are expected to have two copies. We classified 86.2% of genes (1843) as duplicates, whereas only 11.8% (252) were identified as unique single-copy genes. Only 13 genes (0.6%) from the saccharomycetes_odb10 gene set were absent from our predicted list. The genome annotation, CDS, and protein sequences are available at https://github.com/BioHPC/Saccharomyces-bayanus.
We annotated 95% (10,985) of the total predicted genes using eggNOG-mapper (Cantalapiedra et al. 2021), which assigned key functional information, such as descriptions of biological functions, orthologous genes in S. cerevisiae, Gene Ontology, KEGG pathway, and Pfam domains (Supplemental Data S1). The combination of functional annotation and BUSCO assessments confirms that our annotation results are comprehensive, providing a solid foundation for our further analysis.
Inference of parental genomic regions and major genomic events after hybridization
We next sought to determine the origin of genomic regions in each S. bayanus CBS380 chromosome, which would be useful for the identification of major genomic events that have occurred since hybridization, such as recombination, chromosomal rearrangements, and LOH (see Methods). Our first approach used nonoverlapping blocks of 5000 bp for every chromosome in a BLAST search against the genomes of S. eubayanus and S. uvarum, with the origin of each genomic block determined by as the best hit of BLAST search (Supplemental Fig. S6). Each haplotype chromosome contains regions that originated from both S. uvarum and S. eubayanus (Fig. 3A), suggesting the recombination between the two orthologous chromosomes of the two subgenomes creates mosaic chromosomes composed of genomic regions of heterozygous origins. However, the proportions of each subgenome vary substantially across different chromosomes. For example, segments of S. eubayanus origin make up 83% of Chr IVa, whereas they make up only 22% of Chr IVb (Supplemental Table S4). In addition, nine of the 16 chromosome pairs have a high degree of homozygosity, with >80% of genomic regions derived from the same parental species (Supplemental Table S4), meaning that the genomic origin and recombinants are very similar between haplotypes a and b, showing that heterozygosity was quickly lost after hybridization.
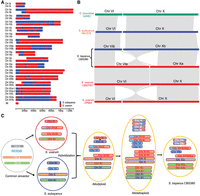
The origin and evolution of S. bayanus chromosomes. (A) Origin of genomic regions of each chromosome in the S. bayanus genome based on BLAST searches of nonoverlapping 5000 bp blocks. Genomic regions that originated from S. eubayanus are shown in blue, and regions inherited from S. uvarum are shown in red. (B) Synteny block of Chr VI and Chr X between S. cerevisiae, S. eubayanus, both S. bayanus haplotypes, S. uvarum strain CBS7001, and S. uvarum strain ZP964. (C) An evolutionary model of S. bayanus chromosomes. For simplification purposes, only four chromosomes are shown, representing different patterns of chromosome inheritances. Translocation between Chr VI and Chr X occurred in S. eubayanus prior to its hybridization with S. bayanus. Recombination and whole-genome duplication occurred in the hybrid S. bayanus genome. Subsequent genome reduction, probably by sporulation, created some heterozygous chromosomes, such as Chr III, and some homozygous chromosomes, such as Chr XI.
To validate the accuracy of inference of parental genomic regions by the BLAST approach, we mapped the raw sequences reads that were assigned to S. uvarum and S. eubayanus by sppIDer to haplotype assemblies, respectively (see Methods) (Supplemental Fig. S7). For each mapping file, we calculated the average mapping depth of every 5 kb region, and a minimum of 30× coverage depth was considered as support of origin. The mapping results show that reads assigned to S. uvarum and S. eubayanus were mapped to nonoverlapping regions of chromosomes, supporting the reliability of this approach. Similar to the first method, the second method reveals the occurrence of recombination between the two parental genomes in most chromosomes, and the locations of recombination breakpoint are highly consistent (Supplemental Fig. S7). In addition, the Pearson correlation coefficient for the percentages of genomic content is 0.99 (Supplemental Table S4), supporting the robustness of our inference of the origin of genomic regions.
We identified a total of 55 major recombination breakpoints in the 16 pairs of CBS380 chromosomes (Supplemental Table S5). To determine if any of these recombination breakpoints were generated by assembly errors, we examined read coverages of ±5 kb regions flanking all major breakpoints. Based on the percentage of supported reads, breakpoints were classified as high support (>90%), moderate support (60%–90%), and low support (<60%). Our results show that 53 out of 55 breakpoints are high support, whereas two were classified as medium and low support, respectively (Supplemental Table S5). Accumulating evidence suggests that dispersed repeated elements, such as Ty elements that contain long terminal repeats (LTRs), may have facilitated recombination between different chromosomes (Fischer et al. 2000; Mieczkowski et al. 2006). We then identified all LTR sequences in the hybrid genome and examined their chromosomal distribution. We found that LTR retrotransposons are significantly enriched within 5 kb regions flanking the breakpoints of recombination in both subgenomes (Fisher's exact test: P-value < 2.2 × 10−16 for subgenome-a and P = 7.5 × 10−05 for subgenome-b) (Supplemental Fig. S8). These results suggest that the recombination between parental subgenomes might have been facilitated by LTR retrotransposons.
