
An optimized protocol for quality control of gene therapy vectors using nanopore direct RNA sequencing
- Kathleen Zeglinski1,
- Christian Montellese2,3,
- Matthew E. Ritchie1,
- Monther Alhamdoosh4,
- Cédric Vonarburg2,3,
- Rory Bowden1,
- Monika Jordi2,
- Quentin Gouil1,5,
- Florian Aeschimann2,3,5 and
- Arthur Hsu4,5
- 1Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria 3052, Australia;
- 2CSL Behring, Research, CH-3014 Bern, Switzerland;
- 3Swiss Institute for Translational Medicine, sitem-insel, 3010 Bern, Switzerland;
- 4Research Data Science Group, R&D, CSL, Parkville, Victoria 3000, Australia
Abstract
Despite recent advances made toward improving the efficacy of lentiviral gene therapies, a sizeable proportion of produced vector contains an incomplete and thus potentially nonfunctional RNA genome. This can undermine gene delivery by the lentivirus as well as increase manufacturing costs and must be improved to facilitate the widespread clinical implementation of lentiviral gene therapies. Here, we compare three long-read sequencing technologies for their ability to detect issues in vector design and determine nanopore direct RNA sequencing to be the most powerful. We show how this approach identifies and quantifies incomplete RNA caused by cryptic splicing and polyadenylation sites, including a potential cryptic polyadenylation site in the widely used Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element (WPRE). Using artificial polyadenylation of the lentiviral RNA, we also identify multiple hairpin-associated truncations in the analyzed lentiviral vectors (LVs), which account for most of the detected RNA fragments. Finally, we show that these insights can be used for the optimization of LV design. In summary, nanopore direct RNA sequencing is a powerful tool for the quality control and optimization of LVs, which may help to improve lentivirus manufacturing and thus the development of higher quality lentiviral gene therapies.
Recent years have seen considerable advances in the field of lentiviral gene therapy, with several clinical trials delivering stable, long-term transgene expression to treat disease (Dunbar et al. 2018; Papanikolaou and Bosio 2021). However, several challenges remain before lentiviral gene therapies can be routinely implemented in the clinic. These include that a substantial proportion of lentiviral RNA can be nonfunctional. As a consequence, the physical titer (number of virus particles) is typically much higher than the infectious titer, which is the number of infectious virus particles (Han et al. 2021). Although a low functional titer is not unusual for lentiviruses; it has been estimated that as few as 1% of HIV virus particles are capable of infection (Dimitrov et al. 1993), this must be improved in order to facilitate cost-effective, large-scale manufacturing of lentiviral gene therapies.
An important cause of nonfunctional lentivirus is the presence of incomplete lentiviral RNA, which can occur for a range of reasons such as anomalous splicing, premature transcription termination at cryptic poly(A) sites, or insufficient processivity of the polymerase due to the length of the construct (Han et al. 2021; Sertkaya et al. 2021). In addition to lowering the infectious titer, this may reduce therapeutic efficacy due to an incomplete transgene and can also pose a safety risk due to loss of safety elements such as chromatin insulators or other regulatory components designed to limit the potential for oncogene activation (Almarza et al. 2011). These issues can be solved by mutating or removing problematic sites or by reducing the length of the vector itself which can remove potentially problematic sites and also increase transcriptional efficiency (Han et al. 2021; Sertkaya et al. 2021). It is, therefore, important to have an assay in place that can identify not only the abundance, but also the causes of lentiviral RNA truncation. RNA sequencing using Illumina (Han et al. 2021) and Oxford Nanopore Technologies (ONT) direct cDNA (Sertkaya et al. 2021) approaches have both been used to demonstrate improvements to lentiviral vectors (LVs), and sequencing has also been applied to other nucleic acid-based therapeutics, such as mRNA vaccines (Gunter et al. 2023). However, there is currently no clear consensus on which sequencing technology is most effective for quality control (QC) of LVs, and current cDNA sequencing approaches which rely on reverse transcription (RT) of the lentiviral RNA can introduce bias (Schulz et al. 2021).
Nanopore direct RNA sequencing is a relatively new technology that enables the direct sequencing of long, potentially full-length, native RNA. Thus, it may present an unbiased, faster, and more efficient way to assess LVs. Sequencing starts from the 3′ end of the RNA, so the ends of sequencing reads correspond directly to the 3′ ends of the physical RNA molecules which allows for precise identification of truncated vector sequences. As such, it has proven successful during the recent pandemic, with the high-throughput sequencing of the SARS-CoV-2 viral genome (Bull et al. 2020; Pater et al. 2021; Barbé et al. 2022). Other long-read approaches such as ONT and PacBio also have the potential to capture the entire lentiviral RNA in a single read, enabling analysis at a single-molecule level.
To facilitate QC of LVs early in development, there is a need for a quick, efficient, and cost-effective sequencing assay. Thus, to determine the most appropriate sequencing technology for LV QC, this study aimed to compare nanopore direct RNA sequencing with cDNA sequencing approaches (from ONT and Pacific Biosciences [PacBio]). We also considered modifications to the ONT direct RNA sequencing protocol to improve the proportion of vector reads and to identify incomplete RNAs that are not naturally polyadenylated. Finally, we aimed to improve our lentiviral constructs by modifying the sequences we identified as problematic.
