
Modeling and predicting cancer clonal evolution with reinforcement learning
Abstract
Cancer results from an evolutionary process that typically yields multiple clones with varying sets of mutations within the same tumor. Accurately modeling this process is key to understanding and predicting cancer evolution. Here, we introduce clone to mutation (CloMu), a flexible and low-parameter tree generative model of cancer evolution. CloMu uses a two-layer neural network trained via reinforcement learning to determine the probability of new mutations based on the existing mutations on a clone. CloMu supports several prediction tasks, including the determination of evolutionary trajectories, tree selection, causality and interchangeability between mutations, and mutation fitness. Importantly, previous methods support only some of these tasks, and many suffer from overfitting on data sets with a large number of mutations. Using simulations, we show that CloMu either matches or outperforms current methods on a wide variety of prediction tasks. In particular, for simulated data with interchangeable mutations, current methods are unable to uncover causal relationships as effectively as CloMu. On breast cancer and leukemia cohorts, we show that CloMu determines similarities and causal relationships between mutations as well as the fitness of mutations. We validate CloMu's inferred mutation fitness values for the leukemia cohort by comparing them to clonal proportion data not used during training, showing high concordance. In summary, CloMu's low-parameter model facilitates a wide range of prediction tasks regarding cancer evolution on increasingly available cohort-level data sets.
Cancer results from an evolutionary process during which somatic mutations accumulate in a population of cells. This process results in a tumor composed of multiple subpopulations of cells, or clones, with varying sets of mutations (Nowell 1976). As different clones within the same tumor harbor different sets of mutations, they have varying phenotypes and fitness (Yates and Campbell 2012). Moreover, although each cancer results from a separate evolutionary process, there are repeated patterns of evolution that recur across cancers (Hanahan and Weinberg 2000). The key challenge in comparative cancer phylogenetics revolves around identifying such repeated evolutionary patterns or trajectories (Turajlic et al. 2018). This is a challenging problem and requires a mathematical model that accurately captures the somatic evolutionary process of cancers.
There are many existing methods for modeling cancer evolution with single-nucleotide variations (SNVs), which can be distinguished into three classes. The first class of methods is based on tree generative models, which include TreeMHN (Luo et al. 2023) and HINTRA (Khakabimamaghani et al. 2019). A generative model restricted to paths and trained using inverse reinforcement learning was considered by Kalantari et al. (2020). The second class of methods is consensus tree models, which seek a small number of consensus trees that summarize common patterns among patient trees. Methods such as REVOLVER (Caravagna et al. 2018), RECAP (Christensen et al. 2020), CONETT (Hodzic et al. 2020), and MASTRO (Pellegrina and Vandin 2022) fall into this category. The third class of methods uses statistical tests to evaluate patterns of co-occurrence and mutual exclusivity of mutations across trees without trying to fully model the evolutionary process. GeneAccord is one such method (Kuipers et al. 2021). Although consensus tree methods have the advantage of being able to detect complicated patterns in tumor evolution, tree generative models must be carefully designed to accommodate complex multimutation effects. An example of such an effect is when mutations s and t only increase the probability of mutation r when they are present together. Conversely, tree generative methods have the advantage of being able to directly model how a clone's mutations affect the probability of new mutations occurring.
A second distinguishing feature of current methods is how the number of model parameters scales with an increasing number m of mutations. Having the number of parameters scale too rapidly with the number of mutations can lead to greatly overfitting on real data sets, as well as a prohibitive running time. One such example is HINTRA (Khakabimamaghani et al. 2019), in which the number of parameters grows exponentially in m, and consequently, it can only accommodate a handful of mutations. On the other hand, models that directly measure relationships between pairs of mutations or fit trees with edges determined by pairs of mutations have their parameters grow quadratically in the number of mutations, reducing overfitting and running time (Caravagna et al. 2018; Christensen et al. 2020; Hodzic et al. 2020; Kuipers et al. 2021; Luo et al. 2023).
Finally, current methods can be distinguished by the prediction tasks they support. The majority of current methods, spanning both tree generative (Khakabimamaghani et al. 2019; Luo et al. 2023) and consensus tree methods (Caravagna et al. 2018; Christensen et al. 2020; Hodzic et al. 2020), aim to identify evolutionary trajectories, which correspond to ordered sequences or trees of mutations reflecting repeated patterns of evolution. Additionally, several methods use signals from across patients to resolve uncertainty in phylogeny inference within a single patient (Caravagna et al. 2018; Khakabimamaghani et al. 2019; Christensen et al. 2020; Hodzic et al. 2020). Another task is the prediction of causality between pairs of mutations. This can be performed in a signed manner, distinguishing between causal and inhibitory relationships, as well as in a bidirectional manner, distinguishing between a mutation s causing t and vice versa. Although TreeMHN (Luo et al. 2023) supports both signed and bidirectional causal relationships, GeneAccord (Kuipers et al. 2021) only supports signed causal relationships. On the other hand, consensus tree methods are unable to support inhibitory causal relationships (Caravagna et al. 2018; Christensen et al. 2020; Hodzic et al. 2020).
