
High-throughput and genome-scale targeted mutagenesis using CRISPR in a nonmodel multicellular organism, Bombyx mori
- Sanyuan Ma1,
- Tong Zhang1,
- Ruolin Wang1,
- Pan Wang1,
- Yue Liu1,2,
- Jiasong Chang1,3,
- Aoming Wang1,
- Xinhui Lan1,
- Le Sun1,
- Hao Sun1,
- Run Shi1,
- Wei Lu1,
- Dan Liu1,
- Na Zhang1,
- Wenbo Hu1,
- Xiaogang Wang1,4,
- Weiqing Xing1,
- Ling Jia1 and
- Qingyou Xia1
- 1Integrative Science Center of Germplasm Creation in Western China (CHONGQING) Science City, Biological Science Research Center, Southwest University, Chongqing 400716, China;
- 2Medical Center of Hematology, Xinqiao Hospital, Army Medical University, Chongqing, 400037, China;
- 3Key Laboratory of Cellular Physiology, Ministry of Education, Shanxi Medical University, Taiyuan, 030001, China;
- 4China Chongqing Key Laboratory of Chinese Medicine & Health Science, Chongqing Academy of Chinese Materia Medica, Chongqing 400065, China
Abstract
Large-scale genetic mutant libraries are powerful approaches to interrogating genotype–phenotype correlations and identifying genes responsible for certain environmental stimuli, both of which are the central goal of life science study. We produced the first large-scale CRISPR-Cas9-induced library in a nonmodel multicellular organism, Bombyx mori. We developed a piggyBac-delivered binary genome editing strategy, which can simultaneously meet the requirements of mixed microinjection, efficient multipurpose genetic operation, and preservation of growth-defect lines. We constructed a single-guide RNA (sgRNA) plasmid library containing 92,917 sgRNAs targeting promoters and exons of 14,645 protein-coding genes, established 1726 transgenic sgRNA lines following microinjection of 66,650 embryos, and generated 300 mutant lines with diverse phenotypic changes. Phenomic characterization of mutant lines identified a large set of genes responsible for visual phenotypic or economically valuable trait changes. Next, we performed pooled context-specific positive screens for tolerance to environmental pollutant cadmium exposure, and identified KWMTBOMO12902 as a strong candidate gene for breeding applications in sericulture industry. Collectively, our results provide a novel and versatile approach for functional B. mori genomics, as well as a powerful resource for identifying the potential of key candidate genes for improving various economic traits. This study also shows the effectiveness, practicality, and convenience of large-scale mutant libraries in other nonmodel organisms.
In the past centuries, the widely application of model organisms has helped to reveal the basics of molecular biology. However, some current model organisms are too simple and thus cannot answer more complex questions nor explain some specific phenomena (Fields and Johnston 2005). With the transition in biology from descriptive to mechanistic understanding, especially with the completion of genome sequencing of massive organisms, more nonmodel systems are emerging for tackling questions across the whole spectrum of biology or for exploiting the unique biological features of a special organism to address questions of general importance (Goldstein and King 2016). For example, axolotl (Echeverri et al. 2022), killifish (Beck et al. 2022), volvox (Umen and Herron 2021), ashbya (Wendland and Walther 2005), and diatoms (Jang et al. 2013) have been used to investigate regeneration, aging, evolution, phase transition, and nanobiotechnology, respectively. The silkworm, Bombyx mori, is another example, with advantages in showing the biology of Lepidoptera insects, which comprise the vast majority of agricultural and forest pests (Xia et al. 2014; Meng et al. 2017a,b). B. mori is also an economically important insect, with sericulture forming a pillar industry in the rural economy of many developing countries.
Currently, a major gap in methodologies and resources between the selected few models and other organisms greatly hinder researchers in choosing and retaining the use of nonmodel organisms (Russell et al. 2017). In B. mori, only a few hundred mutant lines with limited genetic background information have been preserved (Goldsmith et al. 2005; Tong et al. 2022). Functional genomic approaches are lagging, with the first transgenic silkworm obtained in 2000 (Tamura et al. 2000), 17 years after the first transgenic plant (Herrera-Estrella et al. 1983). Widely used RNAi technology has been shown to be extremely inefficient in B. mori and many other lepidoptera insects (Terenius et al. 2011; Kolliopoulou and Swevers 2014).
The emergence of CRISPR-Cas9 has expanded genetic toolboxes in a wide range of systems, including B. mori (Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013; Ma et al. 2019). Its simplicity and modularity have led to the successful construction of large CRISPR libraries for genome-wide functional genomics and drug target discoveries in mammalian cell systems (Shalem et al. 2014; Wang et al. 2014; Qi et al. 2017; Ford et al. 2019). Large-scale mutant libraries have also been constructed for four model organisms: rice (Oryza sativa L.) (Lu et al. 2017; Meng et al. 2017a,b), rapeseed (Brassica napus) (He et al. 2023), zebrafish (Danio rerio) (Sun et al. 2020), and fruit fly (Drosophila melanogaster) (Meltzer et al. 2019; Port et al. 2020; Zirin et al. 2020). CRISPR-based mutants have simple and clear genotype–phenotype correlations, and mutant libraries provide better opportunities to interrogate gene functions than previous libraries, such as those generated through radiation mutagenesis, chemical mutagenesis, random transgenic insertion, or transgenic RNAi (Venken et al. 2014).
