
DNA-m6A calling and integrated long-read epigenetic and genetic analysis with fibertools
- Anupama Jha1,8,
- Stephanie C. Bohaczuk2,8,
- Yizi Mao2,
- Jane Ranchalis2,
- Benjamin J. Mallory1,
- Alan T. Min3,
- Morgan O. Hamm1,
- Elliott Swanson1,
- Danilo Dubocanin4,
- Connor Finkbeiner1,
- Tony Li1,
- Dale Whittington5,
- William Stafford Noble1,6,
- Andrew B. Stergachis1,2,7 and
- Mitchell R. Vollger2
- 1Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA;
- 2Division of Medical Genetics, University of Washington, Seattle, Washington 98195, USA;
- 3Department of Statistics, University of Washington, Seattle, Washington 98195, USA;
- 4Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA;
- 5Department of Medicinal Chemistry, University of Washington, Seattle, Washington 98195, USA;
- 6Paul G. Allen School of Computer Science and Engineering, University of Washington, Seattle, Washington 98195, USA;
- 7Brotman Baty Institute for Precision Medicine, Seattle, Washington 98195, USA
-
↵8 These authors contributed equally to this work.
Abstract
Long-read DNA sequencing has recently emerged as a powerful tool for studying both genetic and epigenetic architectures at single-molecule and single-nucleotide resolution. Long-read epigenetic studies encompass both the direct identification of native cytosine methylation and the identification of exogenously placed DNA N6-methyladenine (DNA-m6A). However, detecting DNA-m6A modifications using single-molecule sequencing, as well as coprocessing single-molecule genetic and epigenetic architectures, is limited by computational demands and a lack of supporting tools. Here, we introduce fibertools, a state-of-the-art toolkit that features a semisupervised convolutional neural network for fast and accurate identification of m6A-marked bases using Pacific Biosciences (PacBio) single-molecule long-read sequencing, as well as the coprocessing of long-read genetic and epigenetic data produced using either the PacBio or Oxford Nanopore Technologies (ONT) sequencing platforms. We demonstrate accurate DNA-m6A identification (>90% precision and recall) along >20 kb long DNA molecules with an ∼1000-fold improvement in speed. In addition, we demonstrate that fibertools can readily integrate genetic and epigenetic data at single-molecule resolution, including the seamless conversion between molecular and reference coordinate systems, allowing for accurate genetic and epigenetic analyses of long-read data within structurally and somatically variable genomic regions.
Highly accurate long-read single-molecule DNA sequencing has revolutionized the comprehensive assembly of phased genetic architectures, enabling the first complete human genome assemblies (Wenger et al. 2019; Vollger et al. 2020; Nurk et al. 2022). In addition, single-molecule long-read DNA sequencing natively identifies endogenously modified DNA bases, such as m6A and 5-methylcytosine (5mC), permitting the co-analysis of both genetic and DNA methylation features at single-molecule resolution (Clark et al. 2012; Marks et al. 2012; Murray et al. 2012; Loman et al. 2015; https://github.com/PacificBiosciences/jasmine). Furthermore, using exogenous DNA methyltransferases to add DNA base modifications, such as in the context of single-molecule chromatin fiber sequencing (Abdulhay et al. 2020; Lee et al. 2020; Shipony et al. 2020; Stergachis et al. 2020; Cheetham et al. 2021; Altemose et al. 2022), permits the co-analysis of genetic, DNA methylation, and chromatin epigenetic features at single-molecule and single-nucleotide resolution.
Specifically, single-molecule chromatin fiber sequencing leverages nonspecific methyltransferases to selectively stencil chromatin protein occupancy patterns directly onto their underlying DNA molecules in the form of modified bases. Modified bases along the individual DNA molecules are then directly identified using PCR-free single-molecule sequencing. For example, during single-molecule, real-time (SMRT) sequencing, the identity of each base is determined by the fluorophore-labeled nucleotide that is incorporated as the polymerase replicates the base. In contrast, the modification status of each base is determined by signature changes in polymerase kinetics at and surrounding that base as it is replicated by the polymerase, such as elongation of the interpulse duration (IPD) owing to polymerase pausing at modified bases (Fig. 1A; Supplemental Fig. S1; Flusberg et al. 2010). Recently developed tools leverage these polymerase kinetic parameters to identify 5mC within specific sequence contexts (Tse et al. 2021; https://github.com/PacificBiosciences/jasmine), genomic positions with consistent m6A signal across multiple sequencing reads (Clark et al. 2012; Marks et al. 2012; Murray et al. 2012), total adenine methylation levels along very short sequencing reads (Kong et al. 2022), and m6A-modified bases at single-molecule resolution along only short (∼2 kb) DNA molecules (ipdSummary, SAMOSA-ChAAT) (Clark et al. 2012; Abdulhay et al. 2023). However, accurately identifying DNA-m6A along multikilobase reads is largely unsolved, as existing approaches either have poor sensitivity/specificity (Supplemental Figs. S2 and S3), require excessive compute and storage resources (Supplemental Fig. S4), and/or are reliant on input files (i.e., subreads) no longer available with modern sequencing chemistries.
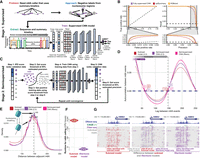
Accurate identification of m6A with supervised machine learning (ML) and refinement with semisupervised ML. (A) Methodology for generating training data and identifying m6A modifications using PacBio HiFi (for details, see Methods). (B) Receiver operating characteristic and precision-recall curves for the CNN (purple), XGBoost (orange), and ipdSummary (red) models. Dashed lines indicate the performance of a random classifier. (C) Methodology for semisupervised ML (for details, see Methods). (D) Autocorrelation between m6A calls made by the subread (red), semisupervised (purple), and Revio (pink) models. (E) Density of the distance between adjacent m6A on the same chromatin fiber (10,000 reads) for the same data sets and models as in D. (F) CPU hours used by fibertools (purple) and subread-based GMM model (red) for individual SMRT cells. fibertools was run with GPU acceleration (NVIDIA A40), which is unavailable for the GMM model. (G) Visualization of m6A calls in the HMBS locus that are unique to fibertools (purple), unique to the subread GMM model (red), or shared by both (gray). Reads are sorted by the number of CCS passes (low to high). DNase-seq (ENCODE July 2012 Freeze) and CAGE signals are shown above.
