
A statistical learning method for simultaneous copy number estimation and subclone clustering with single-cell sequencing data
- 1Department of Epidemiology and Biostatistics, Arnold School of Public Health, University of South Carolina, Columbia, South Carolina 29208, USA;
- 2Department of Environmental Health Science, Arnold School of Public Health, University of South Carolina, Columbia, South Carolina 29208, USA;
- 3Department of Quantitative Sciences, Baylor College of Medicine, Houston, Texas 77030, USA;
- 4Department of Biostatistics, College of Public Health and Health Professions and College of Medicine, University of Florida, Gainesville, Florida 32603, USA
Abstract
The availability of single-cell sequencing (SCS) enables us to assess intra-tumor heterogeneity and identify cellular subclones without the confounding effect of mixed cells. Copy number aberrations (CNAs) have been commonly used to identify subclones in SCS data using various clustering methods, as cells comprising a subpopulation are found to share a genetic profile. However, currently available methods may generate spurious results (e.g., falsely identified variants) in the procedure of CNA detection, thereby diminishing the accuracy of subclone identification within a large, complex cell population. In this study, we developed a subclone clustering method based on a fused lasso model, referred to as FLCNA, which can simultaneously detect CNAs in single-cell DNA sequencing (scDNA-seq) data. Spike-in simulations were conducted to evaluate the clustering and CNA detection performance of FLCNA, benchmarking it against existing copy number estimation methods (SCOPE, HMMcopy) in combination with commonly used clustering methods. Application of FLCNA to a scDNA-seq data set of breast cancer revealed different genomic variation patterns in neoadjuvant chemotherapy-treated samples and pretreated samples. We show that FLCNA is a practical and powerful method for subclone identification and CNA detection with scDNA-seq data.
In cancer, a small population of cancer stem cells evolves into a malignant mass of tumor cells, which then diverge and form distinct subclones, contributing to intra-tumor heterogeneity (ITH). Increased ITH levels are associated with tumor progression and resistance to clinical treatments (Stanta and Bonin 2018). Intrinsic mechanistic processes, such as inherent genomic variation, clonal competition, and tumor–host interactions, also contribute to ITH (Yachida et al. 2010; Gerlinger et al. 2012; Vogelstein et al. 2013; Polyak 2014). Therefore, an accurate assessment of ITH and the identification of subclones are essential to understand the mechanisms of tumor progression and resistance to therapy (Dagogo-Jack and Shaw 2018).
Most existing studies characterize clonal diversity using bulk DNA sequencing (Oesper et al. 2013; Ha et al. 2014; Li and Li 2014; Zhang et al. 2014; Deshwar et al. 2015), which is limited to reporting only an average signal from complex populations of cells (Navin 2015). Single-cell sequencing (SCS) technology enables the assessment of ITH on a single-cell basis (Navin et al. 2011; Wang et al. 2014) using either single-cell DNA or RNA sequencing (scDNA/RNA-seq) to reveal cellular evolutionary relationships (Tang et al. 2019). Subclonal populations can be identified within tumor tissues using SCS, allowing for the inference of tumor evolution and providing insights into future developments of targeted therapy (Jiang et al. 2016).
Subpopulations of cancer cells share genetic characteristics (Cariati et al. 2019), with chromosomal copy number aberration (CNA) being one of the most important types, involving the gain or loss of DNA segments. CNAs stimulate the stemness of other tumor cells, leading to the formation of new cancer stem cells and ultimately giving rise to clonal evolution in cancers (Dai and Liu 2021). Consequently, CNAs serve as good biomarkers to assess ITH and identify subclones. Principally, the detection of CNAs has been performed by identifying changes at the boundaries of copy number regions within chromosomal segments. However, it remains challenging to detect CNAs in scDNA-seq data owing to shallow sequencing and uneven depth of coverage (Mallory et al. 2020). The method HMMcopy, which uses a hidden Markov model to detect copy number variations, was designed for data from array comparative genomic hybridization (aCGH) and high-throughput sequencing (Shah et al. 2006; Ha et al. 2012), although it has been extensively applied in SCS data (https://rdrr.io/bioc/HMMcopy/; Laks et al. 2019). A single-cell specific method, SCOPE (Wang et al. 2020), was recently developed for copy number estimation using a Poisson latent factor model for normalization and an expectation–maximization (EM) algorithm. Studies based on these methods have typically used an independent statistical model to first detect the copy number profile, followed by classical clustering methods (e.g., hierarchical) (Bridges 1966) for subclone identification in downstream analyses. However, this two-step framework risks generating spurious results owing to the carry-over of noise in the copy number profiling step into the subclone clustering.