We established a genome-wide CRISPR screen platform in cultured B. mori cells and showed its simplicity and efficiency in identifying genes essential for cell viability under normal conditions (Chang et al. 2020a), as well as for screening resistance genes for both biotic and nonbiotic stresses (Liu et al. 2021). However, the application of such powerful genome-scale mutagenesis systems at the whole-animal level in nonmodel organisms has been unsuccessful to date. Here, we report the construction of a high-throughput CRISPR-Cas9 mutant library for B. mori, a useful resource for silkworm research and breeding. We show its application for gene function interrogation, as well as its potential use for genetic improvement of economic traits. In silkworm, the most efficient germline transformation platform is piggyBac system, which has been proved and widely used for over 20 years (Ma et al. 2014a; Chang et al. 2020a). piggyBac transposon can drive gene delivery in a wide range of eukaryotes, so our strategy and results also lay the foundation for applying genome-scale mutagenesis to other nonmodel organisms.
Results
Design and construction of the CRISPR single-guide RNA library
Based on our previously reported, highly efficient, CRISPR-Cas9-mediated B. mori targeted mutagenesis in the whole animal and the pooled library in cultured cells (Ma et al. 2014a; Chang et al. 2020a), we sought to explore the ability of the CRISPR system to generate a library of targeted loss-of-function mutants at the whole-animal level. In B. mori, genetic transformation is primarily achieved through embryonic microinjection (Ma et al. 2014b). This process is extremely labor intensive and equipment dependent and requires highly skilled technicians; thus, to maximize the efficiency, we designed a high-input/low-output pooled strategy (Fig. 1A). As in the pooled strategy, the target gene of an individual mutant is indicated by the integrated single-guide RNA (sgRNA) sequence itself, we used a binary CRISPR-Cas9 system (Xu et al. 2017) to avoid misediting of nontarget genes during the early stage of injection. This system expressed Cas9 protein and sgRNA in two separate transgenic lines that use the ubiquitous IE2 and eye-specific 3XP3 promoter-driven EGFP as transformation markers, respectively (Ma et al. 2014a; Xu et al. 2017), and knockout was achieved by crossing them (Fig. 1A).
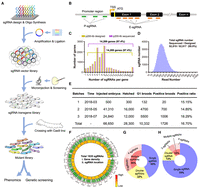
Design and construction of the CRISPR single-guide RNA (sgRNA) library. (A) The strategy of the mutant library construction. (B) The schematic diagram of sgRNA design that targets the promoter region and exon around whole genome of B. mori. The sgRNAs targeting the promoter region were named by P-sgRNA, whereas sgRNAs targeting the exon1 or exon2 were named by E-sgRNA. (C) The sgRNA distribution for the p200-lib of designed and deep sequenced. (D) Distribution of the deep sequenced reads and sgRNA numbers. (E) Summary of microinjection information for B. mori. (F) Distribution of sgRNAs detected by deep sequencing over the B. mori whole genome. (G,H) The number of sgRNAs detected in individuals.
To construct the mutant library, we designed a strategy shown in Figure 1A. The whole genome of B. mori was searched for all exons and ∼500 bp upstream of transcription starting site (Fig. 1B), and 3,489,264 sgRNAs were found (Supplemental Fig. S1). sgRNAs targeting exons were ranked using criteria described previously (Chang et al. 2020a). sgRNAs targeting promoters were ranked using an additional criterion; only sgRNAs targeting the sense strand were selected owing to previous findings that the DSBs were repaired in a direction-biased manner (Chang et al. 2020b; Ma et al. 2021). A total of 92,917 sgRNAs were chosen (about eight E-sgRNAs and about two P-sgRNAs per gene) and synthesized on a microarray chip (Supplemental Table S1). These sgRNAs are designed to target 14,645 genes, and the distribution of these targeted genes across the genome is random (Supplemental Fig. S1), with these genes accounting for ∼86.76% of all genes in the genome. The rest of the genes, 13.24%, was not included because we failed to design proper sgRNAs. The synthesized sgRNA oligonucleotide pool was elongated by polymerase chain reaction (PCR) and ligated into p200 to form a plasmid library (p200-lib). Deep sequencing of the library showed that 99.9% of the designed sgRNAs were successfully cloned; >97.4% of the genes had more than one sgRNA (Fig. 1C), and 99.9% of sgRNAs had 36–4109 reads (Fig. 1D). These results indicate sufficient coverage, accuracy, and diversity of p200-lib for subsequent experiments.
Pooled microinjection and generation of large-scale sgRNA transgenic lines
To test the usability and efficiency, a small fraction of p200-lib was coinjected with a piggyBac transposase expression vector (A3-Helper) into 500 G0 embryos. Approximately 300 embryos hatched and were reared to adulthood. Twenty broods positive for 3Xp3-EGFP expression were obtained from 132 G1 broods. The transformation rate was 15.15% (Fig. 1E), which was the average of our previous individual transgenic experiments (Ma et al. 2013). We crossed the 20 transgenic lines with the Cas9-expressing transgenic line (Fig. 1A), and observed clear phenotypic changes in the progenies of the two lines among the 20 F1 lines (Supplemental Fig. S2). We tested the sgRNA of the two lines through simple genomic PCR and found that both phenotypes were theoretically related to their predicted functions (Paine et al. 2019; Schimmel et al. 2021). The target regions of Peanuts (KWMTBOMO05973) and DNA primase large subunit (KWMTBOMO01805) were amplified and subjected to deep sequencing. Sequencing results showed that 46% of 12,149 reads and 82% of 22,422 reads were edited (Supplemental Fig. S2). We concluded that the strategy is suitable for the construction of a genome-wide mutant library, and the established p200-lib plasmid library is suitable for efficient large-scale germline transformation.