Furthermore, although extensive tooling exists for processing short-read epigenetic data relative to reference coordinates (BEDOPS, BEDTools, etc.) (Neph et al. 2012; Quinlan 2014), comparable tools for processing long-read epigenetic data are limited in their ability to leverage the rich epigenetic and genetic data embedded within long-read sequencing data (Razaghi et al. 2022). Specifically, tools for processing long-read epigenetic data need to operate in four dimensions: (1) they must process multiple types of genetic and epigenetic information present on a single read (e.g., DNA base, mCpG, DNA-m6A, inferred epigenetic marks); (2) they must capture this information across multiple reads mapping to a given reference position; (3) they must capture how this information co-occurs along each read in both reference and molecular coordinates (the original positions from the sequenced read, without adjustments for alignment to the reference sequence) in order to accurately display and detect the impact of structural or somatic variation on epigenetic marks; and (4) they must capture all of this information across the various haplotypes mapping to the same position within a reference.
Here, we introduce a semisupervised machine learning approach for accurately identifying DNA-m6A in Pacific Biosciences (PacBio) sequencing along multikilobase reads that permits the accurate learning of modified DNA bases from noisy training data, a common occurrence with single-molecule sequencing data owing to inherent biological heterogeneity in DNA methylation status between individual DNA molecules. Furthermore, we introduce a comprehensive toolkit for coprocessing long-read genetic and epigenetic data designed for use across sequencing platforms.
Results
Building an accurate tool for DNA-m6A identification requires a training data set of multikilobase reads with both methylated and unmethylated adenines across diverse sequence contexts (i.e., all possible 7-mers containing a central adenine) and methylation density contexts (i.e., isolated or clustered DNA-m6As). Because creating such a data set is not achievable using synthetic DNA or fully methylated and unmethylated samples, we leveraged DNA from single-molecule chromatin fiber sequencing reactions (i.e., Fiber-seq) as the basis for training. Specifically, Fiber-seq uses nonspecific m6A-MTases to selectively mark sites of protein occupancy along individual DNA molecules via m6A-marked bases. Because protein occupancy is highly heterogeneous across chromatinized DNA (Supplemental Fig. S5), each DNA molecule contains methylated adenines within diverse sequence and methylation density contexts (Supplemental Fig. S6). Furthermore, we can employ chromatin features, such as nucleosome occupancy, to bolster training and validation, making Fiber-seq well suited for training a general-purpose DNA-m6A caller given labeled data.
To generate initial positive and negative labels, we used a previously published DNA-m6A caller (referred to here as the “subread model”) (Dubocanin et al. 2022). The subread model improves upon ipdSummary by using ipdSummary's IPD normalization for sequence context (ipdRatios), followed by a Gaussian mixture model (GMM) to identify adenines with ipdRatios that significantly deviate from the expected distribution of unmethylated adenines (i.e., m6A-modified bases) (Supplemental Fig. S7; Dubocanin et al. 2022). These calls are then used to identify m6A-modified bases (positive labels). Negative labels are drawn from regions with extended stretches devoid of m6A corresponding to inferred nucleosome-occluded regions (Methods) (Fig. 1A).
All existing DNA-m6A callers (i.e., ipdSummary, SAMOSA-ChAAT, and the “subread model”) require subreads. However, modern SMRT sequencing does not output subreads and only produces summary kinetic information, making these existing tools largely obsolete. Consequently, we designed fibertools using a two-staged training approach. In stage 1 of training (Fig. 1A,B), we used a fully supervised training regime to validate that m6A calls can be generated using only summary kinetics, bypassing the requirement of all other PacBio m6A callers for individual subread kinetics. Using the data set described above, we independently trained two machine-learning models, XGBoost (Chen and Guestrin 2016) and a fully supervised convolutional neural network (fully supervised CNN), and evaluated their performance on a held-out data set from a separate sequencing experiment (Methods) (Fig. 1A,B). Both the fully supervised CNN and XGBoost models maintained high precision and recall, with an average precision >97% (Supplemental Table S1). As a comparison, we benchmarked the models against ipdSummary (we excluded SAMOSA-ChAAT from benchmarking as it was only optimized for a single deprecated polymerase chemistry). Both our CNN and XGBoost models outperformed ipdSummary in terms of both area under the precision-recall curve (AUPRC) and area under the receiver operating characteristic (AUROC). At a 95% precision threshold, the fully supervised CNN model has a recall of 89.6%, whereas the recall for ipdSummary is only 47.4%. Thus, the fully supervised CNN nearly doubles the number of m6A identifications over ipdSummary at this precision threshold. Notably, the fully supervised CNN and XGBoost models are ∼1000× faster than the subread model used to generate the training data set. Overall, the CNN model had the best performance, and we used it as the basis for stage 2 of training as well as subsequent improvements and validation.
In stage 2 of training, we sought to polish the architecture of our supervised CNN beyond the capabilities and accuracy limitations of existing tools and training data sets using a semisupervised training regime inspired by well-established methods in the field of proteomics (Käll et al. 2007; Fondrie and Noble 2021). To do this, we trained a new semisupervised model initiated with the architecture weights of our stage 1 model to overcome the imperfect training labels derived from existing models. The resulting semisupervised model (referred to onward as “fibertools”) allows for the possibility that the positive labels in the training data set are incorrect (Methods) (Fig. 1C). Using evaluation metrics (e.g., AUPRC/AUROC) based on training/test labels in semisupervised learning violates the inherent assumption that the labels may be incorrect; therefore, to evaluate this model, we established a series of biological validations to test the performance of the semisupervised training.