Previously, Rojas and Wahlberg (2014) proposed a fused lasso method for general change-point detection problems; applied here, this approach converts the problems of detecting chromosomal breakpoints to a convex optimization problem whereby we propose to simultaneously identify subclones. We herein developed FLCNA, a CNA detection method based on the fused lasso model, to achieve accurate subclone clustering and copy number profiling without needing an intermediate step to estimate copy number states. Our approach not only yields more precise clustering results but also increases the accuracy of CNA detection in each individual cell by borrowing information across cells within the same subclone. This study represents the first exploration of the fused lasso model in copy number profile–based subclone clustering.
We benchmarked FLCNA against existing copy number profile detection methods (i.e., SCOPE and HMMcopy) (https://rdrr.io/bioc/HMMcopy; Wang et al. 2020), coupled with classical hierarchical (Bridges 1966) and k-means (James and others 1967) clustering methods. The performance of each approach at subclone clustering and CNA detection was evaluated through extensive simulations. We also applied FLCNA to a breast cancer data set and successfully clustered subclones based on shared breakpoints of cells in three patients. In conclusion, we have developed a robust and powerful tool for subclone clustering and CNAs detection in SCS data.
Results
Overview of the method
To capture the biological heterogeneity between potential subclones, we developed the FLCNA method based on a fused lasso model (Rojas and Wahlberg 2014), which can simultaneously identify subclones and detect breakpoints in scDNA-seq data. The framework of the FLCNA method is summarized in Figure 1. First, data sets undergo preprocessing procedures, including quality control and normalization. Subclone clustering and breakpoint detection are then achieved simultaneously using a Gaussian mixture model (GMM) combined with a fused lasso penalty term. Finally, based on the shared breakpoints in each cluster, candidate CNA segments for each cell are clustered into different CNA states using a GMM-based clustering strategy (Xiao et al. 2019).
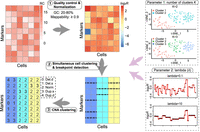
Analysis workflow of FLCNA. FLCNA first implements a quality-control procedure based on GC content and mappability. A two-step median normalization approach sequentially removes the effect of each bias. Data are then scaled by their cell-specific mean and undergo a logarithm transformation (log2R). With log2R, FLCNA clusters subclones and simultaneously detects shared breakpoints using a Gaussian mixture model with a fused lasso penalty term. Finally, based on these shared breakpoints in each cluster, segments for each cell are clustered into five different CNA states: deletion of double copies (Del.d), deletion of a single copy (Del.s), normal/diploid (Norm), duplication of a single copy (Dup.s), and duplication of double copies (Dup.d). There are two hyperparameters in the FLCNA model, the number of cell clusters K and a tuning parameter λ, which controls the number of breakpoints.
Evaluation of FLCNA via spike-in simulations
We first conducted spike-in simulations to assess the clustering performance of FLCNA compared with two other copy number estimation methods (SCOPE and HMMcopy) (https://rdrr.io/bioc/HMMcopy/; Wang et al. 2020) coupled with different clustering methods (hierarchical or k-means). Using a mixture of four different copy number states, including deletion of double copies (Del.d), deletion of a single copy (Del.s), duplication of a single copy (Dup.s), and duplication of double copies (Dup.d), FLCNA outperformed the other approaches in all scenarios having different numbers of clusters (five or three) and CNAs in a cluster (Fig. 2; Supplemental Figs. S1, S2). Specifically, with five predefined clusters and a fixed number of CNAs (i.e., 50) in each cluster, FLCNA's clustering performance incrementally improved as the within-cluster CNA sharing proportion increased. FLCNA outperformed the other methods based on adjusted Rand index (ARI) at larger CNA sharing proportions (>40%) (Fig. 2; Supplemental Table S1), although at a CNA sharing proportion of 20%, almost all methods failed to provide desirable clustering performance (ARI < 0.50).
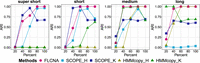
Accuracy of clustering in simulated data with five clusters and mixed CNA states. Clustering results from FLCNA were compared with existing methods (i.e., SCOPE and HMMcopy) coupled with different clustering methods. For each of five clusters, we added 50 CNA segments with varied lengths (supershort: two to five markers; short: five to 10 markers; medium: 10 to 20 markers; and long: 20 to 35 markers) and varied CNA proportions (20%, 40%, 60%, 80%, 100%), respectively, to the background signal. Signals of mixed CNA states (i.e., Del.d, Del.s, Norm, Dup.s, and Dup.d) were spiked in. (ARI) Adjusted Rand index, (SCOPE_H) SCOPE_hierarchical, (SCOPE_K) SCOPE_K-means, (HMMcopy_H) HMMcopy_hierarchical, and (HMMcopy_K) HMMcopy_K-means.