Results
ONT direct RNA sequencing is an ideal long-read RNA sequencing approach for lentiviral vector QC
To compare different approaches for QC of LV integrity, we decided to perform an initial experiment with “Globin LV,” a gamma globin-expressing vector that had shown low functional titers. In this vector, gamma globin is expressed from a β-globin promoter (jointly labeled “Transgene” in Fig. 1D) adjacent to regulatory elements (HS2, HS3, and HS4). In addition, a short hairpin RNA (shRNA) targeting human hypoxanthine phosphoribosyltransferase 1 (HPRT1) is expressed from a human RN7SK RNA Pol III promoter (7SK), allowing for positive selection of transduced cells using 6-thioguanine (6-TG) (Choudhary et al. 2013). A 400 bp extended core element of the chicken hypersensitivity site 4 (cHS4) β-globin chromatin insulator (“insulator” in Fig. 1) is inserted in the 3′ long terminal repeat (LTR) as a safety and antisilencing element. This insulator was previously shown to contain a potent cryptic splice acceptor site, which was identified as the cause for clonal dominance in progenitor and myeloid lineages of patients in clinical trials (Cavazzana-Calvo et al. 2010; De Ravin et al. 2022). A rabbit β-globin polyadenylation signal is added downstream from the 3′ LTR to enhance cleavage and polyadenylation of the vector transcript and to reduce transcriptional readthrough.
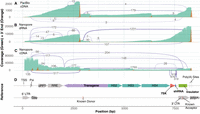
Comparison of three different long-read RNA sequencing approaches for lentiviral vector QC. Sequencing coverage (green), location of the 3′ ends of reads (orange), and splicing patterns (purple lines connecting splice donors and acceptors, with numbers indicating how many reads support each splicing event) for three different long-read sequencing technologies. (A) PacBio cDNA sequencing data. (B) ONT direct RNA sequencing data. (C) ONT direct cDNA sequencing data. (D) Reference sequence of the Globin LV, with various features annotated.
RNA was isolated from concentrated and purified lentiviruses that were produced using stable producer cell lines, which were then subjected to sequencing by ONT direct RNA, ONT direct cDNA, and PacBio cDNA sequencing approaches to compare the three technologies. Read numbers varied by technology, ranging from 491,270 reads for ONT direct cDNA to 273,029 reads for ONT direct RNA (both ONT methods were performed using MinION flow cells) and 50,159 reads for PacBio cDNA sequencing. In all cases, the percentage of vector-aligned reads was relatively low (<30%) (Supplemental Table S1). The remaining reads aligned to the human genome, indicating that most of the sequenced RNA originates from the producer cells rather than the lentivirus and has been copurified during lentivirus purification and RNA isolation.
Sequencing coverage showed a step-like pattern of sharp changes which slowly decay from high to low coverage moving toward the 5′ end of the vector sequence in the PacBio cDNA and ONT direct RNA data (Fig. 1, green track). This decay can be explained by RT (for PacBio cDNA) and sequencing (for ONT direct RNA) beginning at the 3′ end of the RNA, and not extending all the way to the 5′ end. Sharp changes in coverage, such as the one at ∼3000 bp, indicate sites in the vector leading to incomplete transcripts. In contrast, the ONT direct cDNA data (Fig. 1C) shows a different pattern: Its coverage is highest at the 5′ end, with several sharp coverage changes that do not appear in the analysis of the other two approaches. These coverage changes correlate with internal poly(A) tracts in the vector reference sequence, indicating that the 5′ coverage bias is due to internal priming during the RT step leading to the formation of fragments not reflecting the state of the lentiviral RNA.
Plotting of the reads’ 3′ ends revealed the intended poly(A) sites at the 3′ end of the reference sequence as well as three commonly used cryptic poly(A) sites (Fig. 1, orange track) which coincide with many of the sharp coverage changes noted above. Closer examination of the 3′ end positions near the intended poly(A) sites confirmed that they match the cleavage sites downstream from the lentiviral poly(A) signal (smaller peak, within the 3′ LTR) and the cleavage site of the rabbit β-globin polyadenylation signal (main peak). The most significant of the cryptic poly(A) sites, located at ∼3000 bp was used by 36.82% of the reads in the PacBio cDNA sample (as compared to 33.62% of reads that terminate at the 3′ end of the vector) (Fig. 1A) and 16.44% of reads in the Nanopore direct RNA sample (while 59.85% terminate at the 3′ end of the vector) (Fig. 1B). Identification of cryptic poly(A) sites in the Nanopore direct cDNA data is complicated by the large number of internal priming sites, which also produces 3′ end peaks that are indistinguishable from those that originate from truncated RNA (Fig. 1C).
Importantly, numerous splicing patterns were identified in the Globin LV (Fig. 1, purple lines). While the Nanopore sequencing approaches picked up more splice events than the PacBio approach, likely due to the significantly higher number of reads enabling the detection of rarer splicing patterns, several events were consistently observed across all three approaches. In particular, a splicing event involving the known splice acceptor site in the insulator (Cavazzana-Calvo et al. 2010; De Ravin et al. 2022) had the highest number of supporting reads with all three sequencing technologies. This splicing event leads to the formation of an incomplete lentiviral RNA that contains the packaging signal (Psi), which allows these incomplete lentiviral transcripts to be packaged (Poletti and Mavilio 2021). These transcripts, however, lack a substantial portion of the therapeutic globin cassette and are, therefore, unlikely to confer a therapeutic benefit.