Identification of chromosome aneuploidy and intrachromosomal rearrangement
Single-chromosome aneuploidy could be overlooked based on flow cytometry experiments. We performed read depth analysis to detect if any chromosome is aneuploid in the CBS380 genome. We first mapped MinION sequencing reads to each of the haplotype chromosomes and normalized their read depths by the genome-wide average to allow for comparison across different genomic regions. Our analysis shows that, although most chromosomes have a similar read depth with the genome-wide average, the coverage depth of Chr XV is ∼1.5× of the diploid genome average, suggesting the presence of three copies of Chr XV (Supplemental Fig. S9). We aligned the Chr XV of S. uvarum to Chr XV of S. eubayanus to identify unique SNPs. We then mapped our ONT reads to a reference consisting of the combined Chr XV sequences of S. uvarum and S. eubayanus and computed the read depth at the distinguishing loci. Read support at these positions in Chr XVa tends to have about twice the number of supported reads compared with those in Chr XVb, suggesting that the third copy of Chr XV was likely generated by a recent whole-chromosome duplication of Chr XVa, possibly owing to nondisjunction during mitosis (Fig. 3A).
We found significant polymorphism in the chromosome lengths of Chr VI and Chr X between the two subgenomes (haplotypes) in S. bayanus CBS380 (Figs. 2, 3A; Supplemental Table S2). Specifically, Chr VIa is ∼277 kb longer than Chr VIb (544 kb vs. 267 kb), whereas Chr Xa is ∼244 kb shorter than Chr Xb. Our analysis of syntenic regions between the two haplotypes shows that a significant portion of Chr VIa has syntenic regions to Chr Xb. Gene collinearity analysis suggests it was likely generated by a translocation that exchanges an ∼270 kb segment at the left end of Chr X with an ∼30 kb region at the right end of Chr VI (Fig. 3B). Electrophoretic karyotypes of Saccharomyces “sensu stricto” species have identified a translocation of ∼355 kb between Chr VI and Chr X in S. uvarum CBS7001 (Fischer et al. 2000). Thus, it is reasonable to postulate that the observed translocation between Chr VI and Chr X was likely inherited from S. uvarum.
To determine when the translocation occurred during the evolution of S. uvarum, we conducted a chromosomal collinearity analysis for Chr VI and Chr X for all 32 whole-genome shotgun (WGS) contigs of S. uvarum strains at the NCBI WGS database. Considering that many genome assemblies of S. uvarum strains are incomplete, our collinearity analysis focused on ±100 kb regions flanking the breakpoint of translocation in Chr VIa. The translocation was supported by all assemblies of S. uvarum strains, except for strain ZP964 (Fig. 3B; Supplemental Fig. S10). The chromosome architectures of Chr VI and Chr X in S. uvarum ZP964 are very similar to those of Chr VIb and Chr Xb in CBS380. Previous phylogenetic analysis showed that ZP964 belongs to the Australasian clade, which is the most divergent clade of S. uvarum (Almeida et al. 2014). These results suggest that the translocation between Chr VI and Chr X occurred at the very early stage during the evolution of S. uvarum. However, we cannot exclude the possibility of assembly errors in ZP964. Nevertheless, the conclusion that the translocation occurred before divergence of most S. uvarum strains remains unaffected.
A model of allotetraploid origin of S. bayanus and mechanisms of LOH
Our analysis of origins of genomic regions in S. bayanus CBS380 demonstrates that only seven pairs of chromosomes maintained heterozygous status, and LOH occurred in most of genomic regions in the other nine chromosome pairs (Fig. 3A). In these nine chromosomes, LOH extends to most parts of chromosomes, resulting in chromosomal-level LOH. The most parsimonious way to achieve chromosomal-level LOH in most chromosomes is probably chromosomal duplication followed by loss of heterozygous chromosomes. These events might happen individually by nondisjunction. If this is the case, we would expect extensive chromosome aneuploidy, similar to what was observed in another hybrid species S. pastorianus (Salazar et al. 2019). However, chromosome aneuploidy was only observed in Chr XV. Alternatively, all chromosomes might be doubled simultaneously by WGD if cells replicate their genomes without division (endoreplication), which generates a temporary allotetraploid (Shu et al. 2018). Because interspecies hybrids are usually infertile, genome doubling is a simple way to restore their fertility (Marcet-Houben and Gabaldón 2015). Subsequently, the allotetraploid genome would have been reduced into a diploid genome by sporulation, a process of spore formation via meiosis. Assuming segregation of four copies of chromosomes is random, the probability to observe nine pairs of homozygous chromosomes is the second most likely outcome (17.5%). Therefore, endoreplication followed by sporulation provides the most plausible explanation for the patterns, and it is also the most economical mechanism (Fig. 3C). In summary, our observations suggest that endoreplication/sporulation plays a main role in the rapid and chromosomal level of LOH in the genome of CBS380, whereas gene conversion and mitotic recombination also contributed to LOH at some local regions.
Inferring the age of the S. bayanus CBS380 strain
To estimate the age of S. bayanus (CBS380), we analyzed the single-nucleotide polymorphisms (SNPs) in the regions that have become homozygous following LOH events (see Methods). Our analysis shows an average SNP density of 4.244 × 10−4 SNPs per base pair in homozygous regions. Based on the estimated mutation rate of S. cerevisiae and the typical generation type of budding yeasts, we inferred the CBS380 strain was likely generated ∼300−400 years ago. These results align well with historical records of lager brewing, which started in the fifteenth century and became popular in the nineteenth century (Libkind et al. 2011).