In this work, we identify three additional tasks that no current evolutionary trajectory method supports. First is determining the fitness of a mutation, namely, assessing how having that mutation impacts the rate at which a clone develops. Second, although previous work has focused on identifying co-occurrence and mutual exclusivity of mutations at the patient level without directly considering downstream mutation evolution (Leiserson et al. 2015; Dao et al. 2017; Kim et al. 2017; Kuipers et al. 2021), we introduce the task of determining sets of interchangeable mutations that have similar impacts on subsequent mutation evolution. Third is determining collections of evolutionary trajectories between sets of interchangeable mutations. To accommodate these new and all previous tasks, we introduce clone to mutation (CloMu). Underlying CloMu is a tree generative model, which uses a low-parameter neural network trained using reinforcement learning, resulting in fewer parameters than all current models while maintaining high flexibility to model complex multimutation effects. In summary, this study aims to introduce a method for accurately performing a wide variety of prediction tasks on cohort-level cancer phylogeny data.
Results
Overview of CloMu
We take as input tumor phylogenies of n patients with m total mutations. Because of uncertainty in tree inference from sequencing data (Qi et al. 2019), each patient p has a set 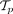 of multiple possible trees
of multiple possible trees 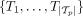 . Similarly to HINTRA (Khakabimamaghani et al. 2019) and TreeMHN (Luo et al. 2023), we make the independent clonal evolution assumption that the event of a clone acquiring a new mutation only depends on the genotype of that clone. Each clone can
be represented as a binary vector c ∈ {0, 1}m, where cs = 1 if the clone harbors mutation s and cs = 0 otherwise. The goal is to identify a model that best describes the causal relationship between clones c and acquired mutations [m] = {1, …, m} under our assumption for the observed data
. Similarly to HINTRA (Khakabimamaghani et al. 2019) and TreeMHN (Luo et al. 2023), we make the independent clonal evolution assumption that the event of a clone acquiring a new mutation only depends on the genotype of that clone. Each clone can
be represented as a binary vector c ∈ {0, 1}m, where cs = 1 if the clone harbors mutation s and cs = 0 otherwise. The goal is to identify a model that best describes the causal relationship between clones c and acquired mutations [m] = {1, …, m} under our assumption for the observed data  . We model this using the function
. We model this using the function 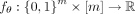 such that
such that 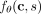 is the logarithm of the rate at which mutation s occurs on clone c (Fig. 1A). Starting with an initial tree T(0) with just a single node corresponding to the normal clone
is the logarithm of the rate at which mutation s occurs on clone c (Fig. 1A). Starting with an initial tree T(0) with just a single node corresponding to the normal clone 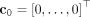 , we sample the mutation that occurs next, yielding a new tree T(1) with an additional clone c1 that introduces one mutation w.r.t. c0. Repeating this process yields a new partial tree T(k) after each mutation is introduced, as shown in Supplemental Figure S1. Note that any tree can be generated through this process.
, we sample the mutation that occurs next, yielding a new tree T(1) with an additional clone c1 that introduces one mutation w.r.t. c0. Repeating this process yields a new partial tree T(k) after each mutation is introduced, as shown in Supplemental Figure S1. Note that any tree can be generated through this process.

Overview of CloMu. (A) Using the independent clonal evolution assumption, our model determines a log rate 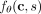 of any clone c ∈ {0, 1}m acquiring a mutation s ∈ [m]. (B) This in turn enables us to compute probabilities P = [pi,s] that the next mutation to occur on a tree T is mutation s at node/clone ci. The resulting Independent Clonal Evolution problem seeks model parameters θ that maximize the data probability
of any clone c ∈ {0, 1}m acquiring a mutation s ∈ [m]. (B) This in turn enables us to compute probabilities P = [pi,s] that the next mutation to occur on a tree T is mutation s at node/clone ci. The resulting Independent Clonal Evolution problem seeks model parameters θ that maximize the data probability  of a cohort of trees for n patients. (C) CloMu represents
of a cohort of trees for n patients. (C) CloMu represents 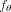 using a low-parameter, two-layer neural network that is trained via reinforcement learning. We use the model for five prediction
tasks: (D) tree selection for each patient, (E) determination of mutation fitness, (F) causality inference for pairs of mutations, (G) identification of interchangeable mutations, and (H) identification of their evolutionary pathways.
using a low-parameter, two-layer neural network that is trained via reinforcement learning. We use the model for five prediction
tasks: (D) tree selection for each patient, (E) determination of mutation fitness, (F) causality inference for pairs of mutations, (G) identification of interchangeable mutations, and (H) identification of their evolutionary pathways.
Our goal is to find the model parameters θ* that maximize the probability of observing the input data, namely, 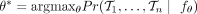 . To do so, we must estimate the probability of any input tree T denoted by
. To do so, we must estimate the probability of any input tree T denoted by 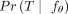 , noting the fact that there are multiple ways of generating each tree (Fig. 1B). This leads to the following problem, which we describe in more detail in the Methods.