Overall, between the compared approaches, we found the ONT direct RNA sequencing technology to be the most appropriate for lentiviral RNA QC given the simplicity of the workflow and the comprehensive overview of identified splice events and cryptic poly(A)sites. While ONT direct cDNA sequencing provides more reads and higher coverage at the 5′ end of the vector, internal priming bias can severely confound the identification of cryptic poly(A) sites. Similarly, for PacBio sequencing, the lower number of reads and high cost per base limited the identification of rarer splicing events.
ONT direct RNA sequencing identifies cryptic poly(A) and splice sites
To further evaluate the utility of nanopore direct RNA sequencing for LV QC, we applied the technology for the analysis of four Wiskott–Aldrich syndrome (WAS) LVs, differing from each other only in their insulator sequences (described below). All four-vectors express human WAS (“Transgene” in Fig. 2) from a strong synthetic promoter. The 3′ untranslated region (UTR) consists of a Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element (WPRE) that is commonly included in LVs to increase expression (Zufferey et al. 1999). As for the Globin LV, a second cassette expresses a shRNA to target HPRT1 under the control of a 7SK promoter. All WAS vectors contain a 650 bp version of the cHS4 insulator (Urbinati et al. 2009), with an identical core sequence to the 400 bp cHS4 insulator and containing the same known splice acceptor site (Cavazzana-Calvo et al. 2010; De Ravin et al. 2022). While the first vector “WAS LV1” harbors an unmodified 650 bp insulator (in reverse orientation), the insulator sequence was modified in the three other vectors to inhibit splice acceptor activity (Supplemental Table S2). Vectors “WAS LV2” and “WAS LV3” contain two and three A-to-T point mutations in “AG” splice acceptor motifs, respectively. Mutated in both vectors are the known splice acceptor site (Cavazzana-Calvo et al. 2010; De Ravin et al. 2022) also present in the Globin LV, and an additional splice acceptor site predicted in silico using NetGene2 (Brunak et al. 1991), located outside the core region and thus specific to the 650 bp insulator version. Vector “WAS LV3” contains an additional mutation in a third “AG” motif located 10 bp away from the known problematic splice acceptor site (Cavazzana-Calvo et al. 2010; De Ravin et al. 2022), consistent with a published vector (De Ravin et al. 2022). A fourth vector “WAS LV4” contains an inverted cHS4 650 bp insulator (i.e., in forward orientation) with the purpose of decoupling splicing and polyadenylation, processes which can costimulate each other (Kaida 2016). In this vector, the known and predicted splice acceptors are in the opposite orientation relative to the lentiviral poly(A) signal.
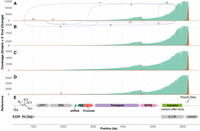
Using ONT direct RNA sequencing to assess WAS vectors. Plots showing the sequencing coverage (green), location of the 3′ ends of reads (orange), and splicing patterns (purple lines connecting splice donors and acceptors) for four different WAS vectors sequenced by ONT direct RNA sequencing: (A) WAS LV1, (B) WAS LV2, (C) WAS LV3, (D) WAS LV4. (E) Reference sequence of the WAS vector, with various features annotated.
Between 445,076 and 743,182 reads were generated for each sample, of which ∼1.5%–3% aligned to the vector reference sequence (Supplemental Table S3). This is much lower than for the Globin LVs and can be explained by the differences in the purification process used. RNase treatment to degrade unpackaged, copurified RNAs did not lead to an improvement in the retrieval of lentiviral RNA (Supplemental Fig. S1; Supplemental Table S4).
The sequencing coverage of WAS vectors (green, Fig. 2) shows a strong pattern of 3′ to 5′ decay, with fewer sharp changes than for the Globin LVs, and a higher percentage of full-length vector (Fig. 1). Overall, the data for the four vectors was very similar, highlighting the robustness and reproducibility of this assay across different LV preparations.
Analysis of the location of 3′ read ends of WAS vectors showed that while ∼80% terminated in the expected position at the end of the reference sequence (tall orange peak, Fig. 2), ∼10% terminated ∼4600 bp, in close proximity to the end of the WAS coding sequence (small orange peak, Fig. 2). This coincides with a sharp change in coverage that occurs at the same position consistently across all four vectors, and we hypothesized that this might correlate with the presence of a cryptic poly(A) site. In addition to the poly(A) site at the 3′ end of the vector, the canonical poly(A) signal motif, AATAAA, was identified at four additional locations within the vector, although none were near 4600 bp. Instead, the only poly(A) motif identified near this site was the noncanonical ATTACA motif, ∼10–20 bp upstream of where the reads terminate, which could potentially lead to premature polyadenylation. Alternatively, given that the putative poly(A) site is located close to the end of the WAS coding sequence and the large proportion of RNAs other than the lentiviral RNA in the sample (purple arrow, Fig. 2E), we investigated whether these transcripts may represent endogenous WAS transcripts expressed from the cells used for lentivirus production. Endogenous WAS differs from the vector sequence by its 265 bp 3′ UTR, as well as by two single-nucleotide variants introduced in the coding sequence of the transgene. None of the reads terminating at ∼4600 bp aligned to the WAS 3′ UTR and the two-point mutations were observed in 99% and 97% of reads across all samples, respectively. This indicates that the transcripts terminating at ∼4600 bp are being transcribed from the vector and terminate prematurely, potentially due to usage of the alternative 3′ ATTACA poly(A) site.