First, we assessed the accuracy of fibertools for identifying nucleosome footprints along Fiber-seq data. Using single-molecule m6A calls with a predicted precision of >95% (Methods), we performed an autocorrelation analysis. Compared with the subread and fully supervised models, fibertools more accurately recapitulated the exact length of nucleosomes (147 bp) (Fig. 1D; Luger et al. 1997) and showed an overall higher amplitude autocorrelation, consistent with higher-quality identification of m6A with nucleosome patterning characteristic of human chromatin. In addition, comparing the distance between adjacent m6A methylation marks in human Fiber-seq data demonstrated clear oscillatory patterns suggestive of nucleosome breathing (Polach and Widom 1995; Anderson and Widom 2000; Hall et al. 2009), further indicative of high-quality m6A identification (Fig. 1E,G).
Second, we evaluated the false-positive rate (FPR) of fibertools using whole-genome-amplified (WGA) DNA that lacked m6A (Fig. 2A). Our findings indicated an FPR of 0.23% at the model-predicted precision level of >95% (Fig. 2B; Supplemental Fig. S8). Notably, given this FPR, fibertools is not suited for identifying nonspecific genomic m6A events within species with low-level endogenous m6A (Kong et al. 2022; Debo et al. 2023).
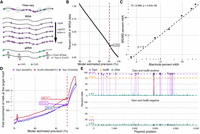
Biological validation of the semisupervised m6A caller. (A) Description of biological samples used for validation of fibertools. (B) Percentage of methylated adenines called by fibertools relative to all adenines in a whole-genome-amplified (WGA) negative control as a function of the estimated precision reported by fibertools. This serves as an estimate of the false-positive rate. The red line marks the default threshold used by fibertools. (C) Percentage of m6A as determined by UHPLC–MS/MS (y-axis) and fibertools (x-axis) at the default precision level for WGA samples with varying levels of m6ATP spiked-in. The text (upper left) indicates the value of the Pearson correlation coefficient and the P-value from a two-sided t-test without adjustment for multiple comparisons. (D) Enrichment of m6A calls within targeted motifs of three motif-specific methyltransferases (Dam [purple], EcoRI [red], and TaqI [blue]) as a function of fibertools estimated precision. (E) Methylation percentage at recognition sites for Dam (purple), HsdM (orange), and other sites (green) among all sequencing reads of a plasmid grown in a dam+/hsdM+ Escherichia coli strain (top) compared with a dam−/hsdM− negative control (bottom). Dotted lines show the average across each category.
Third, we evaluated the ability of fibertools to accurately quantify the total amount of m6A within a sample. Specifically, we spiked varying levels of m6ATP into a WGA reaction (Fig. 2A) and employed ultra-high-performance liquid chromatography tandem mass spectrometry (UHPLC–MS/MS) to determine the percentage of methylated adenines with respect to all adenines (Methods). We then sequenced these samples, applied fibertools, and found a strong correlation (Pearson = 0.998, P-value = 5.2 × 10−6) between our method and mass spectrometry (Fig. 2C).
Fourth, because the above validations demonstrate that m6A calls from fibertools recapitulate bulk chromatin features and total m6A content measured by UHPLC–MS/MS, we next evaluated the precision of fibertools for identifying isolated m6A events, which is relevant for m6A calls within small internucleosomal regions. Using fibertools to predict m6A on genomic DNA treated with motif-specific methyltransferases, we found that m6A calls were enriched by 415-fold, 586-fold, and 407-fold in motifs specific to Dam, EcoRI, and TaqI, respectively (Fig. 2D), consistent with precise m6A calls. The ability to call single m6A events is biologically relevant for Fiber-seq as 10%–25% of all internucleosomal linker regions (i.e., methyltransferase-sensitive patches [MSPs]) within a Fiber-seq data set contain only a single m6A separating adjacent nucleosomes (Supplemental Fig. S9).
Fifth, the precision of fibertools in identifying single m6A events indicated it might be useful for identifying endogenously m6A-modified bases within bacteria, a context that also provides a true-positive validation set as ∼100% m6A methylation can be expected within methyltransferase-specific motifs. Notably, DNA isolated from bacteria expressing both the Dam and HsdM methyltransferases exhibited m6A at 94.1% of the target Dam sites for these methyltransferases, indicating a false-negative rate of <6% in this sequence context (Fig. 2E). In contrast, DNA from bacteria lacking these methyltransferases (Anton et al. 2015) exhibited m6A at <1% of adenines, consistent with our prior FPR estimate.
Sixth, we evaluated the accuracy of fibertools for quantifying m6A-marked chromatin architectures along multikilobase reads. Current SMRT cell chemistries target sequencing reads ∼20 kb in length, yet traditional subread-based m6A models were designed for reads of only ∼2 kb in length. We demonstrate that in comparison to existing subread-based models, fibertools substantially reduces false-negative methylation calls along increasingly longer reads (Fig. 3; Supplemental Fig. S10), enabling the accurate quantification of m6A-marked chromatin architectures along reads >25 kb in length (Fig. 3A,B). The increased false-negative rate of previous callers is owing to a reliance on many subread passes, which becomes less likely with increasing insert size. This limitation is avoided in the fibertools model, which can call m6A on HiFi reads with any number of subread passes.
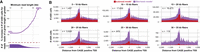
Increased m6A calling on long reads (>20 kb) via fibertools. (A) Percentage increase in fibertools m6A calls over the GMM model as a function of the minimum read length of the underlying sequencing data. The histogram below shows how many reads were used to calculate each percentage increase. (B) Comparison of fibertools and the subread model for m6A calling over CAGE-positive TSS in K562 cells across the genome, separated by read length. Reads are matched between fibertools and the subread model, and the number of Fiber-seq reads used in the calculation of each size range (n) is indicated.
Collectively, these biological validations provide strong evidence that fibertools is highly accurate and specific in identifying m6A events using PacBio HiFi data. Importantly, the semisupervised training design of fibertools enables it to readily adapt to new sequencing chemistries, which often contain updated polymerases that may differ in their kinetic values (Supplemental Fig. S1). For example, we used calls from the model for the PacBio Sequel II 2.2 chemistry as the initialization point for training a semisupervised model for the PacBio Revio chemistry, demonstrating that this new Revio model is similarly highly accurate in identifying m6A events (Fig. 1D,E).