Next, two batches of large-scale microinjections were performed. A total of 66,150 embryos were injected, and 28,000 injected embryos were hatched and were reared to adult G0 moths. By large-scale fluorescent screening the obtained 10,200 G1 broods (about 2,000,000 embryos), 1706 3XP3-EGFP-positive broods, containing one to 100 positive transgenic individuals per brood, were obtained (Fig. 1E). Average hatchability and transformation ratios were 42.46% and 16.70%, respectively. Given that individuals from the same G1 brood might harbor the same sgRNA, we chose one positive individual from each positive G1 brood and reared them in a pooled manner. Adult moths were crossed back with wild types to generate sgRNA transgenic lines, and an sgRNA library containing 1726 lines was established. To determine the sgRNAs in each line, we performed barcoded deep sequencing and revealed that 1726 lines contain a total of 1835 sgRNAs targeting 1615 genes (Fig. 1F). Over half of the individuals harbored more than one sgRNA, indicating multicopy piggyBac-mediated transformation (Fig. 1G). To generate as many single sgRNA lines as possible, we performed two continuous rounds of backcrossing of these sgRNA lines with wild types and increased the percentage of single sgRNA lines from 43% to 75% (Fig. 1H).
Genotypic characterization and editing outcome of the mutant library
After generating 1726 sgRNA lines, we investigated the potential of these lines to generate a genome-edited mutant library. As a proof-of-principle demonstration, we randomly selected 300 sgRNA lines, which contained 243 gRNAs targeting 188 genes, and crossed them with Cas9-expressing lines. A total of 150,000 hybrid progenies were reared on fresh food and screened under fluorescent microscopy for mutants. Individuals with both 3Xp3-EGFP- and IE2-EGFP-positive signals were considered as mutants. To determine editing efficiencies and patterns, we randomly chose three to six mutant silkworms from each line and performed deep sequencing of the amplified target regions. Of the 1436 tested silkworms, 89.8% showed detectable editing events (Supplemental Fig. S3), with >64% showing editing efficiency >50% (Fig. 2A). Such efficiency is sufficient for functional investigation of target genes, and the results encouraged us to improve it. We analyzed the potential parameters that may affect editing efficiency. The GC content was higher in positive than in negative sgRNAs (Fig. 2B). In positive sgRNAs, GC content was 54%, 49%, and 48% in the high-, medium-, and low-efficiency sgRNA groups (Fig. 2C), respectively, with this tendency more significant in the 2–10 nt of the sgRNA (distal to the PAM) region (Fig. 2D), indicating a positive contribution of GC content to editing efficiency (Sun et al. 2020). The first base of the PAM sequence has no effect on editing efficiency (Fig. 2E). We also found that G at the 3′ end (Supplemental Fig. S4A), G at the 5′ end (Supplemental Fig. S4B), G/CG/C dinucleotides (Supplemental Fig. S5) within the seed region, and 6mA methylation at the target site (Fig. 2F) are beneficial for efficient editing, whereas A at the 3′ end (Supplemental Fig. S4A), C at the 5′ end (Supplemental Fig. S4B), and A/TA/T dinucleotides within the seed region (Supplemental Fig. S5) had a negative effect. We analyzed editing efficiencies of multiple sgRNAs in a single animal and found that the integrated multiple sgRNAs did not affect each other (Supplemental Fig. S6).
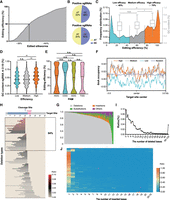
Genotypic characterization and editing outcome of the mutant library. (A) The overall editing efficiency of the edited silkworms. (B) The GC content in the positive and negative sgRNAs. (C) Frequency of distribution of editing efficiency and the GC content in high (editing efficiency > 70%)-efficiency, medium-efficiency, and low-efficiency (editing efficiency < 40%) sgRNA groups: (*) P < 0.05, (**) P < 0.01, (n.s.) not significant; one-way ANOVA with Benjamini–Hochberg post-test multiple comparison. (D) The GC content of different editing efficiency: (*) P < 0.05, (**) P < 0.01, (n.s.), not significant; one-way ANOVA with Benjamini–Hochberg post-test multiple comparison. (E) The editing efficiency of different PAM sequences: (*) P < 0.05, (**) P < 0.01, (n.s.), not significant; one-way ANOVA with Benjamini–Hochberg post-test multiple comparison. (F) The DNA 6mA methylation occupancy around the target center with different editing efficiencies. (G) The overall editing patterns of the edited silkworms. (H) The top 100 deletion types and their percentages. (I) The size of the deletions and their occurrences. (J) The size of the insertions and their percentage.
Large-scale deep sequencing of target sites provided the opportunity to investigate editing outcomes in vivo, the patterns of which were previously found to display distinct differences between cultured cells of B. mori and that of other species (Chang et al. 2020b; Ma et al. 2021). Of the total 61,720,266 reads, we detected 28,927,046 mutated reads containing 1,387,421 mutation types. Most target sites showed editing patterns mainly composed of deletions (86.50%) with a small fraction of insertions (7.39%) and substitutions (3.28%), similar to that of previous observations (Fig. 2G). A few target sites showed a preponderance of insertion or substitution, whereas deletion accounted for only a small part (Fig. 2G). More than 84% of the deletions occurred upstream of the cleavage sites (Fig. 2H), which further confirmed the distinct unidirectional feature of DSB repair in B. mori (Chang et al. 2020b; Ma et al. 2021). Deletion sizes were generally negatively correlated with their occurrence, with significant occurrence peaks at each 3X base pair (Fig. 2I), indicating a potential beneficial selection mechanism during DSB repair in B. mori. Within the insertions, 56.66% and 14.14% were 1-bp and 2-bp insertions, respectively (Fig. 2J). Most insertions were from templated repair (Chang et al. 2020b); 75% of the 1-bp insertions were duplications of −4 nt of the sgRNA sequence; and 80% of the 2-bp insertions were duplications of −5/−4 dinucleotides of the sgRNA sequence (Supplemental Fig. S7).