Little splicing was observed in the WAS vectors, suggesting that splicing events are not a major cause of incomplete lentiviral RNA for these vectors (Fig. 2). The original vector, WAS LV1 showed a small amount of splicing, with <100 out of ∼18,000 vector-aligned reads originating from splicing events (Fig. 2A). Most of these splicing patterns involved the known canonical acceptor site (Cavazzana-Calvo et al. 2010; De Ravin et al. 2022), located in the chromatin insulator region (Fig. 2), which was also observed in the Globin LV (Fig. 1). As described above, we attempted to modulate splicing through modification of the vector either by introduction of point mutations in the splice acceptor or by inversion of the insulator. These modifications appear to be successful in reducing splicing into this site, as WAS LV2 and WAS LV3 showed no splicing at all, while WAS LV4 had only a single splicing event, involving a different acceptor site. This demonstrates the potential for ONT direct RNA sequencing to identify problematic sites in LVs, to inform the design of optimized vectors, and to verify that introduced modifications reduce truncated lentiviral RNA.
Artificial polyadenylation reveals additional sites of truncation
We then explored the use of artificial polyadenylation to capture incomplete lentiviral RNA that may not be naturally polyadenylated, such as those originating from incomplete transcription or that are degraded or fragmented during library preparation. For this analysis, we used the WAS vector with the wild-type insulator sequence containing the detected splice acceptor sites (WAS LV1). A second vector, WAS LV2, was also analyzed using this method (see Supplemental Fig. S2). Compared to the original, nonpolyadenylated samples (Supplemental Table S3), the median read lengths of polyadenylated reads were decreased from ∼700 to ∼300 bp and a smaller percentage of them aligned to the vector (Supplemental Table S5). This can be explained by the abundance of copurified short RNA species (e.g., rRNA, snRNA) that can be sequenced once polyadenylated.
Despite having fewer mapped reads, the polyadenylated samples show a similar pattern of sequencing coverage toward the 3′ end of the vector, whereas at the 5′ end coverage is increased (green, Fig. 3A), coinciding with the presence of sharp coverage changes and 3′ end peaks (orange, Fig. 3B) in this region. These peaks, making up 11.26% and 8.61% of reads, respectively, are more pronounced than the one representing the potential cryptic poly(A) site close to the end of the WAS transgene (4.44% of reads), suggesting that these sites may play a significant role in the generation of incomplete WAS vector RNA. Upon closer examination of the vector reference sequence in these locations, both 3′ peaks occur around RNA stem–loop structures: the shRNA sequence targeting HPRT1 at ∼2300 bp and several conserved stem–loops (SL1–SL4) present within the packaging signal (Psi) at ∼500 bp (Fig. 3C). It is possible that these sites in the lentiviral RNA near stem–loops are being cleaved by endonucleases such as DROSHA, which is known to recognize such structures as they may resemble those of primary miRNA transcripts (Jin et al. 2020). In the case of the shRNA, another possibility is cleavage mediated by AGO2, a component of the RNA-induced silencing complex (RISC), which can occur if the mature siRNA targets its complementary sequence in the lentiviral RNA (Herrera-Carrillo et al. 2017). AGO2-mediated cleavage seems to be the main mechanism behind the cleavage at the shRNA sequence, as the largest drop in coverage occurs in the middle of the siRNA complementary sequence (Supplemental Fig. S3). Overall, these findings demonstrate that artificial polyadenylation of lentiviral RNA before ONT direct RNA sequencing can facilitate the identification of a broader range of truncations.
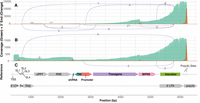
Artificially polyadenylated ONT direct RNA sequencing data for a WAS vector. Plots showing the sequencing coverage (green), location of the 3′ ends of reads (orange), and splicing patterns (purple lines connecting splice donors and acceptors) for WAS LV1 vectors sequenced by ONT direct RNA technology, with and without artificial polyadenylation (performed according to Crabtree et al. [2019]). (A) Standard library preparation protocol (no artificial polyadenylation). (B) Artificially polyadenylated sequencing data. (C) Reference sequence of the WAS vector, with various features annotated.
In addition to identifying additional types of incomplete lentiviral RNA, artificial polyadenylation enables a more precise estimate of the percentage of complete and incomplete RNA as all RNA fragments originating from the LV should be captured. From counting the number of reads that terminate around the suspected cryptic poly(A) site (4.44%) and aforementioned stem–loop structures (19.88%), combined with the 1.31% of reads found to be spliced, we can estimate that at least a quarter of lentiviral RNA reads are incomplete in some way. In addition, ∼15% of read 3′ ends are spread evenly along the lentiviral genome rather than in the 3′ end peaks in Figure 3 (henceforth referred to as “spread reads”). These may result either from insufficient polymerase processivity or instead correspond to lentiviral RNA that has been degraded or fragmented during the isolation and sequencing library preparation processes. Given that 25% of reads are aberrantly spliced or truncated, if all of the 15% spread reads are incomplete due to poor polymerase processivity, then the percentage of full-length WAS LV1 RNAs would be ∼60%. Instead, if all 15% were originally full-length RNAs that were degraded or fragmented during sample preparation, then 75% of WAS LV1 RNAs would be full-length. In reality, the true percentage likely falls somewhere between these two numbers.
