
Global characterization of somatic mutations and DNA methylation changes during vegetative propagation in strawberries
- Shaoqiang Hu1,
- Xiangguo Zeng2,
- Yuguo Liu1,3,
- Yongping Li4,
- Minghao Qu5,
- Wen-Biao Jiao1,3,
- Yongchao Han2 and
- Chunying Kang1,3
- 1National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, Huazhong Agricultural University, Wuhan 430070, China;
- 2Hubei Key Laboratory of Vegetable Germplasm Enhancement and Genetic Improvement, Institute of Industrial Crops, Hubei Academy of Agricultural Sciences, Wuhan 430063, China;
- 3Hubei Hongshan Laboratory, Wuhan 430070, China;
- 4School of Breeding and Multiplication, Hainan University, Sanya 572025, China;
- 5Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Plant Germplasm Research Center, Wuhan Botanical Garden, Innovative Academy of Seed Design, Chinese Academy of Sciences, Wuhan, Hubei 430074, China
Abstract
Somatic mutations arise and accumulate during tissue culture and vegetative propagation, potentially affecting various traits in horticultural crops, but their characteristics are still unclear. Here, somatic mutations in regenerated woodland strawberry derived from tissue culture of shoot tips under different conditions and 12 cultivated strawberry individuals are analyzed by whole genome sequencing. The mutation frequency of single nucleotide variants is significantly increased with increased hormone levels or prolonged culture time in the range of 3.3 × 10−8–3.0 × 10−6 mutations per site. CG methylation shows a stable reduction (0.71%–8.03%) in regenerated plants, and hypoCG-DMRs are more heritable after sexual reproduction. A high-quality haplotype-resolved genome is assembled for the strawberry cultivar “Beni hoppe.” The 12 “Beni hoppe” individuals randomly selected from different locations show 4731–6005 mutations relative to the reference genome, and the mutation frequency varies among the subgenomes. Our study has systematically characterized the genetic and epigenetic variants in regenerated woodland strawberry plants and different individuals of the same strawberry cultivar, providing an accurate assessment of somatic mutations at the genomic scale and nucleotide resolution in plants.
Every somatic cell in both animals and plants can carry new mutations during growth in response to either inherent or environmental stresses, called somatic mutations. Most horticultural plants are propagated vegetatively over many years and can accumulate a large number of somatic mutations. For example, the strawberry cultivar “Beni hoppe” has been propagated asexually in China since its release in 1999 (Takeuchi et al. 1999; Chang et al. 2018). Its propagation relies on the daughter plants produced on runners. In addition, in vitro tissue culture technology is often used to produce virus-free plantlets or to improve propagation efficiency, which can lead to even more mutations (somaclonal mutations). The application of genome editing technologies requires tissue culture to carry out genetic manipulation in plants. During these processes, the induced mutations can provide new genetic diversity for crop breeding (Wang et al. 2021; Liu et al. 2022b), but can also cause unfavorable phenotypic variation. It is, therefore, important to systematically analyze the characteristics of somatic mutations in plants.
Somaclonal variation can often cause phenotypic variation, as demonstrated in Arabidopsis (Jiang et al. 2011). Somaclonal variation occurs at multiple levels, including cytological abnormalities, DNA sequence changes, and epigenetic alterations (Abu-Qaoud and Sami 2010). In cotton, the genome-wide analysis revealed thousands of single nucleotide variants (SNVs) and hundreds of insertions/deletions (indels) in each regenerated plantlet after transformation (Li et al. 2019). In contrast, much fewer SNVs (<200) and indels (<100) were found in rice transformants (Tang et al. 2018). When the genome size is taken into account, the mutation frequencies in cotton and rice are still quite different. Somatic mutations in plants growing under natural conditions have been relatively well analyzed at the genomic level (Ossowski et al. 2010; Wang et al. 2019; Hofmeister et al. 2020). However, a comprehensive understanding of the mutational characteristics of the regenerated plants derived from tissue culture is lacking.
DNA methylation in the context of CG, CHG, and CHH (H = A, T, or C) can affect gene expression levels and silencing of transposons or repeat sequences, etc. Methylated cytosine is also a major source of point mutations during DNA replication (Phillips et al. 1994; Jeltsch 2010; Lu et al. 2021). DNA methylation change has been identified as an important type of epigenetic alteration in tissue culture (Miguel and Marum 2011; Neelakandan and Wang 2012). A well-known example is the loss of Karma methylation caused by tissue culture, resulting in mantled floral organs in oil palm (Ong-Abdullah et al. 2015). Genome-wide increases in DNA methylation levels, particularly at CHH sites, were detected in cultured samples during somatic embryogenesis in soybean (Ji et al. 2019). In sweet orange, the global CHH methylation levels were significantly decreased at the callus induction stage and increased to higher levels after prolonged culture time, suggesting a rapid loss and gain of DNA methylation during callus induction (Wang et al. 2022). In woodland strawberry (Fragaria vesca), slight increases in CG and CHG methylation and decreases in CHH methylation were observed in callus tissue compared to leaf explants (Liu et al. 2022a). In another strawberry species Fragaria nilgerrensis, the DNA methylation levels alternately decreased and increased at six stages during the tissue culture process (Cao et al. 2021). These results suggest that DNA methylation levels are highly dynamic and show a high degree of variation in different species during tissue culture.
Cultivated strawberry (F. × ananassa), an allo-octoploid (2n = 8 ×= 56), is a perennial herbaceous fruit crop of high economic and nutritional value. Woodland strawberry is a diploid ancestor contributing to the dominant subgenome of cultivated strawberry, and is widely distributed around the world with highly divergent traits (Liston et al. 2014; Edger et al. 2019). Here, taking advantage of the simpler and more homozygous nature of the woodland strawberry genome, the inbred plants of the accession Hawaii 4 (H4) were used to simulate the tissue culture process of cultivated strawberry to reveal features of somaclonal variation. DNA mutation and methylation changes were analyzed at single base resolution in regenerated plants obtained at two hormone levels and two culture times. We chose the strawberry cultivar “Beni hoppe” to assemble a high-quality genome and then characterize the somatic variation among the propagated individuals. These data provided an accurate assessment of both in vitro and spontaneous somatic variation during vegetative propagation in strawberry.