Inference of introgressed genes from other species
Our analysis of raw sequencing reads of the hybrid strain suggests the presence of introgressed genomic regions in CBS380 from other species, such as S. mikatae and S. cerevisiae (Supplemental Fig. S4). To identify introgressed genes, we first grouped all annotated protein-coding genes from S. bayanus, S. uvarum, S. eubayanus, S. cerevisiae, S. paradoxus, S. mikatae, and S. kudriavzevii into 6732 orthologous groups (OGs) (Fig. 4A; Supplemental Data Set S4). A total of 3872 OGs are present in all species examined, representing the most conserved groups of genes in the genus of Saccharomyces. Seven hundred forty-three OGs contains member genes from S. bayanus, S. uvarum, and S. eubayanus, which is the second most common type of OGs (Fig. 4A), representing a group of genes that are specific to the lineage of S. uvarum and S. eubayanus.
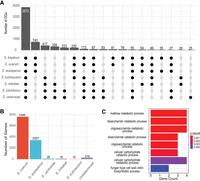
Inference of introgressed genes in the S. bayanus CBS380 genome. (A) Classification and distributions of OGs based on their member species. Only those types with at least 25 OGs are shown in this figure. A black-filled dot represents the presence of member genes in an OG from a specific species, and a gray dot indicates the absence of member genes. (B) The number of genes in S. bayanus CBS380 from each parental genome. (Undetermined) Genes have a P-distance > 0.025 with any orthologous genes, and their origin cannot be confidently determined. (C) Results of GO enrichment of introgressed genes from S. cerevisiae.
We noticed that 78 OGs are only present in the genome of S. bayanus, with a total number of 175 genes. As a large number of de novo gene births or gene loss are extremely unlikely given the short evolutionary history of S. bayanus, we speculated that it is because of genome misannotation in other species. We conducted BLAST searches using the 175 S. bayanus genes as queries against all Saccharomyces genome assemblies in NCBI. The presence of orthologous sequences was defined as a minimum of 95% sequence identities, and it covers at least 50% of query sequences. Based on this threshold, orthologous sequences were identified for all these genes (Supplemental Data Set S5). Specifically, 163 genes have the best hits in the genomes of S. uvarum (110 genes) and S. eubayanus (53 genes). In addition, three of them have the best hits in S. cerevisiae strain AMM/SJ5L and 12 of them from another hybrid strain S. pastorianus, suggesting that these genes could be introgressed. The potential functions of these genes were annotated with “eggnog-mapper” (Cantalapiedra et al. 2021). GO enrichment analysis based on their annotations shows that these “orphan genes” are enriched in GO terms such as ATP hydrolysis activity, nucleotide binding, and carbohydrate derivative binding (Supplemental Table S6).
If a S. bayanus gene was introduced by introgression from a third species, the gene should have the lowest level of sequence divergence with its donor genes than to genes of their parental genomes. Based on this concept, for each S. bayanus gene, we calculated its sequence divergence (P-distance; the proportion of nucleotide sites at which the two sequences compared are different) to all orthologous genes in the same OG. To prevent bias owing to misannotation or gene loss in other genomes, we estimated the maximum P-distance for introgressed genes based on the distribution of P-distance of all genes. The P-distance values for orthologous genes in S. uvarum and S. eubayanus exhibit two distinct peaks, representing two different origins of S. bayanus genes (Supplemental Fig. S11). The left peak, which is zero or very close to zero, includes genes that are directly inherited from the examined species, and the right peak includes genes that originated from the other parental genome. Therefore, if a S. bayanus gene has the lowest P-distance with a gene from a third species and the P-distance is within the first peak (P < 0.025), it is considered as an introgressed gene. Based on this method, we identified 28 introgressed genes from S. cerevisiae, 16 from S. mikatae, and 10 from S. kudriavzevii (Fig. 4B; Supplemental Data Set S6). In addition, the origin of 319 S. bayanus genes cannot be determined (“undetermined”) because they do not have any orthologous genes that have a P-distance < 0.025 (Fig. 4B).
To infer the functional significance of introgressed genes, we conducted GO enrichment analyses for the 28 S. cerevisiae introgressed genes as they have the most complete GO annotation. As shown in Figure 4C, these genes are significantly enriched in sugar metabolism processes, such as maltose utilization. S. cerevisiae is known to have the ability to efficiently ferment sugars. The introgression of these sugar metabolism genes might have facilitated CBS380's adaptation in sugar rich environments.
Population genomics analysis of seven beer strains of S. bayanus suggests nonrandom retention of S. eubayanus genomic content
Comparative studies of genomic content among S. bayanus strains allow us to infer whether segregation and recombination between parental genomes is a neutral process. The contributions of parental genomic content were shown to vary greatly among S. bayanus strains (Langdon et al. 2019). We first evaluated if the natural habitats of yeasts had any impact on retention of parental genomic regions. We found that the 12 strains with beer as an isolation origin tend to have a significantly higher proportion of genomic content derived from S. eubayanus (Supplemental Fig. S12). In contrast, the strains isolated from cider have the lowest genomic contribution from S. eubayanus (P = 3.52 × 10−6, two-tailed t-test), suggesting that the natural habitats may have a strong influence on the retention of parental genomes.