Insights from sequencing can be used to optimize vectors
Having identified the two most significant contributors to incomplete lentiviral RNA in our WAS vectors (the potential cryptic poly(A) site and hairpin-associated truncations), we sought to improve the vector quality by modifying these sites (Fig. 4C). Based on the WAS LV3 vector, a new vector (WAS LV5) was designed and analyzed in Figure 4A, with both the shRNA cassette (a potential AGO2 cleavage site responsible for 8.61% of reads being incomplete) (Supplemental Table S2) removed and the ATTACA motif (a potential cryptic poly(A) site responsible for ∼4%–10% of reads being incomplete) mutated to ATTTCA. The other potential stem–loop adjacent cleavage site, located in Psi and responsible for 11.26% of reads being incomplete, was left unchanged given the important role of this region in the nuclear export and packaging of the lentivirus. In addition, a new reverse transcriptase enzyme (Induro) was used to stabilize the RNA during library preparation, due to its advantages when dealing with highly structured and modified RNA. This enabled the generation of longer reads and therefore better coverage of the lentiviral RNA when compared to the previous protocol, further improving the direct RNA sequencing approach (Supplemental Fig. S4).
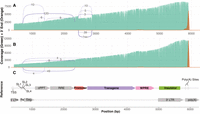
Nanopore direct RNA sequencing of optimized WAS vectors. Plots showing the sequencing coverage (green), location of the 3′ ends of reads (orange), and splicing patterns (purple lines connecting splice donors and acceptors) for two WAS vectors sequenced by Nanopore direct RNA technology (A) WAS LV5 sequencing data (contains a single A-to-T mutation in the potential cryptic poly(A) site). (B) WAS LV6 sequencing data (contains an additional two-point mutations in the promoter to remove splice donor sites). (C) Reference sequence of the WAS vector, with various features annotated.
Mutation of the cryptic poly(A) site did not eliminate premature termination but led to a reduction in the percentage of reads terminating in a 150 bp window around this site to 3.39% in WAS LV5, and to 2.04% in WAS LV6 explained below. The mechanism of reduced but persistent premature termination is still unclear, and further mutations may be required to prevent it completely. Additionally, a new and abundant splicing pattern that removes most of the promoter and a small part of the start of the transgene was observed in these modified vectors; 19.35% of WAS LV5 reads are affected by this splicing pattern, which occurs between the promoter and the transgene. This particular splicing pattern was not observed in any of the previous vector designs (WAS vectors in Fig. 2), although the donor site was used with a different acceptor at very low (<1%) levels. Despite this, splice site prediction with MaxEntScan (Yeo and Burge 2004) gives the donor and acceptor sites identical scores, irrespective of the vector version (8.15 for the donor site, 10.82 for the acceptor site). This splicing event could be eliminated in WAS LV6 by the introduction of two-point mutations in the promoter (Fig. 4B). More specifically, we introduced T-to-A point mutations in two “GT” splice donor motifs occurring within a direct repeat region of the promoter. Introduction of only one point mutation in the observed active GT motif would have likely shifted the activity to the splice donor on the other repeat. We confirmed this result with an additional vector, WAS LV7, identical to WAS LV6 except for one additional (silent) point mutation in the transgene ORF to mutate the splice acceptor observed in Figure 4A (Supplemental Fig. S5; Supplemental Table S2). The overall highly similar results for WAS LV6 and WAS LV7 underline the reproducibility of the assay, also with the Induro RT enzyme. Another set of splicing events can be observed toward the 5′ end of vectors WAS LV5, WAS LV6, and WAS LV7, likely as a consequence of the improved protocol with increased 5′ end coverage. The majority of those splicing events share a splice donor relevant to the virus life cycle termed the major splice donor (MSD) (Ashe et al. 1997). Previous attempts to mutate this known splice donor resulted in lower RNA expression, possibly due to reduced RNA stability (Cui et al. 1999). Thus, in this study, we did not attempt to mutate any of the virus-related parts of the vector to avoid potential negative consequences. Overall, although the complex nature of LVs makes them challenging to optimize, direct RNA sequencing allows for the QC of packaged lentiviral RNA and facilitates the development of improved vectors.
Discussion
Achieving reliable production of intact LVs will have a significant impact on their manufacturing and clinical implementation. To this end, we have evaluated the application of three different long-read sequencing technologies in order to determine the most appropriate approach for LV QC and to allow improvements to LV design. We found ONT direct RNA sequencing to be the most suitable, as it, unlike PacBio cDNA sequencing, which generated relatively few reads, provided high sequencing coverage of the vector without any internal priming as observed in the ONT direct cDNA data. Using this nanopore direct RNA sequencing approach, we were able to identify cryptic splice and poly(A) sites in LVs. The results were also highly reproducible between both sequencing library and lentiviral preparations; the highly similar vectors in Figure 2B–D and Supplemental Figure S5 show almost identical sequencing results, therefore, suggesting that this method is a robust way to perform QC of LVs.