Phenotypic characterization of the mutant library
A major application of mutant libraries is to discover new phenotypes and their causal genotypes. To evaluate this potential in the CRISPR-Cas9 mutant library, we harvested all F1 embryos from 300 randomly selected lines for phenotyping by rearing three broods per line. Thirty-three out of 300 F1 strains showed significant morphological differences compared with the wild type in terms of body size, skin coloration, developmental duration, spot patterns, appendage development, and metamorphic arrest (Fig. 3A,B). We correlated these phenotypes with the corresponding sgRNA and target site sequencing results, finding individuals from all 33 F1 lines had correct sgRNAs and high editing efficiency at target sites (Supplemental Fig. S8). Many mutant phenotypes were consistent with or theoretically reasonable to previously described mutants, either B. mori or other related species, such as Drosophila. For example, genes (VPS11, KWMTBOMO01164; VPS16, KWMTBOMO03319) of two mutants showed an oily skin phenotype and were previously shown to be involved in the transport and accumulation of uric acid in the epidermis (Fujii and Banno 2019; Segala et al. 2019), whereas the developmental arrest mutant gene, neverland (KWMTBOMO06146), was previously shown to be a key enzyme in ecdysone biosynthesis and caused the arrest of both molting and growth during Drosophila development (Yoshiyama et al. 2006; Lang et al. 2012), The wing-deficient mutant gene, Tout-velu (KWMTBOMO02722), was shown to be required for the diffusion of Hedgehog, a key regulator of limb development (Bellaiche et al. 1998). These results indicated that the current library can be used to identify mutants.
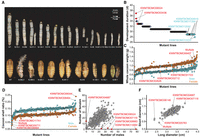
Phenotypic characterization of the mutant library. (A) The body sizes, epidermal coloring, developmental duration, speckle patterns, appendage development, and metamorphosis arrest of 33 F1 lines. These 33 F1 strains showed significant morphological differences among the 300 F1 strains. (B) The development duration of different mutant lines. The genes that may be associated with development extending are highlighted by red; those in light blue are more possibly associated with a shortened development. (C) The whole cocoon weight of different mutant lines. The genes possibly associated with whole cocoon weight improvement are highlighted by red. Each blot represents the average cocoon weight of 50 individuals. (D) The cocoon shell ratio of different mutant lines. The genes possibly associated with cocoon shell ratio improvement are highlighted by red. Each blot represents the average cocoon shell ratio of 50 individuals. (E) The number of males and females in each line. The genes possibly associated with sex differentiation of silkworm are highlighted by red. (F) The short and long diameter of cocoon in each line. The genes that may regulate spinning behaviors are highlighted by red. Each blot represents the average diameters of 30 individuals.
As B. mori is an economically valuable insect in addition to being a research system, we also investigated the economic traits of the mutant lines. Cocoon weight and shell ratio are the two most obvious traits used to evaluate quality of a commercial strain; we measured the cocoon weight and shell ratio of all 300 mutant lines. Most of these cocoons were average; however, we found a few lines that showed a notable higher or lower cocoon weight (Fig. 3C) and cocoon shell ratio (Fig. 3D), including a major component of silk protein (BmFib-L, KWMTBOMO08464) (Fig. 3D). The multibillion-dollar sericulture industry has long sought an easy way to breed only males, because they produce more silk of a higher quality (Kiuchi et al. 2014; Zhang et al. 2018). We counted the males and females in each line and found that progenies of four lines contained only males whereas one line only contained females (Fig. 3E), indicating that knocking out these genes might affect the survival of males or females. We also investigated cocoon shapes of these lines (Supplemental Fig. S9), an important trait that affects postharvesting industrial processes, and found several genes that may regulate spinning behaviors (Fig. 3F). These investigations provide targets for strains with improved traits and further illustrate the viability of our library.