As S. bayanus beer strains tend to have a higher proportion of genomic regions derived from S. eubayanus, we next sought to evaluate if there are any preferences in retention of specific genomic regions using published sequencing data (Langdon et al. 2019). To avoid potential biases, we only used beer strains with at least 10× read depth (Supplemental Data Set S1). We first retrieved raw sequencing reads assigned to S. eubayanus using sppIDer and then mapped these reads to the S. eubayanus FM1318 genome to identify genomic regions in each S. bayanus strain that were derived from S. eubayanus. Each chromosome was divided into 5 kb nonoverlapping regions, and each region was considered as derived from S. eubayanus if it is supported by at least 25% of average genome sequencing depth. We then calculated the number of strains that contained the 5 kb window of S. eubayanus origin. For example, the presence of S. eubayanus genomic content in Chr VII, Chr XII, and Chr XIII are among the lowest among beer strains (Fig. 5A). In contrast, S. eubayanus genomic contents are more likely to be found in Chr III and Chr VI. Only ∼ 2% of S. eubayanus genomic contents are present in all seven beer strains. GO analysis shows that genes within these regions are significantly enriched in several metabolic processes, such as in the maltose metabolic process and sulfur compound metabolic process (Fig. 5B). These observations suggest that the retention of parental genomic content might have been shaped by natural selection.

Shared genomic regions originated from S. eubayanus among seven beer strains of S. bayanus. (A) The number of beer strains with genomic regions originated from S. eubayanus. Each dot represents a 5 kb window. (B) Functional enrichment of genes retained from the S. eubayanus parental genome.
The origin and evolution of genes involved in maltose/maltotriose utilization
A total of 5153 OGs present in S. bayanus and its two parental genomes, and 4475 (86.8%) of them exhibit a 1:1:2 ratio, indicating conservation of gene copy numbers in all three species (Fig. 4A; Supplemental Data Set S4). It is worth noting that 385 OGs contain more than two copies of genes in the diploid genome of S. bayanus CBS380, suggesting an expansion of gene copy number after hybridization. Seven of these expanded OGs are involved in maltose/maltotriose utilization (MAL genes). Considering that introgressed genes are enriched in genes involved in maltose metabolic processes (Figs. 4C, 5B), the expansion of MAL genes could be caused by introgression and retention MAL genes from the S. eubayanus subgenome. S. bayanus CBS380 is mainly found in beer brewing environments, and maltose is the most abundant sugar (∼60%) in brewer's wort (Magalhães et al. 2016). Therefore, the expansion of MAL genes might have facilitated the adaptation of S. bayanus to maltose-rich environments.
MAL genes are classified into three families based on their functions, including maltose transporter (MALT), enzymes that break down maltose (MALS), and genes that regulate the expression of the pathway (MALR). These genes are often organized into clusters and located near the ends of chromosomes (subtelomeric). To elucidate the origins and expansion of MAL genes in S. bayanus, we first identified all MAL genes in S. bayanus and eight other Saccharomyces species. The total numbers of MAL genes vary substantially among nonhybrid species, ranging from zero in Nakaseomyces glabratus to 19 in S. mikatae (Fig. 6A). The diploid genome of S. bayanus contains a significantly higher number of MAL genes (32 in total) compared with seven in S. uvarum and six in S. eubayanus (Fig. 6A), supporting a significant expansion in copy numbers of MAL genes in S. bayanus after hybridization. This is particularly noticeable that the MALS gene family increased from three and six in their parental species genomes to 19 in S. bayanus.
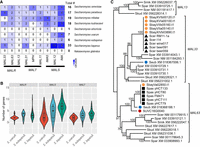
The evolution of gene content in the hybrid genome of S. bayanus CBS380. (A) Evolutionary changes of the three MAL gene families in nine OGs in the Saccharomyces sensu stricto members and Nakaseomyces glabratus. (B) Increased gene copy numbers are observed in each MAL gene family in S. bayanus. (C) A phylogenetic tree of the MALR gene family suggests introgression of MALR genes in S. bayanus. The tree was inferred using the maximum likelihood method with 1000 bootstrap and reported as a percentage on each node. The tree is drawn to scale, with branch lengths denoting the genetic distance.
To better understand the adaptation significance, we evaluated if expansion of MAL genes also occurred in other S. bayanus strains. We conducted searches of homologous genes against 28 other published S. bayanus genomes (Supplemental Data Set S7). By comparing the number of MAL genes, an increased copy number of MAL genes was observed in all three MAL families (Fig. 6B). As the genome assemblies for these hybrid strains were based on Illumina short reads, which were not haplotype-aware, the total number of MAL genes may have been underestimated. Nevertheless, our results support that expansion of MAL genes is a shared pattern among S. bayanus strains.