Having established that fibertools can identify highly accurate m6A events using PacBio HiFi data, we next sought to extend fibertools to enable the simultaneous processing of genetic, cytosine methylation, and adenine methylation data. To accomplish this, we designed fibertools to integrate m6A calls directly into the BAM format using the MM and ML tags (Supplemental Fig. S11). Next, we optimized fibertools using a compiled language, which we provide as a single binary (ft) accessible through bioconda (package “fibertools-rs”). Of note, fibertools can process individual Revio SMRT cells in 15–24 CPU hours and Sequel II SMRT cells in 5–8 CPU hours, an ∼1000-fold increase in speed compared with the previous pipeline when using GPU acceleration (>150-fold increase without GPU) (Fig. 1F; Supplemental Table S2).
We next extended the utility of fibertools to perform fundamental operations necessary for processing single-molecule epigenetic and genetic data produced using either PacBio and ONT sequencing platforms (i.e., fibertools add-nucleosome, fibertools extract, and fibertools center).
Fibertools add-nucleosome enables the identification of stretches of unmethylated adenine bases, which are stored directly in the BAM file using custom flags. It processes 10 million ∼20 kb reads in just 4.6 CPU hours. Importantly, fibertools add-nucleosome works seamlessly with Fiber-seq data sequenced using either a PacBio or ONT instrument. For example, the application of fibertools add-nucleosome to a Fiber-seq library sequenced on an Oxford Nanopore Technologies (ONT) R10.4 flow cell and basecalled using Dorado v0.4.2 (https://github.com/nanoporetech/dorado) enabled the robust identification of clear nucleosomal patterns (Fig. 4A,B), despite the substantial decrease in m6A and DNA base identification accuracy with ONT sequencing (Fig. 4C; Supplemental Fig. S3).

fibertools nucleosome calling with PacBio and ONT Fiber-seq. (A) Data processing pipelines for nucleosome calling with fibertools. (B) Density of nucleosome lengths called by fibertools for PacBio (pink) and ONT (blue) Fiber-seq (n = 500,000 nucleosomes). (C) Visualization of the NAPA and HMBS representative loci for PacBio (top) and ONT (bottom) Fiber-seq. m6A calls from fibertools (PacBio) or Dorado (ONT) are represented by vertical purple dashes, along with nucleosome (gray) and MTase-sensitive patch (MSP; orange) calls from fibertools.
Fibertools extract enables multithreaded conversion of genetic and epigenetic BAM features into plain text formats regardless of upstream tooling with coordinates in either reference or molecular space. For example, nucleosomes, m6A, and 5mC can all be extracted into BED12 format to visualize with the UCSC Genome Browser. Alternatively, all of these features and more can be extracted into a unified table for custom downstream analysis or visualization with both m6A and 5mC base modifications across multiple technologies (Fig. 5).
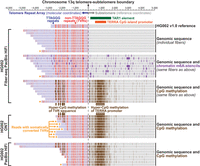
Organization of the HG002 telomere. (Top) HG002 PacBio Fiber-seq with genetic variants, m6A methylation (purple), and CpG-methylation (brown) overlaid for fibers overlapping the maternal telomere of Chromosome 13q (telomeric boundaries were determined with seqtk v1.3). Telomeric sequences (blue), telomeric variants (red), and nontelomeric sequences (gray) are highlighted to show telomeric genetic variation. ONT (Middle) and PacBio (bottom) standard sequencing of HG002 telomeres (Zook et al. 2020) with CpG-methylation overlaid on chromatin architecture.
Fibertools center enables the processing of single-molecule genetic and epigenetic data (i.e., DNA base, mCpG, DNA-m6A, inferred epigenetic marks, etc.) relative to a set of reference genomic coordinates while maintaining how these features co-occur along each read using both reference and molecular coordinate systems, addressing a need that is unique to long-read epigenetic studies. To demonstrate the utility of fibertools center, we applied it to address two fundamental biological questions that require the integration of long-read epigenetic and genetic data: (1) the relationship between somatic DNA variability and overlying altered epigenetic architecture along individual DNA molecules (Fig. 5) and (2) the co-occupancy of transcription factor (TF) binding elements along individual DNA molecules (Fig. 6).
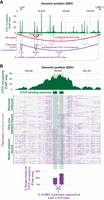
CTCF co-occupancy along the EBV genome. (A) CTCF ChIP-seq (green), significant (red) or insignificant (gray) Hi-C loops (Morgan et al. 2022), and significant CTCF site co-occupancy by Fiber-seq (purple) along the EBV genome. The significance of CTCF site co-occupancy was determined by comparing the expected number of co-occupied fibers to the observed number using Fisher's exact test (for exact counts and P-values, see Supplemental Table S6). (B) Zoom-in of the indicated CTCF peak, which contains two CTCF binding elements. Single-molecule occupancy and co-occupancy from Fiber-seq are shown below.