Use of the CRISPR mutant library for pooled screens
Phenotypes of most mutant lines are not visible to the naked eye but may be responsible for certain physiological changes and exogenous stresses. An important application of the mutant library is pooled screening, which can identify genes responsible for certain fine-designed biotic or abiotic stresses. To explore this potential, we performed a proof-of-principle abiotic stress screening against a common environmental pollutant, cadmium (Cd), exposure to which causes significant economic losses in sericulture areas each year (Liu et al. 2021). We selected approximately 100 embryos (one-fourth of each brood) from each of the 300 mutant lines and pooled them. After hatching, the larvae were reared on a normal artificial diet in a pooled manner until the third instar. They were then fed an artificial diet containing 30 mg/kg CdCl2, which is lethal to the wild-type B. mori. Surviving silkworms were moved to a fresh artificial diet containing CdCl2 to ensure continuous exposure (Fig. 4A). Only 53 individuals survived to the fifth instar. sgRNA and their sequencing revealed that 11 larvae harbored the same sgRNA, targeting Calreticulin (KWMTBOMO12902) (Fig. 4B). Sequencing the target region of this sgRNA confirmed highly efficient mutagenesis of KWMTBOMO12902 (Supplemental Fig. S10). Subcellular localization showed that the gene was located in the endoplasmic reticulum and was involved in calcium ion transport (Lu et al. 2020). Given that the proportion of the sgRNAs in the input pool was principally equal before exposure, we speculated that KWMTBOMO12902 is a Cd stress response gene and that its knockout might cause Cd resistance in silkworms. We checked the performance of this gene in our previous cell line–based genome editing screening results and found that KWMTBOMO12902 was not among the top-ranked positive genes, possibly owing to the diversity of Cd ion exposure and responses between individuals and cells, which emphasized the importance of individual mutant pools for finding new strains. We further challenged silkworm individuals and cultured cells with CdCl2; we observed that KWMTBOMO12902 was significantly up-regulated in the midgut, fat body, and B. mori embryonic cell line BmE cells of silkworm larvae after Cd exposure (Fig. 4C). An independent CdCl2 tolerance test using third instar larvae on a larger scale showed that the mutant line KWMTBOMO12902 had a significantly higher tolerance to CdCl2 than the wild type (Fig. 4D). These results indicate that the mutant library in this study could be a powerful resource for identifying new genes responsible for specific physiological functions.
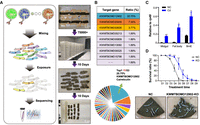
Cadmium (Cd) exposure screening in the mutant library. (A) The schematic diagram of abiotic stress screening in the mutant library with Cd exposure (left) and several scenes during the process (right). (B) The target genes survived from persistent Cd exposure. Individuals with sgRNA targeting KWMTBOMO12902 accounted for the highest proportion. (C) The expression level of KWMTBOMO12902 in midgut, fat body, and BmE cells with or without Cd exposure. (D) The survival rate of normal strain and KWMTBOMO12902-KO strain in Cd stress. The photographs were taken on the fifth day after Cd exposure. Only surviving silkworms were shown in the photograph, as the dead individuals were discarded to avoid cross-infection.
Discussion
Since the early twentieth century, the mutant library has been the main source of major breakthroughs in life science innovation and discovery. Several methods, including physical radiation (X-rays, fast neutrons, and γ-rays), chemical induction (ethyl methane sulphonate and 1,2:3,4-diepoxybutane), random DNA insertion (P-element, piggyBac, and T-DNA), and site-specific mutations (homologous recombination, integrase, RNAi, ZFN, TALEN, and CRISPR-Cas9) have been developed to generate large-scale mutant libraries in a few model organisms such as D. melanogaster, Mus musculus, D. rerio, Arabidopsis thaliana, Nicotiana tabacum, O. sativa L, and B. napus. (Amsterdam et al. 1999; Hrabé de Angelis et al. 2000; Nolan et al. 2000; Gondo 2008; Lein et al. 2008; Xu et al. 2008; Chang et al. 2012; Haelterman et al. 2014; Lu et al. 2017; Meng et al. 2017a,b; Port et al. 2020; He et al. 2023). Here we used the CRISPR-Cas9 system to construct a large-scale mutant library in a nonmodel organism, B. mori, and showed its application in generating novel mutants with important phenotypic changes and in identifying new genes with important physiological functions and economic potential from pooled screening. Research on nonmodel organisms is crucial for solving complex or specialized biological questions; however, they are severely hindered by the lack of efficient genetic tools and mutant resources. We showed the feasibility of generating a large-scale mutant library using a nonmodel organism. Furthermore, because we used piggyBac transposon, which can drive gene delivery in a wide range of eukaryotes and has the largest reported cargo capacity (Wu et al. 2007; Chang et al. 2019; Li et al. 2020) as the delivery strategy, the results in this study also provide a straightforward reference for genome-scale mutagenesis in other nonmodel organisms.
The strategy used in this study has several advantages over previous methods. Traditional mutant B. mori variates were collected from either naturally occurring local or chemically/physically induced mutations (Goldsmith et al. 2005). These have extremely poor phenotype–genotype correlations as the mutations occur randomly across the genomic region, and different mutations have diverse genetic backgrounds (Tong et al. 2022). It usually takes years to map a certain phenotype to its causal genetic alterations (Liu et al. 2010; Atsumi et al. 2012; Yamaguchi et al. 2013). In Drosophila spp. And mice, large-scale mutagenesis can also be achieved through subsequently developed targeted methods such as homologous recombination and RNAi, but both are extremely inefficient in B. mori and many other nonmodel organisms. The CRISPR technology used in our study overcame all the shortcomings of previous methods. First, all mutagenesis was guided by the sgRNA sequences, which were designed to target only the coding regions of the genome, and all mutant lines in the library were truly functionally mutated. Second, the mutated gene and mutant sequences in each line can be tracked through integrated sgRNA sequences by simple targeted PCR amplification and sequencing, meaning all mutant lines in the library have clearer and closer genotype–phenotype correlations than do previous resources. Third, as shown, the efficiency of the CRISPR library for generating large-scale mutants is sufficient for any laboratory with common equipment. Our study also provided many parameters that may further improve this efficiency, such as the high input/low input strategy to avoid repeated sgRNA lines, the backcrossing strategy to reduce multiple sgRNA insertions, and the binary vector system to avoid the loss of growth-deficient lines with lethal genes.