The overall clustering performance of all methods improved in scenarios with three predefined clusters (Supplemental Fig. S1; Supplemental Table S2). For example, with supershort CNAs shared by 60% of samples, the ARI of FLCNA improved from 0.781 in the scenario of five preclusters (Supplemental Table S1) to 0.985 in that of three preclusters (Supplemental Table S2). This improvement was likely because of the increased sample size in each cluster given a fixed total sample size. Compared with the scenario with a fixed number of shared CNAs in each cluster, it was more challenging for all methods to identify subclones when varied numbers of shared CNAs presented, as fewer shared CNAs were generated in some clusters by randomness (Supplemental Fig. S2; Supplemental Table S3). For samples with a single type of copy number state (i.e., Del.d, Del.s, Dup.s, Dup.d), FLCNA still outperformed other methods in clustering subclones (Supplemental Figs. S3–S5; Supplemental Tables S1–S3). All methods resulted in higher ARI for deletions compared with duplications, and for double copy changes compared with single copy, owing to the stronger signals presented in the intensities for the two former cases. FLCNA also clustered cells accurately even for simulations consisting of a single subclone (Supplemental Table S4). Overall, FLCNA had the highest accuracy for identifying subclones in simulated scenarios with commonly shared CNAs.
We also evaluated the accuracy of FLCNA in CNA detection by comparing it to SCOPE and HMMcopy (Fig. 3; Supplemental Figs. S6–S10; Supplemental Tables S5–S7). In comparison, FLCNA showed improved accuracy in identifying CNAs as the proportion of shared variants increased. For example, with a fixed number of CNAs (i.e., 50) shared by all samples in each of five clusters, FLCNA had an F1 score of 0.896, whereas SCOPE only had a score of 0.435 and HMMcopy had a score of 0.783 (Fig. 3; Supplemental Table S5). In general, FLCNA outperformed other methods in detecting supershort and short CNAs.

Accuracy of CNA detection in simulated data with five clusters. CNA calls were generated by FLCNA, SCOPE, and HMMcopy, respectively. For each of five clusters, we added 50 CNA segments with varied lengths (supershort: two to five markers; short: five to 10 markers; medium: 10 to 20 markers; and long: 20 to 35 markers) and varied CNA proportions (20%, 40%, 60%, 80%, 100%), respectively, to the background signal. Deletion of a single copy (Del.s), mixed CNA states (mix), and duplication of a single copy (Dup.s) were spiked in separately. F1 score was used to evaluate the performance of CNA detection for each method.
In summary, FLCNA showed overall great performance in clustering subclones with copy number changes shared by a large proportion of samples within a cluster. In copy number profile detection, FLCNA presented its advantage in detecting short CNAs shared by a relatively large proportion of samples within a cluster, benefiting from the information shared among cells within the same subclone.
Application to a TNBC breast cancer single-cell study
Triple-negative breast cancer (TNBC) remains a critical class of breast cancer, constituting 12%–18% of all breast cancer patients (Foulkes et al. 2010), yet owing to chemoresistance, poor overall survival performance is observed in ∼50% of TNBC patients (Foulkes et al. 2010). Neoadjuvant chemotherapy (NAC) is the standard therapy for TNBC patients who show a low level of the estrogen, progesterone, and ERBB2 (also known as HER2) receptors and are not eligible for hormone or HER2-targeted therapy (Foulkes et al. 2010). Many studies have shown that patients with TNBC harbor high levels of somatic mutations (Wang et al. 2014; Gao et al. 2016), which partially result in extensive ITH. Identifying subclones and detecting mutations in this subpopulation are critical to untangle the molecular mechanisms of chemoresistance in these patients.
To this end, we applied FLCNA to a single-cell study of three breast cancer patients receiving NAC (i.e., KTN126, KTN129, KTN302), resulting in the identification of three clusters in the KTN126 patient and two clusters in the other two patients (Fig. 4; Supplemental Figs. S11, S12). For the KTN126 patient, 64 cells (17 cells with pretreatment and 47 cells with posttreatment) were clustered in cluster A, with nine cells in cluster B and 20 cells in cluster C, all from the pretreatment group (Fig. 4). Similar copy number profile patterns were observed within clusters. Clusters B and C, which included more pretreatment samples, had higher variation in genetic intensities compared with cells in cluster A, indicating that some copy number aberrations were treatment specific. Alternatively, the treatment of NAC may have led to the extinction of tumor cells with these copy number changes in this patient. Similar patterns were observed in the other two patients (Supplemental Figs. S11, S12), and the locations of these treatment-specific copy number changes were consistent across patients KTN129 and KTN302.
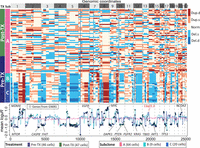
Subclone clustering of the KTN126 patient using FLCNA. Cell clusters and copy number profile with different CNA states (deletion of double copies [Del.d], deletion of a single copy [Del.s], normal/diploid [Norm], duplication of a single copy [Dup.s], and duplication of double copies [Dup.d]) were generated using FLCNA. The mean logarithm transformation of ratio between normalized read counts and its sample-specific mean (log2R) was provided for each cluster. Shared CNAs identified using FLCNA were matched to significant genes from the genome-wide association studies (GWASs) in the NHGRI-EBI GWAS Catalog. (Pre-TX) Pretreatment, (Post-TX) posttreatment.