Results
Experimental design for the production of regenerated plants from shoot tips and leaves in woodland strawberry
To study somatic mutations induced by tissue culture in strawberry, we randomly selected three initial plants (CK1–3) in the woodland strawberry accession H4 to start the culture as planned (Fig. 1A). Specifically, shoot tips from the daughter plants on runners were collected and cultured in the media with different hormone levels. The low hormone level corresponds to 0.05 mg/L NAA (1-naphthaleneacetic acid, auxin) and 0.5 mg/L 6-BA (6-benzylaminopurine, cytokinin), which is the level used for propagation in the industry. The high hormone level (0.25 mg/L NAA and 2.5 mg/L 6-BA) is 5 times higher than the low hormone level with the same auxin/cytokinin ratio. These materials were cultured for short (55 days, a common propagation time in the industry) and long (425 days) periods (Fig. 1B). The regenerated shoots were designated LS (low hormone and short period), LL (low hormone and long period), HS (high hormone and short period), and HL (high hormone and long period) for short. In addition, new shoots were regenerated from leaf-induced callus (CA) in the media containing 0.3 mg/L NAA and 3.0 mg/L 6-BA (hormone levels used for strawberry stable transformation). The regenerated shoots were then placed on the rooting medium to form plantlets, which were subsequently grown in soil for 2 weeks. These plants were quite similar in morphology (Fig. 1C). Whole genome sequencing was performed using the unfolded leaves of the following plants: three initial plants (CK1–3) as controls, six (three from CK1 and three from CK2, except LL all from CK2) individuals for each of the four types (LS, LL, HS, and HL), and three independent CA plants obtained from CK3.
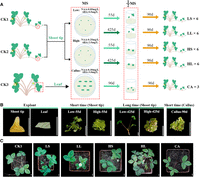
Experimental design of this study and woodland strawberry materials at different stages. (A) Experimental design for the production of regenerated plants. CK, original plant; LS, low hormone levels for 55 days; LL, low hormone levels for 425 days; HS, high hormone levels for 55 days; HL, high hormone levels for 425 days; CA, regenerated plants from callus. LS, LL, HS, and HL have six replicates each and CA has three replicates. (B) Photos showing the explants, callus, and regenerated shoots under different culture conditions. (C) Photos showing the H4 initial plant and the regenerated plants. Scale bars, 1 mm for shoot tip in (B), 1 cm for others.
More mutations are induced by higher hormone levels and longer culture time in the regenerated plants
For the 30 sequenced plants, 48.0–68.7 million clean reads were obtained with an average depth of 38.5× per plant (Supplemental Table 1). On average, 98.47% of the reads could be mapped to the woodland strawberry genome version 4 (Edger et al. 2018). During the analysis, the low-quality mutation sites and the mutation sites close to the simple sequence repeats were filtered out (see “Methods”). The mutations present in the regenerated plants (with a percentage of ≥30% reads in each site) but absent in the control plants were retained. A total of 6479 SNVs and 162 indels were obtained in the regenerated plants compared to the original plants (Supplemental Table 2). The majority (6638 out of 6641) of the mutation sites are heterozygous. To test the efficiency of the analysis, we randomly selected 52 mutations for Sanger sequencing, of which 48 were as expected, resulting in a validation rate of 92.31% (Supplemental Table 3). Accordingly, 7–655 SNVs were identified per plant, resulting in a mutation frequency of 3.3 × 10−8–3.0 × 10−6 (Fig. 2A; Supplemental Table 2). Of note, the mutation frequency in this study indicates mutations per site, except mutation rates (mutations per site per year) shown in Supplemental Figure 1. Much fewer indels were identified, i.e., 1–13 per plant (Fig. 2A; Supplemental Table 2). As the mutation frequency of indels was much lower than that of SNVs, the total mutation frequency was close to that of SNVs (3.7 × 10−8–3.1 × 10−6). For SNVs and total mutations, LS and HS samples had the lowest mutation frequencies, LL samples had higher mutation frequencies, and HL and CA samples had the highest mutation frequencies, indicating accumulative effects of culture time and hormone levels on mutations. However, the indel mutation frequencies did not change significantly across all samples. When comparing mutations per site per year, we found no significant difference between LS and HS for SNVs and total mutations, the mutation rates were significantly higher in HL than LL and highest in CA samples (Supplemental Fig. 1). When comparing mutations between individuals, a large percentage (92.04% for LS, 95.14% for HS, 98.22% for LL, 93.93% for HL, and 99.61% for CA) of the mutated SNVs and indels were present in only one sample (Fig. 2B). In most cases, there were more sample-specific SNVs than indels. These mutations were mostly evenly distributed across the chromosomes with occasional hotspots (Fig. 2C). As the mutations of each individual were pooled together for each plant type, there were fewer mutations in CA (three plants) than in HL (six plants).
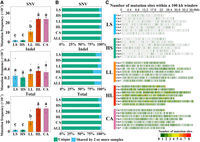
DNA mutations in different groups of regenerated woodland strawberry plants. (A) SNV, indel, and total mutation frequencies (number of mutations per site) were obtained by dividing the number of observed mutations by the number of bases covered in each sample. Data are the mean ± SEM. Different letters indicate significant differences at P < 0.05 using Tukey's test. (B) Percentage of mutations shared by the samples in each group. (ALL) all regenerated plants. (C) Distribution of total mutation sites in each group (three samples for CA and six samples for the others) in the woodland strawberry genome. The bin size is 100 kb. The color key indicates the number of mutations in each bin.