B. mori is one of the most economically beneficial insects and a representative lepidopteran insect because of its crucial role in producing the best-known animal fibers for thousands of years, as well as its finely decoded genomic sequences (Xia et al. 2014; Tong et al. 2022). Systematic functional investigation of B. mori genes is required for both fundamental research and agricultural applications. To achieve this goal, we established multiple genetic manipulation tools, including transgenesis, site-specific recombinases, ZFN, TALEN, and CRISPR-Cas9, as well as a high-throughput CRISPR library screening platform in cultured B. mori cells (Ma et al. 2012, 2013, 2014b; Duan et al. 2013; Liu et al. 2014, 2018; Chang et al. 2020a). However, because the accumulation of genetic resources and research in B. mori relies heavily on the study of orthologous genes in related model organisms, the function of >80% of the B. mori genes remains unclear. We herein present the first high-throughput platform to generate large-scale whole-animal-level mutant libraries and provide a hypothesis-free, cost-effective pipeline to rapidly investigate the function of B. mori genes on a genome-wide scale. Considering the limitations of resources and time, we have decided to focus on protein-coding genes associated with specific mutant phenotypes to gain a better understanding of their functions. As a proof-of-principle demonstration, we designed and constructed a plasmid library containing 92,917 sgRNAs targeting 14,645 protein-coding genes, with high-quality coverage, distribution, and accuracy. A total of 1726 sgRNA lines were generated by microinjection of more than 60,000 embryos, and 300 mutant lines with deciphered mutant sequences containing more than 30 obvious phenotypic changes were obtained. A shortcoming of this study is that we did not identify a sgRNA line that targeted the promoter regions. As more transgenic lines are generated in our future work, it is promising to investigate the phenotypes of promoter-targeting sgRNA lines. These resources provide a powerful platform for functional annotation of B. mori genomes.
We previously established a pooled loss-of-function screening platform in cultured B. mori cells and showed its effectiveness in identifying genes involved in multiple biotic and abiotic stresses (Chang et al. 2020a; Liu et al. 2021). It remains a significant challenge to perform pooled genetic screening at the whole-animal level owing to the lack of mutant resources and an efficient screening method. In large-scale functional screening experiments, it is generally required a high coverage and homogeneity of a library. However, most previous screenings were performed in cultured cells or single-cell systems, whereas this study used a mutant library in the whole organism of higher-order Eukaryote. In the whole-organism-level mutant library construction and screening, it is impossible and also unnecessary to reach a high coverage of sgRNAs. Thus, we performed the first in vivo pooled genetic screening of B. mori using the obtained 300 mutant lines. We showed that the CRISPR mutant library was highly efficient in identifying the genes responsible for stress. The screening results were mutually complemented genome-wide CRISPR screenings in cultured cells, and the selected mutants showed high performance when checked individually. We further identified the KWMTBOMO12902 gene, the absence of which significantly increased the tolerance to Cd exposure. Cd exists extensively in the ecosystem as a typical pollutant. It has been identified as a global threat because it cannot be degraded or resolved, is gradually enriched up the biological chain, and is severely harmful in various animals and plants, including humans (Yang et al. 2022). Studies aiming to decipher signaling and metabolic pathways involved in the pathological effects associated with Cd exposure are urgently needed, but it is difficult to perform large-scale toxicant exposure experiments in mammals. Here we used B. mori for large-scale pooled screens, and with the nature of B. mori being sensitive to environmental pollutants, it provided a potential target to explore organismal detoxification mechanisms (Mao et al. 2019). Environmental Cd exposure causes significant economic damage to the sericulture industry. Results showed the KWMTBOMO12902 gene mutant showed a high tolerance to Cd, even at a dosage much higher than that of environmental Cd, suggesting that KWMTBOMO12902 can be used to breed a Cd-tolerant silkworm strain.
Although the CRISPR-Cas9 system has been a powerful genome editing tool for more than 10 years, designing sgRNAs with high activity remains a major challenge (Chen and Wang 2022). As has been recognized in a number of previous reports and in the current study, not all sgRNAs can cleave their target DNA, and the cleavage efficiencies of different sgRNAs vary significantly (Li et al. 2023). A key parameter that determines the efficiency of sgRNA is its sequence and structural properties. We systematically investigated the effects of sgRNA sequence composition by sequencing 1436 mutant silkworms and found that the GC content in the 2–10 nt of the sgRNA (distal to the PAM) region, C content within the PAM sequences, G at the 3′ end, G at the 5′ end, and G/CG/C dinucleotides within the seed region are beneficial for efficient editing. We also investigated the effect of the local chromosome state on editing efficiency and found that 6mA methylation at the target site has a positive effect. This is an interesting observation, but further investigation in a future study is needed to confirm it and reveal its mechanism. These results provide practical guidance for the design of sgRNA for a successful genome editing experiment in B. mori and increase the understanding of the heterogeneity of sgRNA functions.
In conclusion, we have established the first genome-scale CRISPR mutagenesis platform at the whole-animal level in a nonmodel multicellular organism. A plasmid library containing 92,917 sgRNA-expressing vectors and a collection of 1726 sgRNA transgenic lines was established. By generating and analyzing 300 CRISPR mutant lines, we showed that the CRISPR library is a powerful resource for the rapid discovery of unknown gene functions and for investigating long-standing questions in silkworms and entomology. Applying the established mutant lines, we discovered and highlighted a large set of new genes that can cause divergent visible phenotypic changes, as well as several genes that have promising potential applications in breeding strains, with higher yield or greater tolerance for the sericulture industry. These resources are preserved in the Biological Research Center of Southwest University and are shared worldwide through the silk genome database, SilkDB 3.0 (https://silkdb.bioinfotoolkits.net) (Supplemental Fig. S11; Lu et al. 2020). Furthermore, the number of sgRNA transgenic and mutant lines will continue to increase. We believe that this study lays the foundation for large-scale functional genomics of B. mori and the rapid identification of novel genes involved in diverse biological processes. More importantly, the present study also sheds light on the application of genome-scale mutagenesis to other nonmodel organisms.