FLCNA identified 264, 154, and 156 shared CNAs within a cluster in the KTN126, KTN129, and KTN302 patients, respectively. Consensus CNA-located genes (e.g., DAPK1, TBX3, NCOA3, KRAS) among patients implied the shared common evolutionary path of the tumor cells. Mutations in these genes have been shown to play important roles in evolution (DAPK1, TBX3) (Fischer and Pflugfelder 2015; Zhao et al. 2015) and breast cancer therapy (NCOA3, KRAS) (Burandt et al. 2013; Tokumaru et al. 2020). Overall, the shared CNAs were mapped to 436 out of 575 genes associated with breast cancer risk identified from existing genome-wide association studies (GWASs). These mapped genes were enriched in pathways related to cancer, hormones, immunity, and epithelial–mesenchymal transition (EMT) (Supplemental Fig. S13). Most pathways were shared by all three patients and were consistent with findings from existing studies on breast cancer. For instance, PATHWAYS_IN_CANCER was found to be hyperactivated in human tumor tissue (Seryakov et al. 2021). Pathways related to EMT (e.g., ADHERENS_JUNCTION) (Liu et al. 2016) and immune (e.g., TOLL_LIKE _RECEPTOR) (Shi et al. 2020) play crucial roles in the invasion and metastasis of tumor cells. Various hormone-related pathways were also essential in the occurrence and progression of breast cancer (Subramani et al. 2017). We also observed a novel CNA in 13q31.3 (e.g., LINC01040) among all three patients, with duplications in the KTN126 and KTN129 patients and deletions in the KTN302 patient. This discovery of a new CNA may provide new insights into understanding the progression of breast cancer.
We also evaluated the heterogeneity of CNAs among cells based on the sharing proportion of CNAs per cluster. The mean sharing proportion was 30.3% in the KTN126 patient (18.6% in cluster A, 36.4% in cluster B, and 34.7% in cluster C), 25.7% in the KTN129 patient, and 26.4% in the KTN302 patient (Supplemental Fig. S14). Despite the relatively low sharing percentage for some CNAs, we still observed many CNAs shared by a majority (>60%) of cells within a cluster for the TNBC data set (Supplemental Table S8). For instance, in the case of cluster C in patient KTN126, we found that 18.18% of CNAs were shared by >60% of the cells. Consequently, the priority of our method in data with a large proportion of shared CNAs within clusters will greatly benefit the subclone clustering and CNAs detection in real SCS data. Overall, our method detected CNAs with a wide range of length, varying from two to 1000 bins (Supplemental Fig. S15). In terms of computational speed, on the 93 cells from the KTN126 patient (Supplemental Table S9), FLCNA was significantly faster (1.2 h) than SCOPE (10.5 h) using a high-performance cluster with 12 GB RAM.
Discussion
The importance of copy number changes in modulating human disease is being increasingly recognized, especially concerning response to treatments in cancer. scDNA-seq enables researchers to profile heterogenous tumor tissues at the single-cell level. Accurate detection of CNAs with scDNA-seq data is crucial for identifying copy number profiles at the single-cell resolution to better understand how tumor lineages evolve (Baslan and Hicks 2017; Ren et al. 2018). Numerous scDNA-seq studies (Gao et al. 2016; Hou et al. 2016; Ferronika et al. 2017) have used CNAs to characterize tumor subclones, revealing that most tumors contain multiple subclonal lineages. In our study, we developed FLCNA for simultaneous subclone clustering and copy number estimation based on the fused lasso model. Our method effectively addresses the issue of reduced clustering accuracy caused by false-positive CNAs to which existing two-step methods are susceptible (https://rdrr.io/bioc/HMMcopy/; Wang et al. 2020).
Recent advances in SCS technology have provided emerging tools for computational methods to infer subclones with different genomic variants, including single-nucleotide alterations (SNAs) and CNAs. SAPPH (Xia et al. 2015) estimated CNAs and inferred tumor subclone proportion from paired tumor-normal data. The SCClone (Yu et al. 2022) and SiCloneFit (Zafar et al. 2019) methods clustered subclones using single-cell SNA data to reconstruct the clonal populations and the evolutionary relationship between the clones. Elyanow et al. (2021) overcame the challenges in inferring CNAs from RNA sequencing data by using spatial information of a small group of cells to help identify CNAs and the spatial distribution of clones within a tumor sample. The CONET (Markowska et al. 2022) and SCICoNE (Kuipers et al. 2020) methods were both developed to jointly infer an evolutionary tree on copy number events and copy number profile using scDNA-seq data. A major distinguishing feature of our method is its simultaneous clustering of cells and detection of CNAs, solving the problem of declined clustering performance resulting from falsely identified variants in the stage of variant estimation.