Characterization of SNVs induced by tissue culture
Since SNV is the main type of DNA sequence variation, its characteristics were further analyzed. In general, C:G to T:A was the main type of transitions (Ts), and A:T to T:A was the main type of transversions (Tv), which were more prominent in LL, HL, and CA (Fig. 3A). In total, the mutation frequency of Ts (two types) was slightly lower than that of Tv (four types) (Fig. 3B). As LL had more Ts mutations, its Ts/Tv ratio was higher than other samples (Fig. 3C). We then analyzed the frequency of the flanking bases around the SNV mutation site. Here, capital letters indicate the mutation site, small letters indicate the immediate flanking nucleotides, and the aAg type included both aAg and cTt, the same rule for the other types. Overall, when the neighboring nucleotide in the 3′ direction was t, the mutation frequency would be higher than the other three nucleotides (c, a, or g) in LL, HL, and CA (Fig. 3D; Supplemental Fig. 2A). Specifically, nAt (n indicates a, t, c, or g) had a significantly higher mutation frequency than nCt in HL (Supplemental Fig. 2B).
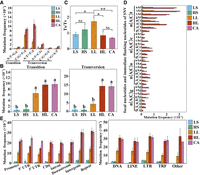
Characterization of the induced SNVs in regenerated woodland strawberry plants. (A) Mutation frequencies (number of mutations per site) of different transition or transversion types in regenerated plants. (B) Mutation frequencies (number of mutations per site) of all transitions and transversions in regenerated plants. Different letters indicate significant differences at P < 0.05 using Tukey's test. (C) Transition/transversion (Ts/Tv) ratios in regenerated plants. (*) P < 0.05, (**) P < 0.01, (NS) not significant, Student's t-test. (D) Neighbor-dependent mutation frequencies (number of mutations per site) at AT and GC bases in regenerated plants. The trinucleotide context-dependent mutation frequency is shown for each treatment. Uppercase indicates mutation site and lowercase indicates immediate flanking nucleotides and n indicates a, t, c, or g. (E) Mutation frequencies (number of mutations/total length of each region type) of SNVs in different genomic regions or transposon families. (Promoter) 2 kb upstream of transcription start site (TSS), (UTR) untranslated region, (Downstream) 2 kb downstream from transcription termination site (TTS), (Repeat) transposable element or tandem repeat sequence, (TRF) tandem repeat sequence, (Other) unclassified transposon. Data are the mean ± SEM.
We then characterized the mutation frequency (number of mutations/the total length of each region type) in different genomic regions. For protein-coding genes, the mutation frequency remained similar in different regions for each sample type, including the promoter, 5′UTR, 3′UTR, CDS, intron, and downstream regions, and was slightly higher in the intergenic and repeat sequences (Fig. 3E). Among SNVs in protein-coding regions, nonsynonymous mutations accounted for the highest percentage, followed by synonymous mutations and stop-gain mutations (Supplemental Fig. 3A). For nonsynonymous mutations, there was no significant difference in mutation frequency between neutral and deleterious mutations (Supplemental Fig. 3B). The lack of bias is likely due to the absence or minimal impact of selective pressure acting on these newly arising mutations. Similarly, the mutation frequency was also comparable for different types of transposons (Fig. 3E). These results further suggest that somaclonal variation occurred randomly in the genome.
DNA methylation changes in the HS, HL, and CA plants
DNA methylation changes are another important type of somaclonal variation that can affect gene expression and nucleotide mutation frequency. Therefore, we performed whole genome bisulfite sequencing (WGBS) to detect DNA methylation levels at the nucleotide resolution for HS (three individuals from CK1 and the other three from CK2), HL (three individuals from CK1 and the other three from CK2), and CA (three individuals from CK3) samples together with CK1–3 as controls. As a result, 54.7–86.0 million clean reads were obtained for each sample, and the bisulfite conversion rates were >99.4%, indicating high data quality (Supplemental Table 4). We found that CK1–3 showed a variation in the average DNA methylation levels, ranging from 51.32% to 56.94% for CG methylation, suggesting a flexible change for the sibling individuals (Supplemental Table 5). Compared to CK, all HL and CA samples showed a decrease in CG methylation levels from 0.71% to 8.03%, while CHG and CHH methylation was mostly downregulated but partially upregulated. Similarly, DNA methylation changes around protein-coding genes and transposons in the CHG and CHH contexts were rather random in all samples (Supplemental Figs. 4, 5). These results from multiple replicates suggest that CG methylation levels are most likely reduced in the regenerated plants, whereas CHG and CHH methylation changes are more random.
Next, the differentially methylated regions (DMRs) were identified in each regenerated plant compared to CK (change >10%, step = 200 bp, window = 200 bp). In general, there were more hypo-DMRs in the CG context but more hyper-DMRs in the CHG and CHH contexts in all samples (Supplemental Table 6). Specifically, HS samples had 2694–9943 hypoCG-DMRs, HL samples had 10,481–21,846 hypoCG-DMRs, CA samples had 15,133–24,853 hypoCG-DMRs, suggesting a significant increase caused by long-term tissue culture (Fig. 4A; Supplemental Table 6). In contrast, the increase in hyperCG-DMRs was not obvious. Furthermore, hyperCG- and hypoCG-DMRs showed obvious sample specificity, i.e., 63.2% of hypoCG-DMRs in HL and even higher in HS and CA samples were present in only one sample (Fig. 4B). Hierarchical clustering of CG-DMRs showed that HS samples were grouped together with CK plants, suggesting more similar DNA methylation profiles between them, whereas HL and CA samples were more distantly grouped, suggesting more changes occurred in these samples (Fig. 4C). Regarding the distribution of hypoCG-DMRs, there were slightly more hypoCG-DMRs in genic regions of protein-coding genes than in intergenic regions or repeat regions (Fig. 4D), and the density (number of DMRs × 200/region length) of hypoCG-DMRs showed an increase in HL and CA samples compared to HS samples. The density of hypoCG-DMRs also showed an increase in HL and CA samples in different types of transposons, to a lesser extent in DNA transposons and long terminal repeat (LTR) transposons (Fig. 4E).