First, we applied fibertools center to resolve the relationship between somatic DNA variability and overlying altered epigenetic architecture along telomeric and subtelomeric regions using Fiber-seq PacBio HiFi, standard PacBio HiFi, and standard ONT sequencing data. Telomere repeat arrays exhibit substantial per-molecule somatic alterations in both their length and sequence content (Dubocanin et al. 2022) and are known to be transcribed into telomeric repeat-containing RNA (TERRA) via a CpG island promoter located within a TAR1 repeat positioned adjacent to the majority of telomere repeats (Azzalin et al. 2007). The substantial molecule-to-molecule heterogeneity in the DNA content of individual telomere repeats originating from the same chromosome arm requires the use of a molecular coordinate system for their appropriate analysis (i.e., the original positions from the sequenced read, without adjustments for alignment to the reference sequence). Application of fibertools center to Fiber-seq data from HG002 cells aligned to the HG002 reference genome enabled the resolution of both genetic and epigenetic architectures of subtelomeric regions in reference coordinates and telomere repeat arrays in molecular coordinates by using the telomere–subtelomere boundary as the centering point. Importantly, by using molecular coordinates to display the telomere repeat array, we were able to identify multiple non-TTAGGG telomere variant repeats (TVRs) (Allshire et al. 1989; Baird et al. 1995) absent from the HG002 reference sequence (Supplemental Fig. S12). Overall, this analysis exposed that the subtelomeric TAR1 element was hyper-CpG-methylated and lacked chromatin accessibility within HG002 cells (Fig. 5), in contrast to CHM13 cells (Dubocanin et al. 2022). Notably, this region of hyper-CpG-methylation extended into TVRs harboring CpG dinucleotides within the telomere repeat array itself (Allshire et al. 1989; Baird et al. 1995), which we validated by applying fibertools to additional genomic ONT and PacBio HiFi sequencing data from HG002 (Zook et al. 2020). TVRs can undergo somatic conversion back into TTAGGG repeats, likely via somatic shortening and subsequent telomerase-mediated elongation (Dubocanin et al. 2022). Integrating the genetic and epigenetic data along individual telomere fibers identified numerous fibers that had undergone such somatic conversions, resulting in the loss of CpG-methylation within that region of the telomere repeat array. However, hyper-CpG-methylation of the TAR1 element and the rest of the telomere repeat array was maintained on these fibers, suggesting that CpG-methylation of TVRs may be a bystander effect of TAR1 hyper-CpG-methylation within HG002 cells.
Second, we applied Fiber-seq and fibertools center to resolve the role of CTCF co-occupancy in guiding higher-order chromatin architectures along the ∼175 kbp Epstein–Barr virus (EBV) genome. CTCF elements within the EBV OriP and LMP loci are known to form a cohesin-dependent loop important for maintaining viral latent cycle gene expression (Arvey et al. 2012; Morgan et al. 2022). Using fibertools, CTCF footprints were resolved on individual DNA molecules (Yin et al. 2017), and we determined that the CTCF ChIP-seq peak within the EBV LMP locus is predominantly mediated by CTCF co-occupying two immediately adjacent CTCF binding elements along the same DNA molecule (Fig. 6). In addition, we observed that EBV nucleoids bound by CTCF within the LMP locus are also preferentially cobound by a single CTCF element within the EBV OriP locus, which is located ∼12 kbp away (Fig. 6A). However, only 13.4% of EBV nucleoids are cobound by CTCF at both the OriP and LMP loci (Fig. 6B), setting an upper limit on the proportion of EBV nucleoids that are directly structured by CTCF-mediated three-dimensional looping between these two loci in a stable manner. Notably, dynamic CTCF-mediated looping has been observed along the nuclear genome (Gabriele et al. 2022), and it is possible that these CTCF cobound EBV nucleoids may represent similarly dynamic CTCF-mediated looping along the EBV genome.
Discussion
In summary, fibertools provides foundational tooling for processing long-read genetic and epigenetic data. Specifically, fibertools enables highly accurate single-molecule DNA-m6A identification using PacBio sequencing with a 1000-fold improvement in speed (Fig. 1F), enabling highly accurate single-molecule chromatin fiber sequencing and endogenous bacterial adenine methylation identification (Figs. 1D, 2E; Supplemental Fig. S13). In addition, fibertools enables the synchronous processing of multiple types of genetic and epigenetic information present within BAM files produced using either the PacBio or ONT sequencing platforms at single-molecule and single-nucleotide resolution (Fig. 4). Furthermore, fibertools enables the seamless conversion between molecular and reference coordinate systems, allowing for accurate genetic and epigenetic analyses of long-read data within structurally and somatically variable genomic regions (Fig. 5). Finally, fibertools is written in a compiled language and is available as a single binary accessible through bioconda (package “fibertools-rs”) and PyPI (package “pyft”), enabling its broad utility within existing computational pipelines for processing long-read data. Recent advances in the cost, accuracy, and throughput of long-read sequencing have enabled the broader adoption of this technology across the genetics community, and we anticipate that fibertools will serve as an integral tool for processing long-read sequencing data.
Finally, the semisupervised training structure that fibertools introduces for identifying m6A-marked bases will likely prove useful for the accurate identification of other base modifications using long-read sequencing (Methods) (Fig. 1C). Specifically, this approach accounts for imperfect training data, which is a core feature of long-read sequencing data owing to the inherent biological per-molecule heterogeneity in the distribution of modified bases. As such, we anticipate that this approach can both improve the accuracy of identifying core base modifications in humans, as well as novel base modifications in bacteria. Furthermore, we demonstrate that models trained using a semisupervised structure can readily adapt to updated sequencing chemistries, making this training approach highly advantageous for long-read sequencing chemistries, which are undergoing frequent iterative development cycles (Fig. 1D,E).
In summary, we present fibertools as a toolkit to facilitate the identification and analysis of long-read genetic and epigenetic sequencing data and establish semisupervised machine learning as an adaptable tool for accurately identifying base modifications using long-read sequencing.
Methods
Initial m6A calling with ipdSummary followed by filtering with a GMM
To initially identify m6A, we use a previously published protocol (Dubocanin et al. 2022) with the following details and modifications. Raw PacBio subread BAM files were converted into circular consensus sequence (CCS) reads using pbccs (v6.0.0; https://ccs.how/) with average kinetics information included. Then subreads were aligned to their respective CCS reads using actc (https://github.com/PacificBiosciences/actc). The resulting alignments were passed to ipdSummary (v3.0; https://github.com/PacificBiosciences/kineticsTools/) to identify positions with m6A modifications with the CCS-specific extracted genomic sequence as the reference and these additional flags: ‐‐pvalue 0.001 ‐‐identify m6A. For each read, we then trained a two-component GMM from the ipdRatios generated by ipdSummary for all adenine bases within the read (push_m6a_to_bam.py). Adenine bases were then classified as m6A modified if the probability that the ipdRatio came from the larger distribution was greater than or equal to 0.99999999. All the code to repeat these steps is available as a single Snakemake (Köster and Rahmann 2012) pipeline on GitHub (https://github.com/StergachisLab/fiberseq-smk) and in the Supplemental Code. K562 and CHM1 PacBio data generated as part of a previous study are available at the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE226394.