The superior clustering performance of FLCNA for scDNA-seq data has been shown in extensive simulations. Our spike-in simulations provided accurate clustering results from FLCNA for the samples with a large proportion of shared CNAs. Moreover, FLCNA outperformed the other two methods in detecting supershort and short CNAs. Notably, identifying supershort and short CNAs is typically more challenging compared with longer-sized CNAs. Using a whole-genome scDNA-seq data set on breast cancer patients, FLCNA successfully clustered subclones containing clearly distinct genomic variation patterns. Our analysis suggests that treatment of NAC may lead to the extinction of tumor cells, despite the possible bias brought by the fact that partial posttreatment samples were collected adjacent to normal tissues. With our method, more CNAs were identified in the previously defined “normal” cells than the original study of this data set (Kim et al. 2018), as well as the SCOPE reanalysis (Wang et al. 2020). In contrast to our approach, these two previous studies predefined “normal” cells as references to calculate signal intensities for change-point detection. FLCNA uses the observed mean value of each cell across bins as a reference, providing increased sensitivity at detecting CNA signals. An advantage of our reference-free normalization method is that it avoids relying on the availability of normal cells, which not all studies contain or which can be mislabeled in cell-type annotation. In addition to prediction accuracy, the high dimensionality of scDNA-seq data sets requires high computational efficiency. Even with 100 kb as the binning size, our method retained computational speed, without compromising the accurate detection of short CNAs.
Like any other studies, there are limitations in our study. First, only shared breakpoints identified in a cluster were used to estimate the underlying copy number profile for each cell, which may not be desirable if the goal is to identify all CNAs at the single-cell level. Additionally, errors owing to doublets in SCS data, in which two or more cells have the same cell barcode label (Zaccaria and Raphael 2021), were not considered in our framework. Although previous studies have shown that doublets can affect the accuracy of copy number estimation (Zaccaria and Raphael 2021), because we only used shared breakpoints for CNA detection, we expect the bias arising from the existence of doublets will be minimal. Additionally, although our method is computationally efficient for a single value of parameters K and λ, selecting the most optimal choice among many candidate value combinations may be computationally intensive.
In conclusion, our FLCNA method offers a novel methodology approach to CNA detection and subclone identification in SCS data. Our method has the potential to extend to other data types, such as scRNA-seq, and to integrate different dimensions of sequencing information (e.g., B-allele frequency, gene expression, spatial information) to improve the identification of subclones and benefit research in cancer outcome-related targeted therapy. For example, we could incorporate spatial information into our model for more comprehensive inference of subclones with scDNA-seq data, as cells located nearby are more likely to share similar genetic patterns and consequently tend to reside within same subclones (Elyanow et al. 2021).
Methods
FLCNA was developed based on a fused lasso model (Rojas and Wahlberg 2014) to cluster subclones and simultaneously detect CNAs with scDNA-seq data. Note that although our framework was motivated by the detection of somatic copy number change, it can also be naturally extended to germline copy number variation detection. For consistency, our framework and evaluation will use the term CNA in describing and evaluating the methods across the paper.
Quality control and preprocessing of scDNA-seq data
In the first step of FLCNA, the read count data for each marker/bin and each cell undergo a binning strategy (Wang et al. 2020), which divides the genome into equally sized windows, and each window is defined as a marker. These data were subsequently
used as input in our model. Next, the GC content and mappability are generated for each marker. To reduce artifacts for copy
number detection, a quality-control procedure removes markers with extreme GC content (<20% and >80%) or those with low mappability
(<0.9) (Wang et al. 2020). Then a two-step median normalization approach (Magi et al. 2013) is used to further remove the effect of biases from the GC content and mappability. Altogether, the normalized read counts
are as follows: 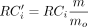 , where RCi are the read counts for marker i, mo is the median read counts of all markers with the same o value (where o = [the GC-content, mappability]) as the ith marker, and m is the overall median read counts of all the markers. In the final preprocessing step, FLCNA computes the ratio of the normalized
read counts to their sample-specific mean, the logarithm transformation of which (i.e., log2R) represents the main signal intensities.
, where RCi are the read counts for marker i, mo is the median read counts of all markers with the same o value (where o = [the GC-content, mappability]) as the ith marker, and m is the overall median read counts of all the markers. In the final preprocessing step, FLCNA computes the ratio of the normalized
read counts to their sample-specific mean, the logarithm transformation of which (i.e., log2R) represents the main signal intensities.