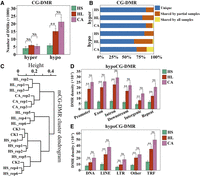
DMRs at CG contexts in regenerated woodland strawberry plants. (A) Number of hyper- and hypoCG-DMRs in HS, HL, and CA samples. (B) Percentage of unique and shared DMRs among the samples in each type. (C) Hierarchical clustering of all the examined samples based on Pearson correlation coefficient of methylation levels for CG-DMRs. (D) Density of hypoCG-DMRs in different genomic regions. (E) Density of hypoCG-DMRs in different transposon families. Data are the mean ± SEM. (**) P < 0.01, (NS) not significant, Student's t-test.
To analyze the biological process that might be affected by DNA methylation changes, we identified the genes with common hypoCG-DMRs localized in the promoters, exonic or intronic regions among HL or CA individuals. For HL, Gene Ontology (GO) enrichment analysis of the 795 common genes revealed several enriched terms, such as “transferase activity,” “transporter activity,” and “response to chemical,” etc. (Supplemental Fig. 6A). Kyoto Encyclopedia of Genes and Genomes (KEGG) functional analysis showed that these genes were enriched in pathways such as “Protein kinases,” “Plant–pathogen interaction,” “Enzymes with EC number,” and “Plant hormone signal transduction” (Supplemental Fig. 6B). For CA, the GO results of the 3307 common genes were similar to those of HL. KEGG analysis showed that these genes were also enriched in pathways such as “DNA repair and recombination proteins” and “MAPK signaling pathway” (Supplemental Fig. 6C,D).
More hypo-DMRs can be inherited by the next generation
To investigate whether variations in DNA methylation derived from tissue culture could be stably inherited by the next generation, we performed WGBS on three self-pollinated progeny of HL_rep5 (Fig. 5A; Supplemental Table 4). Data analysis revealed that 71.8% of CG-DMRs, 48.0% of CHG-DMRs, and 58.7% of CHH-DMRs were retained in at least one sample, indicating that CG-DMRs are more stable than the other two types (Fig. 5B). Furthermore, 38.3% of CG-DMRs, 16.1% of CHG-DMRs, and 14.6% of CHH-DMRs were present in all three samples, suggesting that retention is rather random. In particular, only hypoCG-DMRs had a higher percentage of retained regions, while all the other DMRs had a higher percentage of lost regions (Fig. 5C). We found that 59.9% of the retained hypoCG-DMRs were shared by the three HL_rep5 progeny (Fig. 5D; Supplemental Fig. 7). These results suggest that relatively more tissue culture-induced hypoCG-DMRs could be inherited by the next generation.
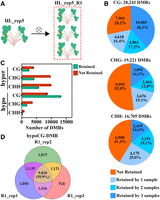
Genetic stability of DMRs in self-pollinated HL plants. (A) HL_rep5 is self-pollinated to generate three R1 plants (HL_rep5_R1). (B) Percentage of retained DMRs at CG, CHG, and CHH contexts in three R1 plants. (C) Number of DMRs at CG, CHG, and CHH contexts retained or lost in R1 plants. Data are the mean ± SEM of three R1 individuals. (D) Number of hypoCG-DMRs shared by three R1 individuals.
Haplotype-resolved genome assembly and characterization for the strawberry cultivar “Beni hoppe”
To characterize somatic mutation in cultivated strawberry, the popular cultivar “Beni hoppe” was chosen because of its widespread distribution and long cultivation history in China. To obtain a good genome reference, its genome was first assembled using the leaves of a single well-grown plant called HY_00. Genome sequencing was performed on the Pacific Biosciences (PacBio) HiFi (101×), Hi-C (202×), Illumina (103×), and ONT ultra-long (57×) platforms, yielding a total of 371.5 G-base data (Supplemental Table 7). The k-mer analysis (k = 21) revealed an estimated genome size of 725.88 Mb and a heterozygosity rate of 1.29% (Supplemental Fig. 8; Supplemental Table 8). The genome assembly yielded three reference genomes: the haploid consensus assembly FaBen (798.77 Mb, contig N50 of 28.73 Mb), the haplotype assemblies hap1 (784.96 Mb, contig N50 of 26.72 Mb), and hap2 (789.17 Mb, contig N50 of 27.91 Mb) (Supplemental Fig. 9; Supplemental Table 9). The FaBen genome has no gaps in all 28 chromosomes, 27 chromosomes consist of one contig, 22 chromosomes have telomeres at both ends (Supplemental Table 10), and all chromosomes have a predicted centromeric region (Supplemental Table 11). The chromosome IDs of FaBen and the two haplotypes were assigned by mapping to the FaRR1 genome (Supplemental Fig. 10; Hardigan et al. 2021). The LTR assembly index (LAI) values ranged from 13.28 to 20.88 for each subgenome (Supplemental Table 12). 46.39%–47.21% of the genome are repetitive sequences, of which the LTR transposons are the most abundant (Supplemental Table 13). Gene annotation revealed 115,961–116,317 gene models with BUSCO completeness scores of 98.0%–98.4% (Fig. 6A; Supplemental Fig. 11; Supplemental Table 14). Based on the gene annotation, the four subgenomes show a high degree of collinearity (Fig. 6B). These results suggest that the assembly and annotation of “Beni hoppe” are of high quality and could serve as a reference for somatic variation analysis.
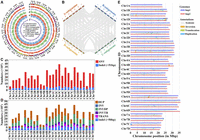
Genome assembly of “Beni hoppe” and variations between hap1 and hap2. (A) Genomic features of the “Beni hoppe” genome FaBen. All statistics were computed for windows of 100 kb. (B) Intergenomic syntenic analysis between A–D subgenomes of FaBen. (C) Number of SNVs and indels (<51 bp) in the same chromosome pair between hap1 and hap2. (D) Number of structural variations in the same chromosome pair between hap1 and hap2. (DUP) duplicated region, (INV) inverted region, (INVDP) inverted duplicated region, (INVTR) inverted translocated region, (TRANS) translocated region. (E) Structural variations in the same chromosome pair between hap1 and hap2.