Hidden Markov model nucleosome calling
To identify nucleosomes initially, we use a previously published protocol (Dubocanin et al. 2022) with the following details and modifications. We used m6A calls to train a two-state (nucleosome, nonnucleosome) hidden Markov model (HMM) with a standardized post hoc correction. The input data for the HMM skipped all G/C bases and considered only the A/T sequence so that GC-rich regions without adenine would not falsely be called nucleosomes. The HMM was then trained using the Baum–Welch algorithm (Baum et al. 1970) using a subsample of 5000 CCS reads per SMRT cell, and the trained model was subsequently applied across all fibers from that SMRT cell using the Python package pomegranate (Schreiber 2017). In tandem with the HMM delineation of nucleosomes, we also applied a simplified approach that looked for unmethylated stretches >85 bp in length (irrespective of A/T content). We used these “simple” calls to refine our HMM nucleosome calls by splitting large HMM nucleosomes that contained multiple “simple” calls. We also refined the terminal boundaries of each nucleosome by bookending it to the nearest m6A call within 10 bp, if present. Nucleosome calling is available as part of a Snakemake pipeline (https://github.com/StergachisLab/fiberseq-smk) and encodes the resulting calls in the custom BAM flags nucleosome start (ns) and nucleosome length (nl), which can be extracted using fibertools-rs.
Generating positive and negative labels for training and validation data
To generate positive labels, we used the previously existing calls made using the GMM-filtered calls from ipdSummary in our Snakemake pipeline. Negative labels were drawn from nucleosome regions as defined by the HMM because we had increased confidence that these regions should be inaccessible owing to the presence of a nucleosome. For each SMRT cell, we selected about 350,000 CCS reads and randomly sampled 5% of the available positive and negative m6A positions within each read to reduce the number of adjacent and, therefore, nonindependent calls in our training data. This resulted in a data set of about 100 million negative labels and about 8 million positive labels for each SMRT cell. We note that by selecting negative labels from within nucleosomes, we allow for the possibility for our models to outperform ipdSummary and the GMM correction even though we draw positive labels from these tools. For each label in our data set, we included a 15 bp window centered around the adenine encoding the sequencing information with one-hot encoding and the CCS kinetics information across the same 15 bases for pulse width and interpulse duration, resulting in a 6 × 15 matrix for each labeled position. The 15 bp window and inclusion of pulse width and interpulse duration were selected based on the ablation study of the supervised model (Supplemental Fig. S14). Code to generate training data from CCS reads is available on GitHub (https://github.com/fiberseq/train-m6A-calling) and in the Supplemental Code.
Training, validation, and testing data sets for machine learning
For the 2.2 PacBio sequencing chemistry, we established three separate data sets, each from a different sequencing run, for developing our models. The three data sets were used for training, validation, and a completely held-out testing data set for determining final accuracies. Data for v2.2 chemistry were generated from previously described samples (K562, CHM1) treated with 200 units of Hia5 for 10 min at 25°C (Dubocanin et al. 2022). Data for v3.2 chemistry were generated from a K562 Fiber-seq sample (described below), and Revio data were provided by PacBio.
Architectures and training of machine-learning models (XGBoost, CNN, and semisupervised CNN)
We trained an XGBoost model with a binary logistic objective function using our training and validation data sets. We selected the following hyperparameters using threefold cross-validation: learning rate of 1 (gamma), maximum tree depth of 8, minimum child weight of 100, and 150 estimators. The code to repeat this training is available on GitHub (https://github.com/fiberseq/train-m6A-calling) and in the Supplemental Code.
Convolutional neural network
We trained a CNN to predict m6A in Fiber-seq HiFi reads. Input to the CNN model is the 6 × 15 matrix described above. The model has three convolutional layers with 30, 10, and five filters of size 5, 5, and 3, respectively. A dense layer of size 25 × 5 and an output layer of size 5 × 2 follow the convolutional layers. All internal layers have ReLU activation, and the output layer has softmax activation. The output layer has two classes, one for m6A and one for unmethylated adenines. Each class generates scores between 0 and 1 for each input matrix, where scores close to 1 denote high confidence that the input belongs to that class. For example, unmethylated adenines score close to 0 for the m6A class, and methylated adenines score close to 1. The CNN optimizes a binary cross-entropy loss function using the Adam optimizer (Kingma and Ba 2015). We trained the model iteratively for 30 epochs, in which the training data were input in random batches of 32 in each epoch. For the size of training and validation data sets from different Fiber-seq chemistries for supervised training, see Supplemental Table S3. The code to repeat this training is available on GitHub (https://github.com/fiberseq/train-m6A-calling) and in the Supplemental Code.
Semisupervised convolutional neural network
We derive m6A labels from GMM-filtered ipdRatios from ipdSummary. These labels can contain false positives. The lack of clean m6A labels makes the supervised training approach less suitable because it assumes that accurate labels are available. Therefore, we developed a semisupervised approach, which assumes that our m6A class has a mixed population of true and false positives and that our non-m6A class is a clean set. Our training approach is derived from the Percolator and Mokapot proteomic tools for identifying peptides from tandem mass spectrometry data (Käll et al. 2007; Fondrie and Noble 2021). This approach yields a classifier with m6A calls at a target precision. The semisupervised algorithm is outlined in the Supplemental Methods and Supplemental Algorithm 1. First, we split our data set into training and validation sets stratified by class labels (for the size of training and validation data sets from different Fiber-seq experiment chemistries for semisupervised training, see Supplemental Table S3). Then our method proceeds in two phases. In the first phase, we use the IPD score of the central base as a classifier and generate an m6A classification score for all examples in the validation set. The classification score ranks the validation examples, and precision is computed at every score threshold. At the end of this phase, we select a score threshold to achieve the target precision. In this work, we use a target precision of 95%. The second phase is iterative, and each iteration consists of three steps. The first step is selecting a high-confidence m6A training set using the current score threshold. The second step consists of training a CNN model on these training data. In the final step, the validation data are rescored using the trained CNN model from the second step, and a new score threshold is generated at 95% precision with the rescored validation data. In the case of a successful second phase training, the number of positives in the validation data identified at target precision increases with every iteration and plateaus when most m6A examples in the validation data have been identified. We define two conditions for convergence, both of which must be satisfied. First, >70% of putative m6A calls from the validation set have been identified. Second, the number of additional m6A calls in a new iteration is <1% of the total putative m6A calls. In practice, it took 12, 11, and three iterations of phase two training to converge 2.2, 3.2, and Revio chemistry Fiber-seq experiments, respectively (Supplemental Fig. S15). The code to repeat this training is available on GitHub (https://github.com/fiberseq/train-m6A-calling) and in the Supplemental Code.