Notations and models
Considering we have N cells in total, let X = (xi, j)P×N be the normalized read count data (i.e., log2R), where xi, j denotes the value of the ith (i = 1, …, P) marker from the jth cell. xj = (x1, j, …, xP, j)T is the data vector for the jth sample. The samples sharing common biological characteristics (e.g., genomic variation) belong to the same cluster. Starting
from a general K-cluster problem with a GMM, the observations xj are assumed to be independent and are generated from a probability density function 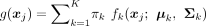 , where the “weights”
, where the “weights” 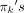 (πk ≥ 0 for each cluster, 1 ≤ k ≤ K and
(πk ≥ 0 for each cluster, 1 ≤ k ≤ K and 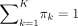 ) are the mixing proportions. For the kth cluster, fk(xj; μk, Σk) denotes the Gaussian density function with mean vector μk = (μ1, k…μP, k)T and covariance matrix Σk. μk denotes the mean genetic intensity of each marker in the kth cluster, and Σk captures the correlations among the markers. In this study, we assume that the covariance matrix
) are the mixing proportions. For the kth cluster, fk(xj; μk, Σk) denotes the Gaussian density function with mean vector μk = (μ1, k…μP, k)T and covariance matrix Σk. μk denotes the mean genetic intensity of each marker in the kth cluster, and Σk captures the correlations among the markers. In this study, we assume that the covariance matrix 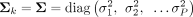 is fixed for all clusters. Parameters including πk, μk, and Σ are unknown and need to be estimated.
is fixed for all clusters. Parameters including πk, μk, and Σ are unknown and need to be estimated.
The problem of copy number profile detection is equivalent to chromosomal breakpoint detection and has been initially explored
in the fused lasso model (Rojas and Wahlberg 2014). Fused lasso was extended from the classical LASSO model to select variables and penalize the difference of successive features.
Its ability in identifying and quantifying significant features is closely related to our problem of breakpoint detection
on locating significant signals from a wide range of constant signals (Rojas and Wahlberg 2014). Using the fused lasso penalty term for change point detection, the penalized log likelihood function in the FLCNA method
is given by
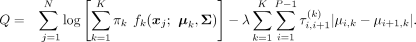 (1)
In the second term of Equation 1, a tuning hyperparameter λ and predefined adaptive weights (Zou 2006)
(1)
In the second term of Equation 1, a tuning hyperparameter λ and predefined adaptive weights (Zou 2006) 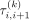 are used to shrink the absolute difference of the mean shift values |μi,k − μi+1,k| in consecutive markers in the kth cluster, ultimately disclosing change points. The tuning hyperparameter λ is used to control the overall number of change points such that fewer change points tend to be generated with larger λ values (Fig. 1). To shrink each pair of consecutive markers with the same weight, the tuning hyperparameter λ is fixed within each cluster; however, such a strategy may decrease the accuracy of penalization and ultimately affect the
clustering performance. To improve the accuracy, an adaptive penalization weight (Zou 2006)
are used to shrink the absolute difference of the mean shift values |μi,k − μi+1,k| in consecutive markers in the kth cluster, ultimately disclosing change points. The tuning hyperparameter λ is used to control the overall number of change points such that fewer change points tend to be generated with larger λ values (Fig. 1). To shrink each pair of consecutive markers with the same weight, the tuning hyperparameter λ is fixed within each cluster; however, such a strategy may decrease the accuracy of penalization and ultimately affect the
clustering performance. To improve the accuracy, an adaptive penalization weight (Zou 2006) 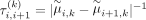 is applied, where
is applied, where 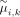 is estimated from the same model without any penalization (λ = 0). This adaptive penalization weight is predefined to dynamically penalize each pair of successive markers. For example,
if there is large difference between
is estimated from the same model without any penalization (λ = 0). This adaptive penalization weight is predefined to dynamically penalize each pair of successive markers. For example,
if there is large difference between 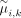 and
and 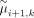 in the model without penalization, a change point is expected to appear between the ith and (i + 1)th marker, and this change point tends to be informative for subclone clustering. In this case, according to
in the model without penalization, a change point is expected to appear between the ith and (i + 1)th marker, and this change point tends to be informative for subclone clustering. In this case, according to 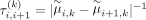 ,
, 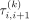 will be small and consequently, the difference between μi,k and μi+1,k in Equation 1 will be lightly penalized and will be more informative for the subclone clustering. Otherwise, the difference between μi,k and μi+1,k will be heavily penalized and will be less informative for the subclone clustering in our FLCNA method. With the focus on
subclone identification and change point detection, our goal is to maximize Equation 1 to estimate the parameter set
will be small and consequently, the difference between μi,k and μi+1,k in Equation 1 will be lightly penalized and will be more informative for the subclone clustering. Otherwise, the difference between μi,k and μi+1,k will be heavily penalized and will be less informative for the subclone clustering in our FLCNA method. With the focus on
subclone identification and change point detection, our goal is to maximize Equation 1 to estimate the parameter set 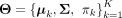 .