To further investigate the mechanism of MAL gene expansion in S. bayanus, we carried out phylogenetic analyses for each of the three MAL families using their amino acid sequences (Fig. 6C; Supplemental Fig. S13). We examined the tree topology and found that the majority of the MAL genes in S. bayanus were not inherited directly from its parental genomes but are more likely acquired through introgression from other species, mostly from S. cerevisiae, followed by multiple gene duplication events. For example, six copies of MALR genes are present in S. bayanus. Five of them form a well-supported clade with genes from different S. cerevisiae strains with identical sequence (branch length = 0) (Fig. 6C), supporting that these S. bayanus MAL33 genes were acquired by recent introgression from S. cerevisiae strains. Examination of chromosomal locations of the five MALR genes showed that they are distributed in three MAL clusters in subtelomeric regions of three different chromosomes (Supplemental Fig. S14). Phylogenetic analysis of other genes in these MAL clusters suggests that the entire clusters were likely introgressed (Supplemental Figs. S13, S14). In addition, the sixth member of MALR in S. bayanus appears to be introgressed from another hybrid strain, S. pastorianus, which is used industrially to produce lager beer (Fig. 6C). Another 10 MALS genes were also derived from introgression of S. pastorianus genes (five MALS genes and five MALT genes). Given that CBS380 was isolated from the same type of habitat, it is not unexpected to observe gene flow from S. pastorianus. In contrast, only five of the 32 MAL genes in S. bayanus were derived from S. eubayanus. These five genes belong to the MALS family and form a monophyletic clade (Supplemental Fig. S13), suggesting that they were likely generated by gene duplications after hybridization.
We noticed that no S. bayanus MAL genes were inherited from S. uvarum (Fig. 6C; Supplemental Fig. S13). Chromosomal locations of MAL genes in S. bayanus also showed that none of these MAL loci are present in S. uvarum–derived regions. It suggests that there was a preferred retention of MAL genes inherited from introgressed donors or a preferred loss of S. uvarum–derived MAL genes. Given that S. uvarum has contributed >60% of the genetic makeup of S. bayanus, the strong exclusion of S. uvarum MAL genes in the S. bayanus genome is not likely owing to random events. One possibility is that MAL genes from S. uvarum might impose selective disadvantages under maltose-rich brewing environments. Future studies on the growth effects of S. uvarum MAL genes may provide new insights into the biased retention of MAL genes.
Discussion
We present the first chromosome-level subgenome assembly and annotations of the hybrid yeast, S. bayanus (CBS380), which will serve as an excellent reference for future studies of this important yeast and other yeast strains. The assembly was completed using only a single MinION flow cell. Thus, we show that the utility of high read depth sequencing is available for moderate costs using this technology. We assessed the assemblies from 15 different de novo assembly pipelines, all run on relatively modestly equipped computer workstations, and concluded that the Flye method (Kolmogorov et al. 2019) outperformed the others in producing an assembly with the fewest contigs and high N50 scores. The successful application of the GALBA pipeline (Brůna et al. 2023) allowed for high-fidelity annotation of the two subgenomes and confirmed the completeness of our genome assembly with a high BUSCO score. This type of sequencing can be carried out in most laboratories without previous sequencing experience or high-performance computational resources.
Our phylogenetic inferences using MIKE (Wang et al. 2024) observed polyphyletic clade formation in hybrid strains in both trees, suggesting multiple independent hybridization events (Fig. 1). However, our findings based on phylogenetic groupings should be interpreted with caution because of frequent recombination between parental subgenomes and differential LOH in hybrid genomes. It has been intensely debated whether S. pastorianus was produced by a single hybridization event or by multiple hybridization events (Gorter de Vries et al. 2017). MIKE is an alignment-free method that calculates the Jaccard coefficient to infer the evolutionary distance between two genomes, which was quantified as the ratio of shared k-mer to the union of two sets of k-mer in raw sequencing reads (Wang et al. 2024), so it provides more accurate phylogenetic inferences if the contributions of parental genome contents are similar. Because the parental genomic contents of different S. bayanus strains vary substantially (Langdon et al. 2019), potential bias on phylogenetic groupings of hybrid genomes owing to variances in parental genomic contributions should be noted. Our results show that genomic regions originating from S. eubayanus parental genomes differ significantly among different S. bayanus strains (Fig. 5A), which provides a different line of evidence supporting that they were likely generated by multiple different hybridizations. As comprehensive phylogenetic inference of S. bayanus strains is not the focus of the study, further studies are needed to fully elucidate the ancestry of these hybrid strains.
We proposed a model of allotetraploid origin of S. bayanus, that is, WGD followed by chromosome losses via sporulation. This model provides plausible explanations for observed chromosomal-level LOH in most chromosomes (Fig. 3C). That being said, the contribution of other genetic mechanisms underlying LOH after hybridization should also be considered, such as gene conversion and mitotic recombination (Marcet-Houben and Gabaldón 2015; Wolfe 2015; Wertheimer et al. 2016). Gene conversion is a common mechanism responsible for LOH (James et al. 2019). Because the track length of gene conversion is usually limited, extending up to ∼2 kb, chromosome-level LOH is unlikely created by gene conversion. We do observe some small tracks of LOH in a few chromosomes, such as in I and IV. Mitotic recombination was found as an important mechanism leading to LOH in diploid yeast (Andersen et al. 2008; Sui et al. 2020). Unlike gene conversion, the size of LOH created by mitotic recombination can be much larger and can extend to the end of the chromosome. Consequently, mitotic recombination creates LOH at one end of the chromosome, whereas the rest of the chromosome remains heterozygous. Among CBS380 chromosomes, this pattern was observed only in Chr IV and Chr XIV, which have a large LOH region at one end of their chromosomes, whereas the rest of the regions maintain heterozygous status (Fig. 3A). Therefore, gene conversion and mitotic recombination may also have contributed to some LOH regions in the CBS380 genome.