, noting the fact that there are multiple ways of generating each tree (Fig. 1B). This leads to the following problem, which we describe in more detail in the Methods.
Given a cohort of tumor phylogenies  for n tumors on m mutations, find model parameters θ such that
for n tumors on m mutations, find model parameters θ such that  is maximized.
is maximized.
To solve the Independent Clonal Evolution problem, we introduce CloMu, which represents the model 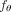 using a two-layer neural network with a small number L = 5 of hidden neurons for the function
using a two-layer neural network with a small number L = 5 of hidden neurons for the function 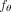 (Fig. 1C). This neural network is trained via reinforcement learning (Methods).
(Fig. 1C). This neural network is trained via reinforcement learning (Methods).
Although we train the model 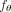 only once for each data set
only once for each data set  by solving a single instance of the Independent Clonal Evolution problem, its outputs can be postprocessed to complete a wide variety of tasks, gaining insights into cancer evolution. The
exact mathematical details of these tasks are discussed in the Methods, but we give a brief summary here. First, one can use
the model to reduce uncertainty in tree inference by determining which phylogenies are most likely to be the true phylogeny
for a particular patient (Fig. 1D). Second, the model can be used to determine the fitness of different mutations based on how they affect the likelihood
of new mutations occurring on a clone (Fig. 1E). Third, the model can be used to infer co-occurrence patterns between pairs of mutations, which we refer to as causal relationships
(Fig. 1F). Fourth, the model can be used to detect interchangeable mutations, namely, mutations with similar downstream mutation
effects that are mutually exclusive or co-occurring, by assessing the values of the L hidden neurons when a clone is taken as an input (Fig. 1G). Fifth, we can determine pathways of interchangeable mutations (Fig. 1H).
by solving a single instance of the Independent Clonal Evolution problem, its outputs can be postprocessed to complete a wide variety of tasks, gaining insights into cancer evolution. The
exact mathematical details of these tasks are discussed in the Methods, but we give a brief summary here. First, one can use
the model to reduce uncertainty in tree inference by determining which phylogenies are most likely to be the true phylogeny
for a particular patient (Fig. 1D). Second, the model can be used to determine the fitness of different mutations based on how they affect the likelihood
of new mutations occurring on a clone (Fig. 1E). Third, the model can be used to infer co-occurrence patterns between pairs of mutations, which we refer to as causal relationships
(Fig. 1F). Fourth, the model can be used to detect interchangeable mutations, namely, mutations with similar downstream mutation
effects that are mutually exclusive or co-occurring, by assessing the values of the L hidden neurons when a clone is taken as an input (Fig. 1G). Fifth, we can determine pathways of interchangeable mutations (Fig. 1H).
Benchmarking on simulated data
We use simulations with known ground truth to assess the performance of CloMu and several existing methods on the prediction tasks (outlined in the section “Prediction tasks”). We consider four sets of simulation instances: (1) simulations to assess tree selection and causality, (2) simulations to assess mutation interchangeability and evolutionary pathways, as well as previous simulations generated in the (3) RECAP (Christensen et al. 2020) and (4) TreeMHN (Luo et al. 2023) papers (Table 1). We include TreeMHN (Luo et al. 2023), RECAP (Christensen et al. 2020), REVOLVER (Caravagna et al. 2018), and GeneAccord (Kuipers et al. 2021) in the benchmarking, and refer to Supplemental Material B.2 for parameter settings.
Characteristics of simulation data sets
We begin by discussing the generation of the first set of simulation instances to assess tree selection, causality, and mutation
interchangeability. We focus on the simplest case with no interchangeable mutations. We considered m = 10 mutations, subdivided into five driver mutations and five passenger mutations. For every ordered pair (s, t) of distinct driver mutations, there is a 50% chance that s causes t. Let X be the resulting set of causal relationship pairs. Note that |X| ≤ 20 as there are 20 ordered pairs (s, t) of distinct driver mutations. For each pair (s, t) ∈ [m] × [m] of mutations, we set the rate multiplier to g(s, t) = 11 if (s, t) ∈ X and to g(s, t) = 1 otherwise. We defined the rate λ(c, t) of a clone c ∈ {0, 1}m acquiring a mutation t as 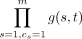 . We define the log rate f(c, t) of a clone c ∈ {0, 1}m acquiring a mutation t as log (λ(c, t)). Given a number n = 500 of patients, we generated one ground-truth tree
. We define the log rate f(c, t) of a clone c ∈ {0, 1}m acquiring a mutation t as log (λ(c, t)). Given a number n = 500 of patients, we generated one ground-truth tree 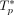 for each patient p ∈ [n] by first drawing the number ℓ of mutations of the tree
for each patient p ∈ [n] by first drawing the number ℓ of mutations of the tree 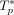 uniformly from {5, 6, 7}. We used the rates λ(c, t) to construct
uniformly from {5, 6, 7}. We used the rates λ(c, t) to construct 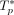 following the generative process with ℓ mutations discussed in the section “Overview of CloMu”—with
following the generative process with ℓ mutations discussed in the section “Overview of CloMu”—with 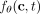 replaced with f(c, t) [or, equivalently,
replaced with f(c, t) [or, equivalently, 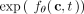 replaced with λ(c, t)]. Although not required by CloMu, we imposed the infinite sites assumption in our simulations. Finally, we simulated five
bulk DNA sequencing samples and performed clonal tree enumeration, resulting in sets
replaced with λ(c, t)]. Although not required by CloMu, we imposed the infinite sites assumption in our simulations. Finally, we simulated five
bulk DNA sequencing samples and performed clonal tree enumeration, resulting in sets  of trees per patient with a varying number of trees per patient as shown in Supplemental Figure S3. We generated a total of 20 simulation instances, denoted simulations I-a. Additionally, to assess the effect of interchangeable
mutations, we generated two additional sets of 20 simulation instances, denoted simulations I-b and I-c, with, respectively,
five pairwise disjoint sets of two and three interchangeable mutations among m = 10 and m = 15 total mutations. Finally, we generated a set of 20 simulation instances, denoted simulation II, to assess evolutionary
pathway identification. Note that each simulation instance is generated independently and thus does not necessarily share
randomly generated properties such as causal relationships with other instances. For further details, see Table 1 and Supplemental Material B.1.