Encoding and selecting precision levels for m6A calling
Using the approach outlined in the semisupervised method, we calculated the empirical precision using the validation data for every score output from the CNN model (see Supplemental Methods; for details, see Supplemental Table S4). These precisions are then multiplied by 256 and rounded into an eight-bit integer following the BAM specification for the ML tag (Li et al. 2009; https://samtools.github.io/hts-specs/SAMtags.pdf). We then chose the first value (244) with a precision >95% (244/256) as the threshold for calling positive m6A events.
Basecalling for Oxford Nanopore
HG002 DNA prepared with the standard fiber-seq protocol (below) was sequenced on a PromethION flow cell (R 10.4.1) with a 5 kHz sampling rate. Alignment to the HG002 or GRCh38 genome and DNA base and base modification calling were performed with Dorado v.0.4.2 with the basecalling model dna_r10.4.1_e8.2_400bps_sup@v4.2.0, 5mC model V2, and 6mA model V3 (https://github.com/nanoporetech/dorado).
Preparation of calcium-competent ER2796 E. coli
A 10 mL culture of the ER2796 Escherichia coli strain (obtained from NEB) was grown overnight without antibiotics. One milliliter of the overnight culture was added to 99 mL of fresh LB media without antibiotics and incubated with shaking for 3–4 h at 200 rpm and 37°C until OD reached 0.4. The culture was separated into two 50 mL falcon tubes and placed on ice for 20 min before being pelleted by centrifugation for 10 min at 4°C and 4000 rpm. The supernatant was discarded, and the cell pellets were resuspended with 20 mL of ice-cold 0.1 M CaCl2 and incubated on ice for 30 min. The cells were then pelleted again by centrifugation, and the supernatant was discarded. The pellets were then combined by resuspending in 5 mL of ice-cold 0.1 M CaCl2 with 15% glycerol. Cells were aliquoted in 50 μL aliquots, frozen in liquid N2, and stored at −80°C.
Plasmid DNA preparation
A pCS2+ plasmid containing FLAG-tagged mgfp5 cloned into the EcoRI/XhoI sites (a gift from Lea Starita) was transformed into two strains of competent E. coli, ER2796 (from NEB, see section “Preparation of calcium-competent ER2796 E. coli”) and NEB 5-alpha (NEB C2987I), using a standard heat shock method. Fifty microliters of the chemically competent cells were thawed on ice and mixed with 50 ng of plasmid DNA. The mixture was placed on ice for 30 min before being heat-shocked for 30 sec at 42°C. After heat shock, the cells were immediately placed back on ice for 5 min. Following incubation, 950 µL of room temperature SOC media (NEB B9020S) was added, and the cells were allowed to outgrow for 1 h in the absence of selection. Fifty microliters of the outgrown cells were diluted in 5 mL of selective media and grown overnight with shaking at 220 rpm and 37°C. Specifically, the NEB 5-alpha cells were grown in LB media with 100 μg/mL ampicillin, whereas the ER2796 cells were grown in LB media with 50 μg/mL kanamycin + 100 μg/mL ampicillin. The following day, plasmid DNA was extracted from the bacterial cells using the Monarch plasmid miniprep kit (NEB T1010L) following the manufacturer's protocol. The elution of plasmid DNA was done with sterile water, and the concentration was measured using the Qubit 1X dsDNA HS assay kit (Invitrogen Q33231).
Cell culture
The following cell lines/DNA samples were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research: GM12878 and GM24385 (HG002). K562 cells were a gift from the Stamatoyannopoulos laboratory. Cells were maintained in suspension in IMDM media supplemented with 10% FBS (HyClone SH30396.03IH25-40) and antibiotic (100 I.U/mL penicillin, 100 μg/mL streptomycin, Gibco 15140122) at 37°C and 5% CO2 in T-75 flasks. Cells were split 1:10 every 3–4 days.
gDNA preparation
Whole-genome DNA was extracted from K562 cells using HMW DNA extraction kit (Promega A2920). gDNA was used as a template for all WGA samples. All DNA samples were rehydrated with 10 mM Tris-HCl (pH 8; Thermo Fisher Scientific J22638.K2) or eluted with ultrapure water unless specified.
WGA preparation
WGA was performed with REPLI-G Mini kit (Qiagen 150023) according to the manufacturer's protocol. Zero, 10, 25, 64, 160, 400, or 1000 µM of N6-methyl-2-dATP (TriLink N-2025; m6dATP) final concentration was spiked into 50 µL of each WGA to generate samples with 0%, 2.56%, 8.46%, 14.45%, 33.42%, 52.27%, and 67.94% m6A samples, as calculated using mass spectrometry according to an 11-point standard quantified every time with the samples during the same run (see below). Samples were purified with ProNex size-selective chemistry (Promega NG2001) by adding a 1:1.3 ratio by volume of sample to magnetic beads. Following incubation for 15 min, the sample was placed on a magnetic rack for 3 min, washed twice with 80% ethanol, and eluted in 50 µL of PacBio elution buffer (PacBio 101-633-500). All m6dATP-spiked samples were purified three times with bead purification, and residual free m6dATP quantified by mass spectrometry of the 10 and 160 µM m6dATP samples treated with Quick CIP only was near background levels.
gDNA methylation with sequence-specific methyltransferases
Samples labeled with the sequence-specific adenine methyltransferases EcoRI (GAATTC; (NEB M0211S), Dam (GATC; NEB M0222S), or TaqI (TCGA; NEB M0219S) were generated as follows: 400 ng of purified gDNA was treated for 1 h with 40 U of EcoRI (37°C), 10 U of TaqI (65°C), or 8 U of Dam (37°C) in supplied buffer and in the presence of 160 μM SAM. All samples were purified once by ProNex size-selective chemistry as described above but with a 1.25:1 ratio by volume of sample to magnetic beads.