Our comparative genomic analyses suggested that the evolution of gene content in the CBS380 hybrid genome may have been shaped by natural selection. A prominent example is the genes involved in maltotriose utilization (MAL genes). MAL genes are highly enriched among introgressed genes in CBS380. MAL genes are also among genes that demonstrated gene number expansions. The increased copy number of MAL genes was likely achieved by both introgression and subsequent gene duplication events based on our phylogenetic inference. These S. bayanus beer strains, including CBS380, were originally isolated from beer, which is brewed from barley wort that consists of 60% maltose and 25% maltotriose (Zastrow et al. 2001). Unlike S. cerevisiae and S. pastorianus strains, wild S. eubayanus isolates have been shown to lack the ability to consume maltotriose (Baker et al. 2015). Therefore, it is reasonable to believe that the introgression and expansion of MAL genes from S. cerevisiae and S. pastorianus strains enable the hybrid to consume maltotriose, providing selective advantages in maltose-rich brewing environments. Despite S. bayanus inheriting most chromosomal segments from S. uvarum, none of the MAL genes in S. bayanus were traced to its S. uvarum progenitor. It is unlikely because of random loss of S. uvarum copies. One possibility is that S. uvarum MAL genes might have antagonistic effects on introgressed MAL genes, resulting in less efficient maltotriose utilization. Further studies can be performed to examine the functional differences of these MAL genes in maltose metabolism between these species, which could provide new valuable information for improving industrial brewing using maltose-rich materials.
Methods
Inference of origin and evolution of S. bayanus strains
Raw sequence reads of 21 S. bayanus strains, 347 S. eubayanus strains, and 27 S. uvarum strains were downloaded from NCBI SRA database (Supplemental Data Set S1). The raw sequencing data of each hybrid strain were aligned to the combination genomes of S. eubayanus strain FM1318 (assembly SEUB3.0) and S. uvarum strain CBS7001 (assembly ASM2755758v1) using sppIDer (Langdon et al. 2018). The sequencing reads assigned to origin of S. uvarum of each hybrid strain were then used to infer their evolutionary relationships with other S. uvarum strains using MIKE (Wang et al. 2024). A phylogenetic tree for S. eubayanus and hybrid strains was reconstructed using reads assigned to S. eubayanus genomes.
Yeast strain, growth condition, genomic DNA isolation
S. bayanus CBS380's cells were grown on YPD medium (1% yeast extract, 2% peptone, and 2% glucose) for 16 h at 30°C. Extraction of high-molecular-weight genomic DNA (HMW gDNA) from S. bayanus cells was carried out by following a protocol described by Denis et al. (2018). In brief, S. bayanus cell wall was first lysed with zymolyase (MP Biomedicals). Spheroplasts were then collected and resuspended in SDS buffer with RNase A. Proteins were precipitated and removed with potassium acetate and centrifugation. The supernatants were used to precipitate DNA with isopropanol. The DNA pellet was then washed with 70% ethanol and dissolved in TE buffer. The quality and quantity of the extracted DNA were determined using Qubit (Invitrogen). HMW gDNA was sheared into 20 kb fragments using g-TUBE (Covaris).
Determination of ploidy
We performed a flow cytometry analysis to determine the ploidy of the S. bayanus CBS380 following the protocol (Todd et al. 2018). We also used a haploid S. uvarum strain YJF1450 (MATα hoΔ::NatMX, derived from CBS7001, a gift from J. Fay laboratory at Rochester University) as a control. Briefly, yeast cells were grown to log-phase (OD = 0.3) in YPD medium on a shaker platform at 30°C by rotation at 225 RPM. Then, cells were fixed in 70% ethanol overnight at 4°C and then sonicated to separate cells. After RNase A (0.5 mg/mL) treatment for 2 h, the cells were stained with 25 μg/mL of propidium iodide overnight at 4°C. Finally, the stained cells were analyzed using BD Accuri C6 plus, and the data were analyzed in FlowJo v10.8.1.
MinION library preparation and sequencing
HMW gDNA were then used to prepare MinION sequencing library using the ONT Rapid Sequencing Kit (SQK-RAD004) following the manufacturer's instructions. Briefly, the sample mix was prepared with 7.5 µL template DNA (∼2 µg) and 2.5 µL fragmentation mix and incubated for 1 min at 30°C and then for 1 min at 80°C. One microliter of rapid adapter was added to the sample mix and incubated for 5 min at room temperature. Priming mix was prepared by adding 30 μL of flush tether and flush buffer. The priming mix was loaded into the flow cell via the priming port. Sequencing mix was prepared with DNA sample mix and was loaded to the flow cell via the SpotON sample port.
Adapter removal
Porechop v0.2.4 (Wick et al. 2017) was used for adapter identification and removal using default thresholds. In all, 179,725 reads had adapters trimmed from their start (15,472,707 bases removed), and 778 reads were split based on middle adapters. (Supplemental Fig. S2). A full list of commands and parameters is available in the Supplemental Materials.