of trees per patient with a varying number of trees per patient as shown in Supplemental Figure S3. We generated a total of 20 simulation instances, denoted simulations I-a. Additionally, to assess the effect of interchangeable
mutations, we generated two additional sets of 20 simulation instances, denoted simulations I-b and I-c, with, respectively,
five pairwise disjoint sets of two and three interchangeable mutations among m = 10 and m = 15 total mutations. Finally, we generated a set of 20 simulation instances, denoted simulation II, to assess evolutionary
pathway identification. Note that each simulation instance is generated independently and thus does not necessarily share
randomly generated properties such as causal relationships with other instances. For further details, see Table 1 and Supplemental Material B.1.
For the tree selection task, we compared CloMu to RECAP and REVOLVER on simulations I-a (Table 1). For each simulation instance, we determined the tree selection accuracy defined as the fraction of patients for which each method correctly identified the ground-truth tree. Figure 2A shows that CloMu achieves the highest tree selection accuracy (median, 0.76), followed by REVOLVER (median, 0.70) and then RECAP (median, 0.65). To assess causality inference, we compared CloMu (using absolute causality) against TreeMHN and GeneAccord, which directly support causality inference (Table 2). Although RECAP and REVOLVER do not directly support this task, we used a simple heuristic based on the frequency of mutation pairs/edges (s, t) among selected trees (Supplemental Material B.2). Figure 2B shows that CloMu and TreeMHN performed near perfectly on this task (median recall and precision of 1.0), followed by RECAP and REVOLVER (respectively, median recall, 0.76 and 0.77; median precision, 1.0 and 1.0) and then GeneAccord (median recall and precision, 0.78 and 0.85). Additionally, we used bootstrapping to assess the confidence intervals on absolute causality predictions, showing that the lower bound for pairs of mutations with a true causal relationship vastly exceeds the upper bound for pairs of mutations without a true causal relationship (Supplemental Material B.3.1; Supplemental Figs. S4, S5). Moreover, we assessed the effect of decreasing numbers of patients and mutations per patient as well as an increased number of passenger mutations and varying causation effect sizes, showing good performance of CloMu in each simulation condition (Supplemental Materials B.3.2–B.3.5; Supplemental Figs. S6–S10).

CloMu outperforms existing methods on several prediction tasks on simulated data with known ground truth. (A) Tree selection accuracy measures the ability to correctly identify the ground-truth tree from a set 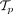 of possible trees generated from simulated bulk sequencing for each patient p. (B) Causality precision and recall measure the ability to determine positive causal relationships between ordered pairs of mutations.
Panels A and B show results for simulations I-a. (C) On simulations I-b and I-c, causality precision and recall measure the ability to identify causation and inhibition between
pairs of mutations in the presence of mutation interchangeability. (D) On simulations II, interchangeability detection shows that the latent representations generated by our model are meaningful,
accurately encapsulating mutation similarity. (E) On simulations II, pathway precision and recall measure the ability to determine evolutionary pathways in the presence of
both interchangeability and complex multimutation interactions.
of possible trees generated from simulated bulk sequencing for each patient p. (B) Causality precision and recall measure the ability to determine positive causal relationships between ordered pairs of mutations.
Panels A and B show results for simulations I-a. (C) On simulations I-b and I-c, causality precision and recall measure the ability to identify causation and inhibition between
pairs of mutations in the presence of mutation interchangeability. (D) On simulations II, interchangeability detection shows that the latent representations generated by our model are meaningful,
accurately encapsulating mutation similarity. (E) On simulations II, pathway precision and recall measure the ability to determine evolutionary pathways in the presence of
both interchangeability and complex multimutation interactions.