Next, hap1 and hap2 were compared to determine the variation between homologous chromosomes. A total of 4,903,553 SNVs and 907,768 indels were obtained. In each chromosome, the number of SNVs was 4.04–6.29 times higher than the number of indels (Fig. 6C). Chr 1A, Chr 5A, Chr 4B, Chr 5B, and Chr 5D had fewer mutations, whereas Chr 3C, Chr 5C, and Chr 1D had more mutations. For structural variants (SVs), the duplicated (DUP, 28,089) and inverted duplicated (INVDP, 22,368) types were the most abundant, while the inverted type (INV) was the least abundant with only 271 (Fig. 6D,E). Similar to SNVs and indels, Chr 1A, Chr 5A, Chr 4B, Chr 5B, and Chr 5D showed fewer SVs, whereas Chr 6B, Chr 2C, Chr 3C, and Chr 1D showed more SVs (Fig. 6D,E). Furthermore, we found that DUP (63.74 Mb) and INVDP (42.30 Mb) types were the longest, while indels (>50 bp, 1.22 Mb) were the shortest (Supplemental Fig. 12). For all mutations, the A subgenome had fewer mutations, whereas the C subgenome had more mutations.
Somatic variants among the 12 “Beni hoppe” individuals
To identify somatic mutations arising from propagation and/or growth in cultivated strawberry, 11 “Beni hoppe” individuals (HY_01–11) randomly collected from different locations in China and HY_00 were analyzed using FaBen as the reference genome. Illumina short reads were generated using unfolded leaves with an average coverage of 30.82× for HY_01–11 (Supplemental Table 1). Due to the high ploidy and heterozygosity of the genome, a rigorous mutation site screening was performed (see “Methods”). As a result, a total of 53,106 SNVs and 11,627 indels were obtained in the 12 “Beni hoppe” individuals (Supplemental Table 2). To test the efficiency of the analysis, we randomly selected 25 mutations for Sanger sequencing, of which 22 were as expected, resulting in a validation rate of 88.0% (Supplemental Table 3). Similar to woodland strawberry, the majority (64,673 out of 64,733, 99.9%) of the mutations were heterozygous. There were on average 4426 SNVs and 969 indels per plant, resulting in a mutation frequency of 6.14–7.76 × 10−6 per site (Fig. 7A; Supplemental Table 2). The number of SNVs in each individual is very close to each other, with the largest difference being 18.56% (Supplemental Table 2). For both SNVs and indels, the A subgenome had the fewest mutations and the C subgenome had the most mutations. Of all mutations, 53.23% were specific to one individual, and 80.95% were shared by five or fewer individuals (Fig. 7B).
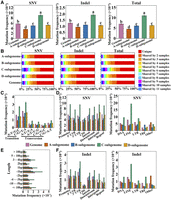
DNA mutations among the 12 “Beni hoppe” individuals. (A) SNV, indel, and total mutation frequencies (number of mutations per site) of the 12 “Beni hoppe” individuals relative to FaBen in the whole genome or each subgenome. Data are the mean ± SEM. Different letters indicate significant differences at P < 0.05 using Tukey's test. (B) Percentage of mutations shared by 12 samples in the whole genome or each subgenome. (C) Mutation frequencies (number of mutations per site) of different transitions or transversions in 12 samples. (D) Mutation frequencies (number of mutations/total length of each region type) of SNVs in different genomic regions of protein-coding genes or transposon families. (E) Mutation frequencies (number of mutations per site) of indels with different lengths. (F) Mutation frequencies (number of mutations/the total length of each region type) of indels in different genomic regions of protein-coding genes or transposon families. (Promoter) 2 kb upstream of transcription start site (TSS), (UTR) untranslated region, (Downstream) 2 kb downstream from transcription termination site (TTS), (Repeat) transposable element or tandem repeat sequence, (TRF) tandem repeat sequence; Other, unclassified transposon.
For SNVs, each of the two Ts types had approximately twofold mutations than the Tv types (Fig. 7C; Supplemental Fig. 13). The mutation frequency of SNVs was the lowest in the CDS region, which is most likely to affect gene function, and lower in TRF transposons (Fig. 7D). Most SNVs resulted in nonsynonymous or synonymous amino acids, while very few mutations caused stop codon gain or loss (Supplemental Fig. 14). For indels, 1 bp insertions or deletions were the most common types, and the mutation frequency of deletions was significantly higher than that of insertions (Fig. 7E; Supplemental Fig. 15). Similar to SNVs, there were also fewer indels in the CDS regions (Fig. 7F). In contrast, the long interspersed nuclear elements (LINE) transposons had a relatively higher mutation frequency of indels compared to other transposons (Fig. 7F). For both SNVs and indels, the C subgenome had the highest mutation frequency in most regions.
Discussion
Somatic mutations, including somaclonal variation, can lead to trait variation in plants, which is particularly important in the vegetatively propagated horticultural crops. Therefore, there is a strong need to study their characteristics. This work analyzed somatic mutations in wild and cultivated strawberries during clonal propagation at genome-wide scale and single nucleotide resolution, providing further insights into this process in strawberry and a valuable reference for other vegetatively propagated crops.
DNA mutations are increased with higher hormone levels and longer culture time in regenerated woodland strawberry plantlets
Woodland strawberry is used as a model to simulate the tissue culture process of cultivated strawberry in this study, due to its small and high-quality genome (∼220 Mb) (Edger et al. 2018). The much higher number of mutations identified in “Beni hoppe” individuals demonstrates that this strategy is effective in revealing the somaclonal mutation characteristics in strawberry (Supplemental Table 2). Hormone levels and culture time are two major factors contributing to the mutation frequency (LoSchiavo et al. 1989; Rodrigues et al. 1998; Fossi et al. 2019), but the exact range and characteristics are unclear. In this study, we set up a rigorous experimental design to evaluate the effects of hormone levels and culture time in woodland strawberry (Fig. 1). The increase in mutation frequency from LS to LL and then to HL regenerants shows that hormone levels and culture time are indeed important for mutation frequency. The total number of mutations in the HL and CA plants is about 10–20 times higher than that in the LS and HS plants, providing a range of mutation frequencies under different conditions. Our mutation rates are in the same order of magnitude as the somaclonal mutation rates (Jiang et al. 2011; Tang et al. 2018; Li et al. 2019; Wang et al. 2022), but two orders of magnitude higher than the spontaneous somatic mutation rates in other species (Ossowski et al. 2010; Yang et al. 2017; Wang et al. 2019). The possible reason for this difference is that the in vitro tissues are subjected to greater external stresses and developmental adaptations during culture. Other factors may also affect the somaclonal mutation rate, such as light intensity and temperature (Belfield et al. 2021; Lu et al. 2021), which could be tested in the same way.