To improve the nanopore direct RNA sequencing approach, we tried to incorporate an RNase treatment to increase the proportion of vector-aligned reads, artificial polyadenylation to investigate whether there were incomplete RNAs without natural poly(A) tails and use of the Induro reverse transcriptase (RT) to increase read length and thereby coverage. The RNase treatment was unsuccessful, as there was only a slight increase in reads aligning to the vector and RNA degradation rendered the data patchy, due to the decrease in read length (Supplementa1 Fig. S1; Supplemental Table S4). We suspect that this may be due to most of the sequenced human RNA coming from within extracellular vesicles, which are often copurified with lentivirus and have been shown to contain human RNA from producer cells (Do Minh et al. 2021). Applying a more rigorous lentiviral purification than the research-grade ultracentrifugation purification (which is known to struggle with the separation of virus and extracellular vesicles) (McNamara and Dittmer 2020) used for this study would, therefore, likely improve this sequencing assay by increasing the percentage of on-target reads. Nevertheless, the output of MinION flow cells is large enough that a sufficient number of lentiviral reads are obtained for research-grade lentiviruses.
Artificial polyadenylation on the other hand produced interesting results, revealing two highly abundant species of incomplete vector RNA, associated with stem–loop structures. Finally, an Induro-based library preparation method (as compared to the standard SuperScript III protocol) delivered significantly longer reads (Supplemental Fig. S3). The Induro reverse transcriptase, which has increased processivity and may perform better with the RNA modifications and secondary structure that are present in lentiviral RNAs, is likely better able to support the RNA strand being sequenced and minimize the formation of RNA secondary structures that can block the nanopores. This highlights the importance of using RT to produce an RNA–cDNA hybrid during direct RNA library preparation of lentiviral samples, due to their secondary structure, and suggests that future improvements to RT may benefit this protocol.
Using this assay, we identified and mitigated several problematic sites contributing to incomplete lentiviral RNAs. In both WAS LV1 and Globin LV, a splice site in the chromatin insulator sequence was identified, and subsequent vectors with point mutations or inversions were not subject to splicing at this site (Fig. 2). We also identified a potential cryptic poly(A) site with a noncanonical ATTACA motif as a probable cause of truncation in up to 10% reads, which is located in the WPRE that is commonly included in LVs to increase expression (Zufferey et al. 1999). By mutation of the potentially underlying ATTACA poly(A) signal, we reduced the usage of the cryptic poly(A) site to about half. Using the artificially polyadenylated sequencing data, we also identified two truncation sites near hairpins within the vector sequence which together result in approximately a fifth of all reads being incomplete. It is likely that the mechanisms behind the observed truncations differ for these two sites. Our results indicate that the truncation within the shRNA sequence is the result of a self-targeting mechanism mediated by RISC (Supplemental Fig. S3). Assembly of RISC with the siRNA originating from the shRNA cassette can lead to cleavage of the complementary sequence within the LV. This is surprising, as it was previously suggested that the stem–loop structure shields the complementary sequence from the siRNA (Herrera-Carrillo et al. 2017). The mechanism behind the second observed truncation, leading to cleavage in the LV packaging signal, is unclear. One possibility is that the hairpins are targeted by endonucleases recognizing stem–loop structures such as DROSHA. This would be consistent with literature showing decreased levels of full-length genomic LV RNA as well as lower titers of LVs containing shRNA hairpin structures, only in the presence of functional DROSHA (Park et al. 2018).
For the purpose of this study, we simply prevented the truncation at the shRNA sequence by removing the shRNA cassette to see if this would improve vector quality. However, the removal of the shRNA cassette introduced a new splicing pattern nearby that removed the promoter in ∼20% of reads. Why this splicing pattern only emerges after the removal of the shRNA is unclear, although one possibility is that the antiviral activity of DROSHA, wherein it binds the stem–loop and confers steric hindrance to prevent viral replication (Aguado et al. 2017) or AGO2, which has been shown to act as a restriction factor against SARS-CoV-2 (Lopez-Orozco et al. 2023) may have been blocking the spliceosome from accessing this site. Although the splicing pattern was subsequently prevented by mutation of the promoter (Fig. 4B), this highlights the difficulty of optimizing complex LV sequences based solely on sequence prediction and design, and the importance of reassessing their quality by sequencing after modification in order to identify any new problems that may have arisen.
As these improvements increase the percentage of full-length RNA, it is assumed that they will translate into an improved functional titer and safety profile. While studies of functional consequences and safety improvements will be key to the development of a clinical vector, for this study, we concentrated on the direct RNA sequencing approach allowing vector quality to quickly be checked, while also providing the information necessary to make further improvements to the vector design.