Genome assembly, postassembly correction, and genome polishing
Draft collapsed-consensus assemblies were generated using Canu v2.2 (Koren et al. 2017), Flye v2.9 (Kolmogorov et al. 2019), Wtdbg2 v2.5 (Ruan and Li 2020), NECAT v0.0.1 (Chen et al. 2021), SMARTdenovo v1.0.0 (Liu et al. 2021), NextDenovo v2.5.0 (Hu et al. 2024), Raven v1.8.0 (Vaser and Šikić 2021), and Ra v0.2.1 (Vaser and Šikić 2019), with both uncorrected and Canu corrected and trimmed reads (Supplemental Table S1). These methods were executed on a general workstation-level computer (36 cores and 128 GB memory), demonstrating the feasibility of ONT-based de novo assembly for small genomes in modestly equipped laboratories.
Complete subgenome-aware de novo genome assembly
Given the diploid nature of our target organism, we aimed to generate a diploid-level representation of each chromosome. We employed long-read sequencing to facilitate the generation of full-length, phased haplotype de novo assemblies, using a suite of assembly tools, as detailed below and in the Supplemental Material.
Haplotype-aware de novo genome assembly
We experimented with haplotype-aware assembly methods such as Flye (with haplotype preservation enabled) (Kolmogorov et al. 2019), Shasta (Shafin et al. 2020), Phasebook (Luo et al. 2021), and CanuTrio (which organizes reads into haplotype-specific bins before assembly) (Koren et al. 2017). These approaches did not yield high-quality assemblies that were both contiguous and reflective of the expected genome size, leading to their exclusion from analysis.
Phasing-based diploid genome assembly
To tackle the complexities of S. bayanus CBS380's diploid genome, we undertook a phasing-based assembly strategy, leveraging the long reads generated from ONT's MinION platform. Prior to phasing, the purge_dups pipeline was used to remove haplotype duplication in the primary assemblies (Guan et al. 2020). To construct a phased diploid genome assembly, we first called variants using Claire (Zheng et al. 2022). The variant calls were processed through the WhatsHap pipeline, which exploits the connectivity between heterozygous variants within individual reads to generate phased haplotypes (Patterson et al. 2015). To generate a haplotype-specific genomic representation, we used BCFtools “consensus” (Danecek et al. 2021). This allowed us to extract the separate FASTA representations for each haplotype, effectively translating the phased information into a coherent, usable format for further analysis. BUSCO was used to assess the assembly's completion (Manni et al. 2021). A comprehensive list of commands and parameters, along with the phased variant calls, are accessible in the Supplemental Materials, offering a resource for future genetic and evolutionary studies.
Genome correction and polishing
For assembly correction and polishing, the raw ONT sequencing reads were split via the “whatshap split” subcommand to segregate the set of unmapped reads according to their haplotypes. This generated two distinct FASTQ files, each corresponding to one of the haplotypes identified within the sample. The assembled contigs were then passed to a series of correction and polishing steps to enhance their accuracy, utilizing Racon (v1.4.3) (Vaser et al. 2017) and Medaka (v1.9.1; https://github.com/nanoporetech/medaka) for error correction and sequence improvement. This correction process was executed separately for each haplotype, utilizing their respective reads. A total of four iterative rounds of correction were iteratively performed with Racon for each haplotype. This cycle involved mapping the haplotype-resolved reads to the assembled contigs using minimap2 (using the ONT-specific “-x map-ont” option), followed by Racon-based correction to refine assembly quality progressively. After completing the Racon correction cycles, a final round of polishing was conducted using Medaka. This step uses a neural network–based approach to correct consensus sequence errors, further enhancing the accuracy of the assembled haplotypes.
Genome annotation
We employed the GALBA pipeline to annotate protein-coding genes for the assembled nuclear genome (Brůna et al. 2023). Specifically, we used amino acid sequences from S. cerevisiae, S. uvarum, and S. eubayanus as inputs. These protein sequences were aligned to both subgenomes of S. bayanus using the Miniprot (Li 2023), followed by gene annotation using AUGUSTUS (Stanke et al. 2006). The output GTF files were processed using AGAT (https://github.com/NBISweden/AGAT) for format cleaning and conversion. The completeness of the gene annotation was evaluated using the BUSCO (version 5.5.0) (Simão et al. 2015), employing the saccharomycetes_odb10 database for assessment. For functional annotation of predicted genes, we utilized the web version of eggNOG-mapper (Cantalapiedra et al. 2021) to upload the S. bayanus protein files. All other parameters were retained as default settings. Visualization of the genome assembly, gene density, and synteny blocks were generated using Circos (Krzywinski et al. 2009).
Inferring the age of the S. bayanus CBS380 strain
To estimate the time since divergence of the S. bayanus CBS380 strain, we aligned genomic sequences from both subgenomes using the NUCmer tool from the MUMmer package (Kurtz et al. 2004) and identified SNPs in homozygous regions that resulted from LOH events. SNP density was calculated for these regions, and this density, along with an estimated mutation rate of 1.84 × 1010 per base pair per generation in S. cerevisiae (Fay and Benavides 2005) and an assumed generation time of ∼90 min for budding yeast (Khmelinskii et al. 2012; Kolmogorov et al. 2019), was used to calculate the time since divergence. This approach provided an estimate of the strain's age, aligning with the historical timeline of brewing practices (Langdon et al. 2019).