Overview of methods for detecting patterns of cancer evolution
Upon observing CloMu and TreeMHN's near perfect performance, we decided to include interchangeable mutations as well as inhibitory relationships (simulations I-b and I-c) (see Table 1). We refer to Supplemental Material B.2 and Supplemental Table S2 for the updated definitions of causality precision and recall for the case of signed causality, which match TreeMHN's definitions. We found that CloMu maintained good performance whereas TreeMHN's performance dropped, especially with increasing numbers of interchangeable mutations (Fig. 2C). This was also the case when all causal relationships between interchangeable mutations were made inhibitory (Supplemental Material B.3.8; Supplemental Fig. S14). We believe the reason TreeMHN performed poorly on these instances is its assumption of independent causal relationships between pairs of mutations. The number m2 − m of causal relationships that TreeMHN must independently determine grows quadratically with the number m of mutations. In contrast, if CLoMu's neural network learns that there are only k ≪ m noninterchangeable types of mutations, it must only detect k2 causal relationships. Specifically, for these simulation instances, there were k = 5 sets of interchangeable mutations among a total of m ∈ {10, 15} mutations (see Supplemental Material B.1), leading to k2 = 25 causal relationships for CloMu to detect and m2 − m ∈ {90, 210} for TreeMHN. One potential concern is that the high performance of CloMu and avoidance of overfitting is only owing to the small number of parameters caused by our choice of L = 5 hidden neurons. However, in Supplemental Material B.3.9, we show that with an increased number L = 20 of hidden neurons, CloMu even gives slightly more accurate causal relationship predictions (Supplemental Fig. S15). Supplemental Material B.3.10 and Supplemental Figures S17 and S18 show CloMu's accuracy on simulations in which mutations have related effects rather than being exactly interchangeable. The effects of mutations are not independent despite not being identical, as shown in Supplemental Figure S16. We show CloMu's ability to accurately identify multimutation causality effects in Supplemental Material B.3.11 and Supplemental Figure S19.
We now focus on simulations II to further assess performance on determining interchangeable mutations and their evolutionary pathways. Figure 2D shows that CloMu accurately distinguishes between pairs of interchangeable and noninterchangeable mutations as assessed by the Euclidean distance on CloMu's latent representation. We note that these simulations contain causal effects that only occur in the presence of specific combinations of mutations on a clone (Supplemental Material B.1). Accurately determining the pathways requires modeling these multimutation effects, as shown in Supplemental Figure S2. To have some baseline for comparison, we applied a heuristic to determine the pathways of interchangeable mutations from the trees selected by RECAP and REVOLVER (Supplemental Material B.2). Briefly, our heuristic measured the number of times each edge occurs in selected trees for this method and then inferred the pathway edges as all edges that occur above some frequency. TreeMHN and GeneAccord could not be adapted to form a baseline because they do not select trees. Additionally, they have no way of modeling effects that only occur in the presence of a combination of mutations. We determined the number of true and false positives, as well as false negatives, by comparing the set of true pathways edges and predicted edges, enabling us to compute pathway precision and recall. We found that CloMu, RECAP, and REVOLVER all achieved a median pathway recall of 1.0 in simulations with only one pathway (Fig. 2E). However, CloMu outperformed the baselines in terms of precision (overall median, 1.0 vs. 0.92 and 0.87 for RECAP and REVOLVER, respectively), and especially for cases with two evolutionary pathways, it additionally outperformed the baseline methods in terms of recall (overall median, 1.0 vs. 0.75 and 0.83 for RECAP and REVOLVER, respectively).
Finally, we ran CloMu on data generated in the RECAP and TreeMHN papers, denoted as simulations III and IV, respectively (Table 1). We found that we matched RECAP's performance (Supplemental Material B.3.12; Supplemental Fig. S20) but that CloMu was outperformed by TreeMHN when using L = 5 hidden neurons but achieved approximately similar performance when using a linear model with no hidden neurons (Supplemental Material B.3.13; Supplemental Fig. S21). It is important to note that this results from the absence of interchangeable mutations or any mutations with shared causal properties in TreeMHN's simulations. Such mutations are present in real data as we will show. As previously stated, our model uses the independent clonal evolution assumption. To show that our model is robust to violations in this assumption, we generated causal relationship simulations with clonal cooperation, showing robustness by maintaining high causal inference performance on these data (Supplemental Material B.3.6; Supplemental Fig. S11). To test robustness to the inclusion of highly incorrect trees, we additionally generated simulations in Supplemental Material B.3.7, achieving highly accurate causal relationship prediction (Supplemental Fig. S12) and tree selection (Supplemental Fig. S13). Naturally, the quality of CloMu's outputs is limited by the quality of the patterns that exist in the input trees. In Supplemental Material B.3.7, we also show that CloMu does not predict causal relationships when all input trees are randomly generated.
In summary, this simulation study shows that CloMu is a versatile model and supports a wide variety of prediction tasks. CloMu's low-parameter neural network affords it the flexibility to model complex multimutation interactions while retaining resistance to overfitting.