.
Parameter estimation using EM algorithm
In FLCNA, the parameter set Θ is estimated using the EM algorithm (Dempster et al. 1977). We initialize Θ with parameters estimated from the model without penalty (λ = 0) and then update these parameters by alternating between the E- and M-steps. In the M-step, given the starting values
of Θ, the probability for the jth sample belonging to the kth cluster is calculated by dividing the density of the kth cluster by the sum of densities from all clusters. Thereafter, an E-step is used to update the estimated values of Θ. Specifically, the “weight” for the kth cluster πk and the variance for the ith marker 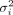 are estimated by taking the first derivative of Q(Θ) w.r.t. πk and
are estimated by taking the first derivative of Q(Θ) w.r.t. πk and 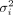 , respectively. The estimates of the cluster means
, respectively. The estimates of the cluster means 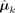 are computed with a local quadratic approximation algorithm (Fan and Li 2001). These updated Θ estimation values were iteratively computed between the E- and M-steps until convergence. We refer readers to the Supplemental Methods for a thorough description of this part of algorithm. After this step, all the parameters of interest
are computed with a local quadratic approximation algorithm (Fan and Li 2001). These updated Θ estimation values were iteratively computed between the E- and M-steps until convergence. We refer readers to the Supplemental Methods for a thorough description of this part of algorithm. After this step, all the parameters of interest 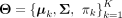 are successfully estimated, based on which cells are clustered according to the estimated cluster weights and copy number
states are assigned by the estimated cluster means (described below).
are successfully estimated, based on which cells are clustered according to the estimated cluster weights and copy number
states are assigned by the estimated cluster means (described below).
Copy number profile identification and hyperparameter estimation
With the estimated cluster means (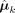 ), we locate and quantify all the shared change points and identify copy number segments within clusters. Based on these identified
shared segments, we assign the most likely copy number state for each segment in each cell. A GMM-based clustering strategy
(Xiao et al. 2019) is implemented for CNA clustering using the normalized read count data (i.e., log2R). Segments sharing similar intensity levels in a cell are identified as the ones with the same copy number states. Each segment
is classified using a five-state classification scheme with Del.d, Del.s, normal/diploid, Dup.s, and Dup.d. We aggregate the
states with more than four copies in the copy number state of Dup.d, considering our primary focus is on the clustering of
cell subclones. Two hyperparameters will be predefined, including the number of clusters K and the tuning parameter λ. To find the optimal values of K and λ, we use a Bayesian information criterion (BIC) (Guo et al. 2010), and the clustering model with the smallest BIC value is selected as the optimal model.
), we locate and quantify all the shared change points and identify copy number segments within clusters. Based on these identified
shared segments, we assign the most likely copy number state for each segment in each cell. A GMM-based clustering strategy
(Xiao et al. 2019) is implemented for CNA clustering using the normalized read count data (i.e., log2R). Segments sharing similar intensity levels in a cell are identified as the ones with the same copy number states. Each segment
is classified using a five-state classification scheme with Del.d, Del.s, normal/diploid, Dup.s, and Dup.d. We aggregate the
states with more than four copies in the copy number state of Dup.d, considering our primary focus is on the clustering of
cell subclones. Two hyperparameters will be predefined, including the number of clusters K and the tuning parameter λ. To find the optimal values of K and λ, we use a Bayesian information criterion (BIC) (Guo et al. 2010), and the clustering model with the smallest BIC value is selected as the optimal model.
Data description
We used two publicly available scDNA-seq data sets for illustration and evaluation of FLCNA in simulations and real data analyses. As described below, the BRCA5 data set was used to mimic real data signals in simulations, and the TNBC data set was analyzed for real data applications.
The BRCA5 data set consists of 10,088 cells from an aggregate of five breast tissue nuclei sections in frozen breast tumor tissue. Cells were sequenced using the 10x Genomics platform (https://www.10xgenomics.com/resources/datasets/aggregate-of-breast-tissue-nuclei-sections-10-k-cells-1-standard-1-1-0), which uses microfluidic droplets to barcode cells and perform library construction. We generated a read depth matrix of 28,760 markers and 10,088 cells from BAM files after binning with a 100-kb bin size (Wang et al. 2020). To save computational time, we randomly selected 220 cells from the data set and used the read counts from the entire genome to mimic real data in our simulations. The TNBC data set consists of data from TNBC patients obtained from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra; accession no. SRP114962) (Kim et al. 2018). TNBC is characterized by extensive ITH and frequently develops resistance to NAC treatment. Three patients (i.e., KTN126, KTN129, KTN302) were used for our analyses, in which tumor cells were only reported in the pretreatment samples. For each patient, cells were sequenced at two time points (pre- and mid/posttreatment) with 93 cells (46 pre- and 47 posttreatment) in the KTN126 patient, 90 cells (46 pre- and 44 posttreatment) in the KTN129 patient, and 92 cells (47 pre- and 45 midtreatment) in the KTN302 patient, respectively. For these samples, FASTQ files were generated with Fastq-dump from SRA-Toolkit (Leinonen et al. 2011) and then aligned to the NCBI hg19 reference genome and converted to BAM files. The raw read depth of the coverage data was generated from the BAM files with a bin size of 100 kb (Wang et al. 2020). We used the hg19 reference genome for alignment in our study because both the original study of this data set (Kim et al. 2018) and the SCOPE paper (Wang et al. 2020) used the same reference genome. Theoretically, our method does not rely on the version of reference genome and can also be applied to data generated with other genome assemblies.