In addition to the increased mutation frequency, we identified other characteristics of the mutations. First, the increase in the mutation frequency during tissue culture was mainly due to the increase in SNVs, while indels did not show any obvious changes (Fig. 2A). Second, the A:T to T:A transversion type had a comparable number to the C:G to T:A transition type (Fig. 3A), which differs from the finding that the transition types were the dominant mutations in the plants without tissue culture period (Hofmeister et al. 2020; Lu et al. 2021). Previous studies have shown that the mutation spectrum can be affected by different stresses. For example, heat stress did not alter the mutation spectrum, whereas salinity stress and fast-neutron irradiation profoundly altered the mutation spectrum (Belfield et al. 2012, 2021; Jiang et al. 2014). In contrast, Arabidopsis plants regenerated in tissue culture had a much lower Ts/Tv ratio than those from seeds (Jiang et al. 2011), highlighting the difference between somaclonal and spontaneous mutations. We speculate that the cultured tissues may induce a different mutation mechanism or have a reduced DNA repair efficiency at the A/T bases. Third, the mutation frequency in intergenic and repetitive sequence regions was slightly higher than that in genic regions (Fig. 3E), consistent with the study in rice (Tang et al. 2018). This might be due to the differences in base composition, local recombination rate, gene density, or DNA repair domain (Baer et al. 2007). Previous studies showed a much higher difference between these regions (Jiang et al. 2011; Tang et al. 2018; Hofmeister et al. 2020), which did not take into account their length. Almost all mutations are heterozygous in the first generation of regenerated plants, so most of them may not be genetically selected. The randomness of somatic mutations has been discussed recently (Monroe et al. 2022; Wang et al. 2023). Our results support the random induction theory of mutations in general.
DNA methylation change caused by tissue culture varies and can be inherited in woodland strawberry
We found that the three CK plants had quite different DNA methylation levels (Supplemental Table 5), suggesting that the DNA methylation level itself is highly dynamic even in the siblings growing under normal conditions, probably due to changes in internal or external factors (López et al. 2022; Zhang et al. 2023). This dynamic change has also been demonstrated in strawberry plants following regeneration (Cao et al. 2021), which may be due to the status of the plant rather than the regeneration process. To avoid randomness, we tested 15 individuals obtained by different methods and compared them with the CK plants, showing that the average CG methylation levels tend to be reduced, but the CHG and CHH methylation changes are more random (Supplemental Table 5). In particular, the number of CHH-DMRs in the regenerated plants is not significantly different between the HS, HL, and CA samples (Supplemental Table 6), a result similar to that found in rice (Stroud et al. 2013), probably because most of the CHH methylation changes were not stable and therefore eliminated after the formation of the regenerated plants. The HL and CA samples were obtained by different culture methods, but the mutation frequency, mutation characteristics, and DNA methylation changes were almost identical, suggesting a common mechanism underlying these processes.
The number of DMRs in the next generation showed a retention of 71.2% of CG-DMRs compared to the first generation in woodland strawberry (Fig. 5). This is much lower than that in rice, where 83.6% of hypoCG-DMRs are stably maintained over four generations (Stroud et al. 2013), but comparable to Arabidopsis with a retention of ∼76% in one generation (Jiang et al. 2014). Another finding is that the hypoCG-DMRs are more stable than other types after sexual reproduction (Fig. 5C). These results also suggest that a considerable proportion of the DMRs would be lost after a few generations. In woodland strawberry, some heat-induced DNA methylation marks can be stably transferred to the daughter plants emerging from runners, especially the early ones (López et al. 2024). This suggests that some DMRs generated in tissue culture could be maintained during asexual propagation, but may be largely lost after a few rounds of asexual production in cultivated strawberries.
DNA mutations among different individuals of the “Beni hoppe” strawberry cultivar
To study DNA mutations in cultivated strawberries, we selected a single “Beni hoppe” plant to assemble a high-quality haplotype-resolved genome (Fig. 6), comparable to recently published strawberry genomes (Hardigan et al. 2021; Lee et al. 2021; Mao et al. 2023). This reference genome is useful for the analyses of somatic variants. The 12 individuals were randomly selected from different sites provided by different vendors, so we cannot trace their ancestry. Nevertheless, it is valuable for assessing the genomic differences in different individuals of the same cultivar. Studies can be carried out in the future on the regenerated plants of this or other strawberry cultivars.
The majority of the mutations in each individual are private with a high validation rate, so the results should be reliable to reveal the bona fide mutation characteristics in cultivated strawberry. Several characteristics of somatic mutations were identified. First, most mutations are heterozygous due to asexual production. Second, the A subgenome has the lowest mutation frequency and the C subgenome has the highest mutation frequency (Fig. 7). The A subgenome is the dominant subgenome in cultivated strawberries (Edger et al. 2019), so more potentially deleterious mutations may have been purged from this subgenome. Third, the two transition types are more frequent than the transversion types (Fig. 7C), which is different from woodland strawberry but consistent with other studies (Hofmeister et al. 2020; Zheng et al. 2022). If the A:T to T:A transversion type is a signature mutation spectrum of tissue culture, the mutations in different “Beni hoppe” plants should be mainly due to somatic mutations during growth rather than tissue culture. Fourth, the mutation frequency in “Beni hoppe” individuals is much higher. These mutations could be caused by multiple rounds of tissue culture or by a long history of growth, especially under unfavorable conditions such as high temperatures or salty soil (Jiang et al. 2014; Belfield et al. 2021; Lu et al. 2021).