Fiber-seq
Nonspecific methyltransferase, Hia5, was purified, and the activity was quantified as previously described (Stergachis et al. 2020). Fiber-seq samples were prepared as previously described (Stergachis et al. 2020).
Library preparation
Samples were sheared in g-TUBEs (Covaris 520079) for four passes at 3200 RPM for 2–4 min in an Eppendorf 5424R centrifuge. Post-shear samples were quantified by Qubit dsDNA high-sensitivity assay (Qubit Q32851) following the manufacturer's protocol. Multiplexed library preparation was performed using the SMRTbell prep kit 3.0 (PacBio 102-141-700) and SMRTbell barcoded adapter plate 3.0 (bc2001-bc2019; PacBio 102-009-200) according to the manufacturer's instructions but with the following modifications: After barcoded adapter ligation, samples were incubated for 10 min at 65°C to heat-inactivate the ligase. Barcoded samples were pooled and purified with ProNex size-selective chemistry as described above, with a 1:1 ratio by volume of sample to magnetic beads. Following nuclease treatment, the library was purified first with a 1:3.1 ratio of sample to 35% v/v Ampure PB beads (PacBio 100-265-900)/PacBio elution buffer. A second purification was performed with a 1:1 ratio of sample to ProNex size-selective chemistry, as described. The sample was loaded onto a single Sequel II SMRT cell (v3.2 chemistry) and sequenced by the University of Washington PacBio Sequencing Services. The full composition and sample barcode IDs of the multiplexed library are listed in Supplemental Table S5. Plasmid DNA was prepared in a separate multiplexed library. Plasmids were linearized with KpnI prior to multiplexed library preparation.
Quantification of m6A/A by UHPLC-MS/MS
Samples for quantification were treated as previously described with minor modifications (Kong et al. 2022). In brief, 30–50 ng of DNA from each sample was mixed with 0.02 U phosphodiesterase I (Worthington LS003926), 1 U Benzonase (Millipore Sigma E1014), and 2 U quick CIP (NEB M0525S) in digestion buffer (10 mM Tris, 1 mM MgCl at pH 8 at RT) for 3 h at 37°C. Single nucleotides were separated from the enzymes by collecting the flow-through of a Nanosep centrifugal filter (MWCO 3 kDa; Pall OD003C33). The UHPLC-MS/MS analysis of adenosine and m6A was performed on an ACQUITY premier UPLC system coupled with XEVO-TQ-XS triple quadrupole mass spectrometer. UPLC was performed on a ZORBAX eclipse plus C18 column (2.1 × 50 mm I.D., 1.8 μm particle size; Agilent 959757-902) using a 10%–90% linear gradient of solvent B (0.1% acetic acid in 100% methanol) in solvent A (0.1% acetic acid in water) within 4 min and a flow rate of 0.3 mL/min. MS/MS analysis was operated in positive ionization mode with 3000 V capillary voltage as well at 150°C and 1000 L/H nitrogen drying gas. A multiple reaction monitoring (MRM) mode was adopted with the following m/z transition: 252.10 → 136.09 for dA (collision energy, 14 eV), and 266.2 → 150.2 for m6A (collision energy, 15 eV). MassLynX was used to quantify the data.
A calibration curve was generated with 11 mixtures containing different ratios of 2-deoxyadenosine (>99%, Fisher Scientific
AAJ6388606)(A) to N6-methyl-2-deoxyadenine (>99%, Fisher Scientific AAJ64961MD)(m6A). A new standard was measured and used for each run. The standard
was fit to a third-degree polynomial (Equation 1) with y as m6A percentage (%) and X as the quantified MS peak area of m6A over the sum of adenosine and m6A peak area: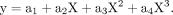 (1)
(1)
Software availability
Code for training (https://github.com/fiberseq/train-m6A-calling), evaluation (https://github.com/mrvollger/Fiber-m6A-figures-and-tables) and execution (https://github.com/fiberseq/fibertools-rs) of fibertools is publicly available on GitHub and as Supplemental Code.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA956114.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Sayeh Gorjifard for organizing the University of Washington Genome Sciences Hackathon, which initiated this work; Michelle Noyes for designing the fibertools logo art; and Tonia Brown for assistance in editing this manuscript. Cell lines obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research include GM12878 and GM24385. A.B.S. holds a career award for medical scientists from the Burroughs Wellcome Fund and is a Pew biomedical scholar. This study was supported by U.S. National Institutes of Health (NIH) grants 1DP5OD029630, UM1DA058220, and OT2OD002748 to A.B.S and R01-HG011466 to W.S.N. M.R.V. and S.C.B. were supported by a training grant (T32) from the NIH (2T32GM007454-46).
Author contributions: Conceptualization and design were by M.R.V., A.J., S.C.B., and A.B.S. Experimental design and execution were by S.C.B., Y.M., J.R., B.J.M., and A.B.S. Preliminary implementation was by M.R.V., A.J., S.C.B., A.T.M., M.O.H., E.S., C.F., and T.L. Final implementation was by M.R.V. and A.J. Supplemental material organization was by M.R.V., S.C.B., and A.J. Display items were by M.R.V., A.J., S.C.B., and A.B.S. Manuscript writing was by M.R.V., S.C.B., and A.B.S. with input from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279095.124.
- Received February 9, 2024.
- Accepted May 21, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