Spike-in simulations
We compared FLCNA to existing copy number profile detection methods, including SCOPE (Wang et al. 2020) and HMMcopy (https://rdrr.io/bioc/HMMcopy/), using spike-in simulations. Because these two methods were only designed for the detection of CNAs without cell clustering,
they were followed by two commonly used clustering methods, hierarchical (Bridges 1966) and k-means (James and others 1967). The simulation mimicked a scDNA-seq data set of frozen breast tissue, the BRCA5 data set (data description), by randomly
selecting 220 cells from the data set and using SCOPE and HMMcopy to remove genetic regions with putative CNAs. Genetic regions
with copy number changes detected by either method were excluded from the analysis, and the remaining sequences were treated
as copy number–free sequences. Among these cells, 20 cells were randomly selected as reference cells for the SCOPE method,
and signals of spiked-in CNAs were added to the remaining cells. To evaluate the robustness of FLCNA, we randomly generated
CNAs of varied sizes (supershort: two to five markers; short: five to 10 markers; medium: 10 to 20 markers; long: 20 to 35
markers) and simulated different numbers of clusters (three or five). We evaluated varied copy number states, including Del.d,
Del.s, Dup.s, and Dup.d, respectively. Moreover, data with a mixture of the above four different copy number states were generated
to mimic real-world data. Because a CNA may not be shared by all the cells in a cluster, different CNA sharing proportions
(20%, 40%, 60%, 80%, 100%) were considered. For each cluster, we added 50 CNA segments to the background sequences. Additionally,
we evaluated the performance on the scenarios with different numbers of CNAs among clusters by assigning random numbers of
CNAs (20 to 80) to each cluster. For each of these scenarios, signals were spiked in by multiplying the background depth of
coverage by c/2, where c is a normal random variable following N(0.4, 0.12) for Del.d, N(1.2, 0.12) for Del.s, N(2.8, 0.12) for Dup.s, and N(4.2, 0.12) for Dup.d (Wang et al. 2020). The ARI (Vinh et al. 2009) was calculated to evaluate the clustering performance of these methods by comparing the identified clusters of each method
to the predefined “true” classes. An ARI near one indicates that the clusters identified are completely consistent with the
ground truth, whereas values closer to zero indicate random clustering. The performance of can detection for these methods
was assessed using the F1 score 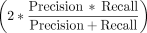 , with precision rate defined as
, with precision rate defined as 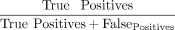 and recall rate defined as
and recall rate defined as 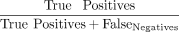 . Notably, for a CNA to be classified as a true-positive call, it must intersect with regions of “true” CNAs and show copy
number states that align with the predefined “true” states.
. Notably, for a CNA to be classified as a true-positive call, it must intersect with regions of “true” CNAs and show copy
number states that align with the predefined “true” states.
FLCNA application to the TNBC data set for breast cancer subcloning
With the data set introduced previously, FLCNA was also applied to the TNBC data set of breast cancer with three unrelated patients (i.e., KTN126, KTN129, KTN302) who have been treated with NAC. For the HMMcopy and SCOPE methods, we adhered to their default parameter settings. Three candidate numbers of clusters K (i.e., two, three, four) and the default value for the tuning hyperparameter λ were used for the FLCNA method. We identified shared CNAs using FLCNA and mapped them to 575 significant genes from GWASs with breast cancer curated in the NHGRI-EBI GWAS Catalog (Sollis et al. 2023). Pathway and network analyses were conducted for these genes; gene set enrichment analysis (GSEA) (Subramanian et al. 2005) was conducted with enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2016). Further, the summary statistics from GSEA were used to generate connection networks for these three patients using the enrichment map implemented in Cytoscape (Shannon et al. 2003).
Software availability
FLCNA is available as an open-source tool at GitHub (https://github.com/FeifeiXiao-lab/FLCNA) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the U.S. National Institutes of Health grant R21 HG010925 (F.X.).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278098.123.
- Received May 15, 2023.
- Accepted January 8, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