CloMu identifies high-fitness mutations and causal relationships in a breast cancer cohort
We applied CloMu to a breast cancer cohort composed of 1918 tumors from 1756 patients that were sequenced using a gene panel
(Razavi et al. 2018). We used the same processing steps as in the RECAP paper (Christensen et al. 2020), restricting our analysis to SNVs that occur in copy-neutral autosomal regions followed by running SPRUCE (El-Kebir et al. 2016) to obtain a set 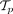 for each patient p. As in the RECAP analysis, we only retained mutations that occurred in at least 100 patients and removed patients that did
not contain any of these recurrent mutations. CloMu can be run on arbitrarily large trees; however, patients with vast numbers
(hundreds or thousands) of possible trees can greatly increase the computational requirements of running CloMu if not removed.
Therefore, we additionally removed patients with more than nine mutations and consequently had a large number of trees. This
left us with a data set with n = 1224 patients and m = 406 mutations. We ran CloMu with default settings and L = 5 hidden neurons. Our RL algorithm took <5 h to train the neural network on a laptop with a 2.4-GHz CPU and 64 GB of RAM
without using a GPU.
for each patient p. As in the RECAP analysis, we only retained mutations that occurred in at least 100 patients and removed patients that did
not contain any of these recurrent mutations. CloMu can be run on arbitrarily large trees; however, patients with vast numbers
(hundreds or thousands) of possible trees can greatly increase the computational requirements of running CloMu if not removed.
Therefore, we additionally removed patients with more than nine mutations and consequently had a large number of trees. This
left us with a data set with n = 1224 patients and m = 406 mutations. We ran CloMu with default settings and L = 5 hidden neurons. Our RL algorithm took <5 h to train the neural network on a laptop with a 2.4-GHz CPU and 64 GB of RAM
without using a GPU.
On the task of determining mutation fitness, we found that only eight mutations have high fitness values ( > 0.003) as shown in Figure 3A. Specifically, the highest fitness mutation was determined to be TP53, a known tumor-suppressor gene (Hollstein et al. 1991). The clone with only TP53 had a fitness (0.0125) over five times the median fitness value (0.00234). Additionally, in order, the next highest fitness mutations were determined to be CDH1, PIK3CA, GATA3, and MAP3K1, with fitness values between 0.00658 and 0.0104. Finally, the third tier consists of ARID1A, ESR1, and KMT2C with fitness values between 0.00384 and 0.00440. All of these are known driver mutations (Sondka et al. 2018).

CloMu determines mutation fitness, uncovers mutation similarity, and interchangeability, and finds relative causal relationships on a breast cancer cohort (Razavi et al. 2018). (A) CloMu identified eight mutations that have a far greater fitness value than other mutations. (B) Among these eight mutations, five mutations had latent representations with significant nonzero magnitudes. This plot shows that CDH1 and PIK3CA, as well as GATA3 and MAP3K1, are interchangeable on these data. (C) Relative causal relationships between mutation pairs (s, t). A value greater than zero (red) is indicative of mutation s (row) causing mutation t (column) in the same clone, whereas a value smaller than zero (blue) is indicative of mutation s inhibiting the occurrence of mutation t in the same clone.
On the task of determining interchangeability and shared mutation properties, we inspected the latent representations of all mutations. We found five mutations with significant magnitude latent representations, corresponding to the five highest fitness mutations: CDH1, GATA3, MAP3K1, PIK3CA, and TP53. Among these mutations, we found two pairs of interchangeable mutations (Fig. 3B). First, CDH1 and PIK3CA had similar latent representations, with similar values (0.580 and 0.491, respectively) in the first component and values of roughly zero (magnitude under 0.03) in the other components. Second, GATA3 and MAP3K1 have similar latent representations, with similar values (0.334 and 0.233, respectively) in the first component and values of roughly zero (magnitude under 0.001) in the other components. In fact, the only high-fitness mutation that our model determined to be highly unique is TP53. We found TP53 to have a separate property expressed by having a different dimension in its latent representation unseen by other mutations (corresponding to the second component). In Supplemental Material C.1.2 and Supplemental Figure S25, these latent representations are also shown to be associated with the hormone receptor status of the breast cancer patient. Additionally, Supplemental Figures S26 and S27 further analyze the latent representations and their association with fitness.
Finally, we analyzed relative causality among the five highest-fitness mutations. Recall that a relative causality value R(s, t) significantly greater than zero is indicative of mutation s causing mutation t in the same clone, whereas a value significantly smaller than zero is indicative of mutation s inhibiting the occurrence of mutation t in the same clone (see section “Prediction tasks”). We identified more negative (11 pairs) than positive (six pairs) causal relationships between pairs (s, t) of mutations (Fig. 3C). This indicates that high-fitness mutations, or drivers, increase the likelihood of passenger mutations by more than they increase the likelihood of other driver mutations. This is especially the case for TP53, which has a median relative causality value of −0.253 for the seven other highest-fitness mutations versus a relative causality value of 0.476 for the remaining mutations. Among the positive causal relationships, we found that CDH1 and GATA3 both cause PIK3CA. This is in agreement with TreeMHN's conclusions that CDH1 causes PIK3CA and that there is some level of co-occurrence between GATA3 and PIK3CA (Luo et al. 2023).