Methods
Plant materials and growth conditions
The woodland strawberry (F. vesca) variety H4 and the strawberry cultivar “Beni hoppe” were used in this study. The original H4 plants and regenerated plants were grown on an ∼2:1 mixture of peat soil and vermiculite under long-day conditions (16 h:8 h, light:dark) at 22°C with 55% relative humidity and a light intensity of 100 μmol m−2 sec−1. “Beni hoppe” plants were grown in the greenhouse of the Hubei Academy of Agricultural Sciences (Wuhan, China).
Tissue culture
The F. vesca H4 daughter plants were rinsed with water for 15 min, treated with 75% alcohol for 1 min and 84 disinfectant solution for 6 min, and then washed 3 times with sterile water in a sterile environment. Shoot tips of 1 mm in length were dissected with forceps and placed on MS medium (pH = 5.6) containing 0.05 mg L−1 NAA and 0.5 mg L−1 6-BA for low hormone level and 0.25 mg L−1 NAA and 2.5 mg L−1 6-BA for high hormone level. For callus cell regeneration, young leaves of sterile seedlings were cut into small strips with scissors and placed on MS medium containing 0.3 mg L−1 NAA and 3 mg L−1 6-BA to induce callus. The cultures were subcultured at 1 month intervals. These seedlings or shoots were placed in 1/2 MS for rooting for 3 months and finally planted in soil for characterization.
DNA extraction and high-throughput sequencing
A total of 3 original plants (CK), 27 regenerated plants under different conditions (LS, LL, HS, HL, CA), and 12 “Beni hoppe” plants were used. For each sample, the young leaves were collected and immediately frozen in liquid nitrogen. Genomic DNA was extracted and purified using a RaPure Plant DNA Mini Kit (Magen, Guangzhou, China). Short-read sequencing was performed on the Illumina HiSeq X Ten platform (Novogene). The PacBio library was constructed and sequenced on a PacBio Sequel II instrument (Novogene). The Hi-C library was constructed from cross-linked chromatin of plant cells using a standard protocol (DpnII enzyme) and sequenced on the Illumina HiSeq X Ten platform (Novogene). The ONT ultra-long reads were generated using the PromethION platform (Novogene).
Read alignment, variant calling, and annotation
Raw reads from the woodland strawberry plants were aligned to the Ver4 genome using BWA-MEM v0.7.17-r1188 (Li and Durbin 2009; Edger et al. 2018). Raw reads from the “Beni hoppe” plants were aligned to the FaBen genome assembled in this study. Low mapping quality reads <20 were removed, and the BAM files were sorted using SAMtools v1.9 (Li et al. 2009). Duplicate reads were removed using GATK MarkDuplicates (McKenna et al. 2010). The SNVs and indels in F. vesca were called using GATK HaplotypeCaller with the parameters “‐‐min-base-quality-score 20 ‐‐minimum-mapping-quality 20” and then filtered with the following parameters: (1) for SNVs, QD < 2.0 || FS > 60.0 || MQ < 20.0 || MQRankSum < –12.5 || ReadPosRankSum < –8.0 || -cluster 2 -window 20; (2) for indels, QD < 2.0 || FS > 200.0 || MQ < 20.0 || ReadPosRankSum < – 20.0 || -cluster 2 -window 20; and (3) mapped reads ≥8 for SNVs and ≥5 for the indels. The heterozygous sites of the control and the mutated sites near the polynucleotide sequence were further filtered out.
For “Beni hoppe” individuals, the initial candidate mutations were called using GATK HaplotypeCaller with the parameters “‐‐min-base-quality-score 30 ‐‐minimum-mapping-quality 40” and then filtered with the following parameters: (1) for SNVs, QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < –12.5 || ReadPosRankSum < –8.0 || -cluster 2 -window 20; (2) for indels, QD < 2.0 || FS > 200.0 || MQ < 40.0 || ReadPosRankSum < –20.0 || -cluster 2 -window 20; and (3) mapped reads ≥10 for SNVs and indels. To obtain somatic mutations, the BAM files of 12 individuals were simultaneously entered into the GATK HaplotypeCaller. The following criteria were used for filtering: (1) a sequencing depth of 33–300 for HY_00 and 10–90 for HY_01–11; (2) the genotypes of the 12 individuals must be 0/0, 0/1, or 1/1; and (3) the mutations that do not have any mutant reads in at least one individual compared to the reference. The variation sites between the subgenomes and the mutated sites near the polynucleotide sequence were further filtered out.
Finally, the percentage of mutant reads for each mutation should be ≥30%. The high-quality SNVs and indels were annotated using ANNOVAR software (Wang et al. 2010). The functional effects of nonsynonymous amino acids were predicted using PROVEAN software (Choi and Chan 2015) and were considered deleterious with a score of <−2.5. SNV and indel positions were extracted using BEDTools v2.29.2 (Quinlan and Hall 2010).
Whole genome bisulfite sequencing and data analysis
The 3 CK plants, 15 HS, HL, and CA plants, and 3 self-pollinated progeny of HL_rep5 were selected for WGBS. Raw reads were mapped to the F. vesca Ver4 genome using Bismark v0.22.3 (Krueger and Andrews 2011). The bismark_methylation_extractor program was used to calculate DNA methylation levels for each cytosine. Methylation levels were calculated as #C/(#C+#T). Only sites with at least fivefold coverage were included. We compared the methylation levels of regenerated plants and three self-pollinated progeny of HL_rep5 with the CK to identify DMRs using the methylKit package in R with the following settings: lo.count = 5 and hi.perc = 99.9 for site filtering; win.size = 200 and step.size = 200 for sliding windows; difference = 10 and q-value = 0.05 for differential methylation (Akalin et al. 2012). DNA methylation at the CG, CHG, and CHH contexts was evaluated separately. The DMR of HL_rep5 is considered heritable if it is present in the three self-pollinated progeny.
GO enrichment and KEGG enrichment
GO terms and KEGG terms of F. vesca proteins were annotated in the public database eggNOG v5.0.2 using eggNOG-mapper v2.1.10 (Huerta-Cepas et al. 2019). GO enrichment and KEGG enrichment analyses were performed using TBtools v2.001 (Chen et al. 2020).