Methods
Silkworm strains and rearing conditions
Two silkworm strains, Dazao and Nistari, were used in the present study. Dazao was used to inject the sgRNA library and Nistari to express Cas9. Dazao was reared in our laboratory, whereas Nistari was provided by Prof. Anjiang Tan from Jiangsu University of Science and Technology, Zhenjiang, China. The silkworms were fed fresh mulberry leaves or artificial diet at 25°C and 80% relative humidity. BmE is a silkworm embryonic cell line preserved in our laboratory. These cells were grown in Grace medium, which was supplemented with 10% fetal bovine serum (FBS) as well as penicillin/streptomycin. The incubation temperature for the cultures was set at 27°C.
Design and construction of the vectors
The piggyBac transposase expression vector A3-helper was obtained from stocks in our laboratory. pUC57-3Xp3-EGFP-sv40 was constructed by replacing DsRed in pUC57-3Xp3-DsRed-sv40 (3DS) (Ma et al. 2014a) with the EGFP sequence. pUC57-U6-gRNA(AarI) was constructed by replacing the BbsI restriction site in pUC57-gRNA (Ma et al. 2014a) with an AarI restriction site. PUC57-3Xp3-EGFP-sv40 was cloned into the AgeI/AscI sites of the pBac-modified vector to generate pB-modified {3Xp3-EGFP-sv40}. pUC57-U6-gRNA(AarI) was cloned into the NheI/AscI site of pB-modified {3Xp3-EGFP-sv40} to generate the vector pB-modified {3Xp3-EGFP-sv40}{U6-gRNA(AarI)}, named p200.
sgRNA library design and synthesis
The silkworm genome assembly and gene models are obtained from SilkBase (http://silkbase.ab.a.u-tokyo.ac.jp). sgRNAs were designed using the CCTop method (Stemmer et al. 2015), which uses CRISPRater values to select gRNAs with potentially higher activity. All sgRNAs selected had 5′ G added to improve the U6 transcription efficiency. The sgRNA library was encoded within 70-nt oligonucleotides and synthesized on 92,917 arrays using the services of the Beijing Genomics Institute (China). The oligonucleotide sequence used was 5′-TACAAAATATCGTGCTCTACAAGTGNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAGTT-3′.
Construction of sgRNA library
The DGP-1 fragment (U6 promoter) was PCR-amplified from p200 using the primers KU-1R and DG-1R. The pool of sgRNA oligonucleotides was amplified by PCR using primers DP-2F and DP-2R and named DGP-2 fragments (multiple sgRNAs). The DGP-3 fragment (sgRNA scaffolds) was amplified by PCR from p200 using the primers DG-3F and KU-1F. These three fragments were linked using overlap PCR to generate DGP-4 fragments (U6-sgRNA library). All PCR were performed using PrimeSTAR max DNA polymerase (Takara). The DGP-4 fragments were then cloned into the AscI/NheI (NEB) site of the p200 vector to construct the sgRNA library (p200 library) according to the following protocol: The gel-purified DGP-4 fragments (200 ng) and p200 vector (200 ng) were mixed and ligated in a 50 µL T4 ligation reaction (NEB) according to the manufacturer's instructions. The ligation reaction (1 µL) was transformed into 50 µL of Escherichia coli HST08 premium electro-cells (Takara) using a gene pulser Xcell (Bio-Rad) according to the manufacturer's instructions. To ensure the diversity of the sgRNA library, 15 parallel electro-transformations were performed with ampicillin selection, which resulted in more than 1000× library coverage. All clones were collected, and the plasmid library was extracted using a QIAprep spin miniprep kit (Qiagen).
Microinjection and positive transgenic screening
After mating, the female moths were placed on paper starched with paste to lay eggs in the dark. Eggs were collected every 0.5 h and arranged on slides (approximately 100 eggs per slide). After the eggs were fixed on slides, plasmids were injected using a microinjection apparatus (Eppendorf). The injection was completed within 60–220 min after laying. The injected plasmid was mixed with sgRNA plasmid:helper plasmid = 1 × 1 (molar ratio of the substance), and the final concentration was 400 ng/μL. Plasmids (10–15 nL) were injected into each egg. Immediately after injection, the eggs were sealed with nontoxic glue (instant strong glue mini). The eggs were maintained at a relative humidity >85% until they hatched. After hatching, the larvae were reared according to the type of vector injected. The G0 generation obtained by self-cross or backcross seed production were detected and screened using a macrosteric fluorescence microscope (Olympus DP73) and fed separately.