To summarize, CloMu agrees with TreeMHN on many causal relationship predictions while also producing interchangeable mutation predictions and fitness predictions that match known driver mutations. We assessed the confidence of our mutation fitness and causality predictions using bootstrapping in Supplemental Material C.1.1 and Supplemental Figures S22 through S24, finding that our predictions are stable across bootstrapped instances. A more systematic comparison of the causal relationship predictions of CloMu and TreeMHN is provided in Supplemental Material C.1.3 and Supplemental Table S3, showing that CloMu and TreeMHN fully agree on a subset of mutation pairs in which one would expect agreement despite our definition differences. We note that, unlike our simulations (Supplemental Material B.3.11), we did not detect any evidence of nonlinear multimutation causality. Finally, we tested CloMu's ability to predict subsequent mutations on subtrees (as explained in Supplemental Fig. S28) in Supplemental Material C.1.4 and Supplemental Figure S29, showing accuracy beyond baseline methods.
CloMu identifies orthogonally validated fitness values in an acute myeloid leukemia cohort
We analyzed a cohort of 123 acute myeloid leukemia (AML) patients that underwent high-throughput single-cell DNA panel sequencing (Morita et al. 2020). Because of the relatively small number of patients, we only analyzed the gene-level data with the exception of the gene FLT3, for which we additionally distinguished an internal tandem duplication mutation in FLT3 denoted as FLT3-ITD. We used the phylogenies inferred by Morita et al. (2020) using SCITE (Jahn et al. 2016). We restricted our analysis to the 77 patients with reported clonal prevalences. Again, for training efficiency reasons for our particular RL implementation (Supplemental Material A.1.4), we removed patients with more than 10 mutated genes. We arrived at n = 75 patients with m = 22 total mutated genes. One reason we chose to analyze this data set is that it includes clonal prevalence data, an independent source of fitness information that we used for orthogonal validation of our fitness predictions. Because we collapsed mutations to the gene level, we disabled the infinite sites mode in CloMu. We used default parameters with L = 5 hidden neurons. Training the neural network took <1 h on a laptop with a 2.4-GHz CPU and 64 GB of RAM without using a GPU.
We used CloMu to predict mutation fitness, interchangeability, and causality. Our model determined the most fit mutations to be NPM1 and DNMT3A, with fitness values of 0.338 and 0.184 relative to a median fitness of 0.019 (Fig. 4A). NPM1 and DNMT3A are well-known driver mutations for AML (Sondka et al. 2018). Corroborating our finding, TreeMHN determined the DNMT3A mutation to cause the largest number of other mutations (eight pairs) while only inhibiting one mutation. In addition, CloMu determined mutations ASXL1, GATA2, and U2AF1 to have high fitness, with values ranging from 0.0394 to 0.0546. On the other hand, we found that FLT3 had a below median fitness of 0.00585. This matches the fact that TreeMHN determined FLT3 to have the largest number of strong negative causal relationships (three pairs).
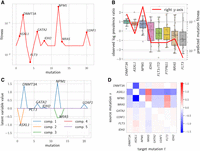
CloMu predicts fitness values validated by clone prevalence measurements and uncovers interchangeability and relative causal relationships on an AML data set (Morita et al. 2020). (A) CloMu identifies seven mutations with far greater fitness values than other mutations and identified one mutation (FLT3) with a substantially lower fitness value. (B) Box plot of log prevalence ratios for the eight mutations with the low standard error in log prevalence ratios (left y-axis), as well as a line showing our predicted mutation fitness values (right y-axis). The results show the validity of our fitness predictions. (C) Latent representations of mutations obtained from the L = 5 hidden neurons. This plot shows that GATA2 and U2AF1 are interchangeable on these data. (D) Relative causal relationships between mutation pairs (s, t). A value greater than zero (red) is indicative of mutation s (row) causing mutation t (column) in the same clone, whereas a value smaller than zero (blue) is indicative of mutation s inhibiting the occurrence of mutation t in the same clone.
To confirm the validity of our fitness predictions, we investigated the clone prevalence data that we did not use for training CloMu. Specifically, for each patient p and mutation s, let γ1(p, s) be the prevalence of the clone in which mutation s was introduced. Additionally, let γ0(p, s) be the prevalence of the parent clone of the clone in which mutation s was introduced. We define the log prevalence ratio as Γ(p, s) = log ((γ1(p, s) + 0.01)/(γ0(p, s) + 0.01)), which adds 0.01 to all prevalence measurements in order to avoid issues caused by dividing by or taking the log of very low numbers. Intuitively, if mutation s is highly fit, it should cause the clone that introduced mutation s to outgrow its parent clone, which would lead to a positive value for Γ(p, s). Conversely, a mutation s that does not increase the fitness of the clone that introduced it would lead to a nonpositive Γ(p, s). We analyzed the eight mutations with the lowest standard error in their Γ(p, s) measurements. Three of these mutations (DNMT3A, ASXL1, and NPM1) were determined to be highly fit by our model, and five were determined to have low fitness (IDH1, FLT3-ITD, PTPN11, NRAS, and FLT3). The log prevalence ratio measurements agreed with our model's conclusion on all eight mutations (Fig. 4B). In fact, the one mutation, FLT3, that our model predicted to have below median fitness also was determined to have the smallest median log prevalence ratio of −0.888. We further investigated the association between fitness and prevalence in Supplemental Material C.2.1 and Supplemental Figures S30 and S31 to ensure the validity of these results. Briefly, we found that although our method may have diminished sensitivity in detecting high fitness mutations that typically occur terminally, we believe that specificity is not affected by mutation timing.