Genome size estimation
K-mers with 21 bp counts were collected from the sequenced Illumina reads using jellyfish v2.2.10 (Marçais and Kingsford 2011), and genome size was estimated using GenomeScope v2.0 (Vurture et al. 2017).
Genome assembly and pseudomolecule chromosome construction
HiFi reads were used to de novo assemble the “Beni hoppe” genome using Hifiasm v0.16.1 with the parameter “-D10” (Cheng et al. 2022), and integrated with the Hi-C data to obtain haplotype-resolved assemblies (hap1.p_ctg.gfa and hap2.p_ctg.gfa) and the best contig sequence of the two sets of haplotypes (p_ctg.gfa). These contigs were anchored to 84 pseudochromosomes (28 for each of FaBen, hap1, and hap2) using ALLHiC v0.9.8 and Juicebox v1.11.08 in combination with the Hi-C reads (Durand et al. 2016; Zhang et al. 2019). Finally, the ONT ultra-long and Illumina short reads were used to fill N-gaps with TGS-GapCloser v1.1.1 (Xu et al. 2020). LTRs were predicted using LTR_FINDER v1.07 with parameters “-D 15000 -d 1000 -L 7000 -l 100 -p 20 -M 0.9” and using LTRharvest v1.6.1 with parameters “-similar 85 -vic 10 -seed 20 -seqids yes -minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1” (Xu and Wang 2007; Ellinghaus et al. 2008). The results were integrated using LTR_retriever v2.9.0 (Ou and Jiang 2018). LAI index was used to evaluate genome completeness (Ou et al. 2018). Telomeres and centromeres were identified by QuarTeT (Lin et al. 2023).
Genome annotation and evaluation
A de novo TE library was constructed for the “Beni hoppe” genomes using RepeatModeler v2.0.1. Repetitive sequences were then identified using RepeatMasker v4.0.9 with the combination of the de novo TE library and Repbase (v20181026) (Tarailo-Graovac and Chen 2009). Protein-coding genes were annotated for the repeat-softmasked genome using ab initio gene predictions, homology searches, and Iso-Seq and RNA-seq data sets. Protein sequences from F. vesca, “Royal Royce,” “Camarosa,” Arabidopsis thaliana, and other plant proteins from UniProt were aligned to the genome using exonerate v2.4.0 with the options ‐‐softmasktarget TRUE ‐‐percent 50 ‐‐minintron 20 ‐‐maxintron 60000 (Slater and Birney 2005). Iso-Seq data from the “Royal Royce” genome assembly and RNA-seq data from 67 F. × ananassa samples were downloaded (Supplemental Table 15). RNA-seq reads were mapped to the genome using STAR v2.7.10a (Dobin et al. 2013). Gene models were assembled by StringTie v2.0 with the parameter “-f 0.2” to remove weakly expressed transcripts (Pertea et al. 2015). The Iso-Seq reads, RNA-seq reads, and other protein sequences were integrated using BRAKER v2.1.6, GeneMarkS-T v5.1, and TSEBRA v1.0.2 to generate initial gene models (Tang et al. 2015; Brůna et al. 2021; Gabriel et al. 2021). Trinity v2.13.2 was used to perform genome-guided and de novo transcript assembly (Grabherr et al. 2011). Transcripts obtained from the RNA-seq reads and Iso-Seq reads were used separately to construct the gene models using PASA v2.5.2 (Haas et al. 2003). Finally, all gene models were combined using EVM v1.1.1 (Haas et al. 2008). Gene models were refined using PASA v2.5.2 by adding alternatively spliced isoforms, adding UTR annotations, and modifying gene structures. Genome completeness was assessed using BUSCO v5.2.2 with the “embryophyta_odb10_v4” database (Simão et al. 2015). Genomic features of “Beni hoppe” were plotted using Circos (Krzywinski et al. 2009). Genomic synteny was plotted using Python JCVI utilities (https://github.com/tanghaibao/jcvi).
Subgenome assignment and identification of variations between haplotypes and between subgenomes
Chromosome sequences of “Royal Royce” were mapped to those of “Beni hoppe” using NUCmer v3.1 (https://sourceforge.net/projects/mummer/) with parameters “‐‐maxmatch -l 80 -c 200” to determine each subgenome. Dot plots of the syntenic blocks were generated using mummerCoordsDotPlotly.R (https://github.com/tpoorten/dotPlotly). The variations between hap1 and hap2 were retrieved using NUCmer v3.1 with the options ‐‐maxmatch -c 100 -b 500 -l 50 and further filtered using delta-filter with the options -m -i 90 -l 100. Finally, the variants were obtained and visualized using SyRI v1.6.3 (Goel et al. 2019). The same method was used for the variation between subgenomes.
Statistical analyses
All statistical analyses were performed using the ggsignif v0.6.3 package in R (Ahlmann-Eltze and Patil 2021) and GraphPad Prism 8 software. Pairwise comparisons were determined by Student's t-test ([*] P < 0.05; [**] P < 0.01; [***] P < 0.001). Multiple comparisons were determined by one-way ANOVA combined with Tukey's test (P < 0.05).
Data access
All raw sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA1050661, PRJNA1050678, PRJNA1053423, and PRJNA1134915. Genome assembly data have been deposited at CNGB (https://db.cngb.org/cnsa/) under accession code CNP0005942. Original data on mutation sites and DMRs are available in Supplemental Data.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2018YFD1000102), the National Natural Science Foundation of China (32172539 and 32372669), the Hubei Seed Industry High-quality Development Funding Project (HBZY2023B00502), and the Fundamental Research Funds for the Central Universities (2662023PY011).
Author contributions: C.K., Y.H., and X.Z. proposed the project and designed the experiments. S.H. performed most of the experiments and data analysis. X.Z. collected the “Beni hoppe” plants. Yg.L. took photographs and manually checked the mutations. Yp.L., W-B.J., and M.Q. helped with data analysis. C.K. and S.H. drafted the manuscript. All authors participated in the discussion and revision of the manuscript. All authors read and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279378.124.
- Received March 19, 2024.
- Accepted September 16, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