The trade-off of using nanopore direct RNA sequencing as opposed to cDNA sequencing with ONT, PacBio, or Illumina is that the number of reads generated is relatively low, especially when combined with the low percentage of vector-aligned reads. Thus, only ∼10,000 LV reads are produced on average from a single MinION flow cell. Nevertheless, many splices, poly(A), and hairpin-associated truncation sites can be identified from the sequencing data. Therefore, a new direct RNA sample barcoding approach (van der Toorn et al. 2024) could be used to reduce sequencing costs by multiplexing several vectors on a single flow cell. Single-nucleotide polymorphisms (SNPs), however, were difficult to determine from our nanopore direct RNA sequencing due to the relatively higher rate of both random and systematic errors in the RNA002 chemistry used (Liu-Wei et al. 2024), as well as the abundance of modified bases in lentiviral RNA, which can lead to miscalling or insertions/deletions during base calling. These issues may be addressed through the recently released ONT SQK-RNA004 kit, which offers higher throughput and sequencing accuracy (Perlas et al. 2024).
In summary, ONT direct RNA sequencing is a powerful tool for the QC of LVs, allowing for the identification of sites that cause truncations. It can be easily implemented using the affordable, handheld MinION device, which allows for rapid, in-house sequencing. To identify all sources of truncation in a vector, including hairpin-associated truncations that are not naturally polyadenylated, we recommend that two nanopore direct RNA sequencing runs are performed: one with and one without artificial polyadenylation. This will enable rapid analysis and subsequently improvement of LVs to increase the proportion of full-length RNA and hopefully facilitate the manufacturing of better LVs in the future.
Methods
Lentiviral vector design
All LVs used in this study (listed in Supplemental Table S6) are derived from a pCl20c vector backbone (Throm et al. 2009) and contain a rabbit β-globin polyadenylation signal (“poly(A)” in Figs. 1⇑⇑–4) downstream from the 3′ LTR to provide a stronger polyadenylation signal for the vector transcript. In the Globin LV (Fig. 1), gamma globin is expressed from a 254 bp β-globin promoter, placed in reverse orientation with respect to the viral transcript. The expression is regulated by a β-globin locus control region (LCR) consisting of hypersensitivity sites (HSs) 2, 3, and 4 elements. A second expression cassette is inserted in forward orientation and downstream from the globin expression cassette. It expresses a shRNA targeting human HPRT1 under the control of a human 7SK RNA Pol III promoter. As a safety and antisilencing element, a 400 bp extended core element of the cHS4 β-globin chromatin insulator is inserted in the 3′ LTR in reverse orientation with respect to the viral transcript. All WAS vectors contain a strong synthetic promoter driving the expression of a transgene encoding for human WAS, in forward orientation (relative to the viral transcript). In the 3′ UTR region downstream from the WAS CDS, the vectors contain a WPRE that harbors six-point mutations to abrogate WHX truncated protein expression (Zanta-Boussif et al. 2009). In some WAS vectors, a human 7SK RNA Pol III promoter drives the expression of a shRNA targeting HPRT1, in reverse orientation to and located upstream of the first expression cassette. Vector “WAS LV1” contains the original sequence of the 650 bp insulator, reconstructed from the public database (NCBI GenBank [https://www.ncbi.nlm.nih.gov/genbank/] accession number GGU78775) and from the description of the cloning strategy used by Urbinati et al. (2009) in reverse orientation. A second vector “WAS LV2” contains two A-to-T point mutations in “AG” splice acceptor motifs in the insulator with the purpose of abrogating splice acceptor activity at these sites. Whereas one mutated “AG” motif corresponds to the known splice acceptor site (Cavazzana-Calvo et al. 2010; De Ravin et al. 2022) also present in the Globin LV, the other mutated “AG” motif (specific to the 650 bp and not part of the 400 bp cHS4 insulator variant) was predicted as a likely splice acceptor site in silico using NetGene2 (Brunak et al. 1991). A third vector “WAS LV3” contains three A-to-T point mutations in “AG” motifs in the insulator. In addition to the two “AG” motifs mutated in “WAS LV2,” a third “AG” motif located 10 bp away from the known problematic splice acceptor site (Cavazzana-Calvo et al. 2010; De Ravin et al. 2022) is mutated, consistent with another published improved vector with a 400 bp cHS4 insulator variant (De Ravin et al. 2022). Vector “WAS LV4” harbors a cHS4 650 bp insulator in forward orientation to decouple splicing and polyadenylation. Vector “WAS LV5” is derived from WAS LV3 with two introduced changes: The shRNA expression cassette is deleted and the ATTACA motif in the WPRE element is mutated to ATTTCA by introducing a T => A point mutation.
Lentiviral production and RNA purification
WAS lentiviruses were produced by transient transfection of transfer plasmids containing the constructs of interest into GPRTG lentivirus producer cell lines (Throm et al. 2009; Wielgosz et al. 2015). Briefly, 152 × 106 GPRTG cells were seeded into a 2-STACK culture chamber (Corning) and transfected 24 h later with 238 µg transfer plasmid using the CalPhos Mammalian Transfection Kit (Takara Bio) according to the manufacturer's instructions. The transfection mix was replaced with fresh medium 4 h post transfection and virus-containing supernatant was collected 2 days later. After filtration through a 0.22 µM filter, 31 mL of the filtrate was transferred to each of six 40PA ultracentrifugation tubes (Himac), underlayed with 5 mL 20% (w/v) sucrose in 100 mM NaCl, 20 mM HEPES, 1 mM EDTA, and centrifuged for 2 h at 25,000 rpm and 4°C. After centrifugation, the supernatant was discarded and the viral pellet was resuspended in 50 µL X-Vivo10 medium (Lonza) supplemented with 2% human serum albumin. Resuspended viral pellets were filtered through a 0.22 µM filter, pooled, and stored at −70°C. For subsequent analysis by sequencing, lentiviral RNA was purified using the QIAamp Viral RNA Mini Kit (Qiagen) according to the manufacturer's instructions. QC of extracted RNA was performed using a fragment analyzer.