Pooled genetic screening
After collecting the eggs from the silkworm mutant library, approximately one-fourth of each brood was gathered from every mutant line, totaling approximately 30,000–50,000 eggs. Subsequently, these larvae were uniformly hatched and fed a normal artificial diet until the third instar. The larvae were fed an artificial diet containing 30 mg/kg CdCl2. Subsequently, dead larvae were removed, and survival was supported with a fresh diet. Ultimately only 53 individuals survived until the fifth instar. They were used as donors for the extraction of genomic DNA and sgRNA sequences.
sgRNA sequencing, target site sequencing, and data analysis
The genome was amplified using primers with distinct barcodes and sequenced using Illumina sequencing technology. Low-quality raw sequencing data of amplicons were filtered out using Trimmomatic (v.0.39; LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50) (Bolger et al. 2014). Paired-ended clean reads were merged using FLASH (v.1.2.11) (Magoč and Salzberg 2011). cutadapt (v.1.18) (Marcel 2011) was used to split the data based on the unique barcode of each sample. Bowtie 2 (v.2.2.5) (Langmead and Salzberg 2012) was used for aligning to identify the sgRNA sequences in each sequenced sample. Based on the detected sgRNA, Primer3 (v.2.4.0) (Untergasser et al. 2012) was used to design primers for each target site, amplify the genomes of different samples using diverse primers with specific barcodes, and conduct second-generation sequencing. The analysis methods for filtering out, merging, and splitting of target site sequences were the same as those used in the sgRNA data analysis, whereas mutation detection of target sites was performed using CRISPResso (v.2.0.45) (Clement et al. 2019). Barcode sequences associated with each sample are listed in Supplemental Tables S2 and S3. The m6A data set of silkworm was downloaded from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA480541.
Cd exposure experiment
To further determine the function of KWMTBOMO12902 in Cd tolerance, we separately treated the KWMTBOMO12902-KO silkworm strain with Cd exposure. Embryos were hatched and fed a fresh artificial diet. Most toxicological studies focus on the third to fifth instar larvae, during which the organs related to detoxification such as the fat body and midgut are fully developed, and the larvae enter a stage of rapid feeding, which facilitates observation and toxicological analysis. Therefore, we choose to conduct exposure experiments starting from the third instar. In the third instar, the artificial diet was supplemented with 30 mg/kg CdCl2. The survival rate was calculated daily. The gRNAs of the surviving individuals were detected from the extracted DNA after Cd exposure. To further investigate the role of KWMTBOMO12902 in Cd tolerance, we exposed KWMTBOMO12902-KO strain silkworms to Cd. Embryos were hatched and fed a fresh artificial diet. In the third instar, the artificial diet was supplemented with 30 mg/kg CdCl2. The survival rate was calculated daily.
Phenotypic characterization
Fourth-instar larvae of 300 strains of the F1 generation were screened using a stereo-fluorescence microscope (Olympus DP73), and all double-positive individuals (the eye with green light and the body with green light) in each brood were selected for subsequent research. Mutants with significant developmental abnormalities (larval development speed and individual size, larval pattern, pupation process, cocoon color and type, and moth-changing process) were selected. The cocoon weight and pupal weight of all double-positive males and females in each brood were measured separately using an analytical balance, and the sex ratio was counted simultaneously. The cocoon shape was measured for length and breadth using ImageJ software (https://imagej.nih.gov/ij/index.html), and 10 female and 10 male cocoons were randomly measured.
Quantitative real-time PCR
The larvae were dissected to obtain different tissues, and the total RNA from BmE cells or different tissues was extracted. For each sample, 1 μg of RNA was subjected to reverse transcription using the one-step gDNA removal and cDNA synthesis SuperMix kit (TransGen Biotech). Quantitative real-time PCR (qPCR) was performed using SYBR Premix Ex Taq II (Takara), and target gene expression was normalized to the reference gene rp49. The test was performed independently at least three times. The primer sequences for the target genes were as follows: KWMTBOMO12902-qPCR-F, TGAAGAGCGAACAAGACGAG; KWMTBOMO12902-qPCR-R, TTCCTCATCGTCAGGCTTTT; Rp49-qPCR-F, CAGGCGGTTCAAGGGTCAATAC; and Rp49-qPCR-R, TGCTGGGCTCTTTCCACGA.
Data access
The raw data generated in this study were submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA901072. The mutant library resources are preserved in the Biological Research Center of Southwest University and are shared worldwide through the Silkworm Knowledgebase (SilkDB 3.0; https://silkdb.bioinfotoolkits.net/). The mutant module in the database contains comprehensive phenotype and genotype data for the mutants (Supplemental Fig. S11). The plasmid library can be obtained through Addgene (https://www.addgene.org/209615/).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Feng Wang, Dr. Zhangchuan Peng, Dr. Wenliang Qian, Dr. Kaiyu Guo, Dr. Qingsong Liu, Xia Zhang, and Xiaoxu Chen for their kind help with the microinjection experiments. This work was supported by grants from the National Natural Science Foundation of China (no. 32122084), the Chongqing Natural Science Foundation (no. cstc2021ycjh-bgzxm0005), the PhD Start-up Foundation of Southwest University (no. SWU120012), and the Fundamental Research Funds for Central Universities (no. SWU-KT22042). None of these funding sources played a role in the design of the study, collection, analysis, and interpretation of data, or in writing the manuscript.
Author contributions: S.M. conceived of the study and designed the experiments. T.Z. designed the sgRNA and performed all bioinformatic analyses. R.W. constructed the plasmid library. Y.L. performed the pooled screening experiments. P.W., L.J., and A.W. generated and maintained the transgenic lines with the help of J.C., L.S., H.S., R.S., W.L., D.L., N.Z., W.H., X.W., and W.X. X.L. performed the genotyping experiments. L.J. and S.M. wrote the manuscript with the help of Q.X. All authors have read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278297.123.
- Received August 2, 2023.
- Accepted November 29, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