Ancestral inference of S. bayanus using a BLAST-based approach
To infer the ancestral parentage of the hybrid yeast, we conducted a comparative genomic analysis using a custom script that performs local BLAST (Camacho et al. 2009) homology searches (see Supplemental Code C1). The hybrid yeast genome was segmented into consecutive, nonoverlapping windows of 5000 bp, which were then individually compared against the genomes of the two parental strains using the BLAST algorithm (Supplemental Fig. S6). This approach allowed for the identification of the closest matching regions between the hybrid and each parent genome, based on the highest bitscore values obtained from the BLAST results. The bitscore, serving as a measure of sequence similarity, was selected as the primary criterion for parental inference. A score threshold of 100 bits was set to distinguish between significant and nonsignificant matches, thereby facilitating the identification of the most probable ancestral parent for each genomic segment of the hybrid yeast.
Inference of origin of S. bayanus genomic regions based on MinION sequencing reads
Given that sppIDer (Langdon et al. 2018) was designed for Illumina sequencing reads, which have fairly uniform read lengths, the algorithm for inference of parental genome contribution may not be suitable for long-read data, which tend to have much more variable read lengths. Thus, we revised the sppIDer pipeline to better suit MinION reads. Specifically, MinION reads were mapped to combination genomes of S. uvarum (CBS7001), S. eubayanus (FM1318), S. cerevisiae (S288c), S. mikatae (IFO1815), S. kudriavzevii (IFO1802), S. arboricola (ZP960), and S. paradoxus (CBS432) using minimap2 (Li 2018). Secondary and supplemental aligned reads and reads with a mapping quality less than three were excluded from analysis. The contribution of genomic content from a species was weighted their total read length, instead of read number.
To infer the genomic regions in CBS380 that were inherited from S. uvarum, we remapped MinION reads that were assigned to S. uvarum based on the approach described above. Each chromosome was divided into nonoverlapping 5 kb windows. A 5 kb window with a support of at least 30 reads (equivalent to 25% genome average read depth) was considered as originated from S. uvarum. A similar method was used to infer genomic region originated from S. eubayanus.
Inference of gene origin in S. bayanus based on P-distance of nucleotide sequences
To delineate the evolutionary origin of genes in S. bayanus, we identified all OGs for all protein-coding genes from S. bayanus, S. uvarum, S. eubayanus, S. cerevisiae, S. paradoxus, S. mikatae, and S. kudriavzevii using OrthoFinder (Emms and Kelly 2019). Multiple sequence alignment of nucleotide sequences for each OGs were generated by MAFFT (Katoh and Standley 2013). Pairwise sequence divergence in each OG was calculated using a custom Python script (Supplemental Code C2).
Comparative genomic analysis and evolutionary study of the MAL gene family in S. bayanus
To elucidate the evolutionary relationships among S. bayanus and closely related species, we analyzed coding sequence (CDS) data sets for S. cerevisiae, S. paradoxus, S. mikatae, S. kudriavzevii, S. arboricola, S. uvarum, S. eubayanus, and N. glabratus. Using OrthoFinder (Emms and Kelly 2019), we identified orthogroups to enable a comparative genomics study. Adopting the protocols from Brown et al. (2010) and Baker et al. (2015), we identified genes belonging to the maltose utilization (MAL) gene families. Sequence alignment was conducted with MAFFT using the L-INS-i strategy (Katoh and Standley 2013). To avoid bias owing to genome miasannotation and strain-specific gene loss, we also searched for MAL homologous genes from genomes of all non–S. bayanus strains. Because of the presence of a large number of significant hits, we only extracted sequences from top five hits to be included for phylogenetic analyses. Phylogenetic trees were generated using the maximum likelihood method using IQ-TREE 2 with 1000 bootstrap tests (Minh et al. 2020). The evolutionary distances were computed using the maximum composite likelihood method and are in the units of the number of base substitutions per site. These trees were visualized and refined with MEGA11 (Tamura et al. 2021).
Data access
Sequencing and genome assembly data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA741321. Assembly files, analysis scripts, and genomic and mitochondrial annotations are available at GitHub (https://github.com/BioHPC/Saccharomyces-bayanus) and as Supplemental Material.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
C.G., C.H., D.D., and T.A. were supported by the National Science Foundation (NSF) under grant no. 1564894, and Z.Lin was supported by NSF under grant no. 1951332. M.J.D. receives funding from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI123407. We thank Dr. Justin Fay for providing S. uvarum YJF1450 strain for ploidy analysis. We are truly grateful for all constructive comments provided by three anonymous reviewers that significantly improved this work.
Author contributions: T.A. and Z.Lin conceived the idea. T.A., Z.Lin, and M.J.D. supervised this study. Z.Lu isolated DNA, prepared libraries, and performed ONT sequencing. Y.Z. performed flow cytometry. C.G., J.C., C.H., and D.D. analyzed the data. All authors wrote the manuscript and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279364.124.
- Received March 17, 2024.
- Accepted September 11, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