Artificial polyadenylation
Artificial polyadenylation was carried out based on a published protocol (Crabtree et al. 2019). Reactions containing 15 µL of RNA (∼500 ng), 2 µL of poly(A) buffer, 2 µL of 10 mM ATP (NEB P0756S), 1 µL of E. coli poly(A) polymerase (NEB M0276S) and 0.5 µL of Murine RNase Inhibitor (NEB M0314S) were incubated for 30 min at 37°C, 20 min at 65°C, and then 5 min at 98°C before being placed on ice. The RNA was then cleaned up with magnetic beads at 1.8× ratio and eluted in 15 µL nuclease-free water ready for the ONT direct RNA sequencing protocol. Given the poorer quality of artificially polyadenylated reads, it might be beneficial to adopt a more gentle approach (Yan et al. 2018).
PacBio cDNA sequencing
PacBio SMRTbell libraries were prepared according to the manufacturer's instructions and sequenced on the PacBio Sequel platform with v3.0 chemistry. The generated subreads were demultiplexed and CCS reads were obtained using v4.2.0 of PacBio's CCS algorithm with default parameters.
ONT direct cDNA sequencing
ONT direct cDNA sequencing libraries were prepared using the SQK-DCS109 kit, according to the manufacturer's instructions. Sequencing was performed using a MinION R9.4.1 flow cell (FLO-MIN106D) on a GridION instrument.
ONT direct RNA sequencing
ONT direct RNA sequencing libraries were prepared using the SQK-RNA002 kit, according to the manufacturer's instructions. For the final set of experiments (Fig. 4), the SuperScript III RT used in the RT step of the protocol was replaced with Induro RT. Reactions containing 2 µL of dNTP, 8 µL of 5× Induro RT Buffer (NEB M0681S), 12.6 µL nuclease-free water, 0.4 µL RNase Inhibitor, and 2 µL Induro RT (200 U/µL; NEB M0681S) were incubated for 15 min at 60°C, 10 min at 70°C, and then cooled to 4°C immediately. Sequencing was performed using MinION R9.4.1 flow cells (FLO-MIN106D) on a GridION instrument.
Bioinformatic analysis
Following basecalling and basic sequencing QC using the SeqKit (Shen et al. 2016) stats function, reads were aligned to both the LV and human genome reference sequences using minimap2 (Li 2018) in splice mode. The SAMtools (Li et al. 2009) software was used to determine alignment statistics (SAMtools flagstat) and coverage of the vector reference sequences (SAMtools depth). Positions of 3′ read endings were extracted from the PAF output file generated by minimap2 (Li 2018) in R (R Core Team 2021) and sashimi plots of the splicing patterns were generated using ggsashimi (Garrido-Martín et al. 2018). This information was then collated in R (R Core Team 2021) and plotted with ggplot2 (Wickham 2016), using the gggenes (https://cran.r-project.org/web/packages/gggenes/index.html) package to plot the vector reference sequence, ggrepel (https://cran.r-project.org/web/packages/ggrepel/index.html) to label the vector elements and cowplot (https://cran.r-project.org/web/packages/cowplot/index.html) to combine multiple plots into a single panel. Cryptic poly(A) sites in the data were predicted based on the motifs described in the PolyASite 2.0 database (Herrmann et al. 2019), and hairpins were modeled using the RNAfold (Gruber et al. 2008) webserver. A detailed bioinformatic tutorial can be found at https://kzeglinski.github.io/lentiviral_qc/bioinf.html.
Data access
The raw sequencing data generated in this study has been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/) under accession number PRJEB72210.
Competing interest statement
One or more of the model vectors used as the basis for this study were published in International Publication Nos. WO2019018383, WO2020139796, or WO2022232191. Authors C.M., C.V., and F.A. are named as inventors. K.Z and Q.G. have previously received travel and accommodation expenses from Oxford Nanopore Technologies.
Acknowledgments
We thank Esther Chen for virus production of the Globin LV, Martina Biserni for support with lentivirus production and RNA extraction, Alban Ramette and the team at IFIK for the Nanopore sequencing service, Genewiz for the PacBio sequencing service, and the CSL/WEHI Translational Data Science Alliance for funding this project.
Author contributions: K.Z.: Conceptualization, data Curation, formal analysis, visualization, writing—original draft preparation; C.M.: Conceptualization, investigation, writing—review and editing; M.E.R.: Formal analysis, supervision; M.A.: Supervision; C.V.: Supervision, resources; R.B.: Supervision; M.J.: Investigation; Q.G.: Conceptualization, formal analysis, supervision; F.A.: Conceptualization, resources, writing—review and editing, project administration; A.H.: Conceptualization, writing—review and editing, supervision.
Footnotes
-
↵5 Joint last authors.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279405.124.
- Received March 25, 2024.
- Accepted September 27, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