CloMu also analyzed shared properties and interchangeability of mutations. We found GATA2 and U2AF1 to be interchangeable with similar latent representations (Fig. 4C). Additionally, we found NPM1 and DNMT3A to have shared properties. Specifically, the first dimension of their latent representation is nearly identical (shown in blue in Fig. 4C). However, NPM1 also has a shared property in the third latent dimension (shown in green in our plot), which is not present in DNMT3A. Finally, ASXL1 was determined to be completely unique, using the second dimension in its latent representation (shown in orange in our plot) unlike any other mutation. These patterns of interchangeability were also reflected in the causality relationships identified by CloMu (Fig. 4D). We provide additional plots and analyses of these latent representations in Supplemental Material C.2.2, Supplemental Figures S32 and S33.
The two main positive causal relationships found are from ASXL1 and NPM1 to NRAS—with R(ASXL1, NRAS) = 0.814 and R(NPM1, NRAS) = 0.505 (Fig. 4D). TreeMHN agrees with our conclusion that NPM1 causes NRAS. Although GeneAccord detects the co-occurrence of NPM1 and NRAS, the method does not have the capability to identify the directionality causal relationships. In our model, we found the strongest negative causal relationship to be from NPM1 to ASXL1, namely, R(NPM1, ASXL1) = −1.57. In addition, we identified a strong negative causal relationship in the reverse direction, namely, R(ASXL1, NPM1) = −0.667. Such bidirectional negative causality is indicative of mutual exclusivity. Indeed, TreeMHN also identified this bidirectional negative causal relationship. Mutual exclusivity between ASXL1 and NPM1 has been described previously in the literature (Pratcorona et al. 2012). Moreover, a strong negative association from ASXL1 to FLT3-ITD has also been reported (Pratcorona et al. 2012). Indeed, CloMu identified a strong negative causal relationship from ASXL1 to FLT3-ITD (R(ASXL1, FLT3 − ITD) = −0.754 and R(FLT3 − ITD, ASXL1) = 0.131). CloMu thus determines known causal relationships not picked up by other methods while also producing orthogonally validated fitness predictions. For a more systematic comparison of the causal relationship predictions of CloMu and TreeMHN, see Supplemental Material C.2.3 and Supplemental Table S4. We did not detect any evidence of nonlinear multimutation causality in this data set. Finally, we evaluated CloMu's ability to predict subsequent mutations on subtrees of these data in Supplemental Material C.2.4 and Supplemental Figure S34.
Discussion
In this work, we introduced CloMu, a tree generative model of cancer evolution, which can be used to perform tree selection; determine mutation fitness, causality, and interchangeability; and detect complex evolutionary pathways composed of sets of interchangeable mutations. Like TreeMHN (Luo et al. 2023) and HINTRA (Khakabimamaghani et al. 2019), CloMu models the generation of trees by independently determining the rate of new mutations occurring on a clone based on the clone's current mutations. CloMu uses a low-parameter neural network, which affords it the flexibility to model complex multimutation interactions while retaining resistance to overfitting. Using simulations without interchangeable mutations, we showed that CloMu matches TreeMHN's performance and outperforms all other baseline methods for the task of detecting causal relationships. Additionally, in the presence of interchangeable mutations, CloMu greatly outperformed TreeMHN as well. Our simulations further showed CloMu's accuracy in performing tree selection and identifying interchangeable mutations and their evolutionary pathways, outperforming competing methods. On real cancer data consisting of a breast cancer cohort (Razavi et al. 2018) and an AML cohort (Morita et al. 2020), CloMu was able to assess mutation fitness, determine interchangeable mutations and find causal relationships. We found that mutations with high fitness typically correspond to known driver mutations and that causal relationships matched previously reported associations and patterns of mutual exclusivity. For the AML data, we performed additional orthogonal validation by comparing predicted mutation fitness values to the prevalences of clones, finding high consistency. These findings show that CloMu is a versatile and effective method for a wide variety of prediction tasks regarding cancer evolution.
There are several directions for future work and expansions of CloMu. First, a limitation of our method is how sampling time affects our predictions. In practice, the time at which the tumor is sampled is nonrandom. If some mutation greatly increases the severity of the tumor as well as symptoms, the patient is more likely to have the tumor sequenced at this time. Consequently, if a clone is sufficiently severe, it may result in the tumor being sequenced before that clone has had time to acquire additional mutations (thus leading to lower fitness per our definition). Such a terminal clone may have high clonal prevalence, a signal that is not used by the model. Thus, beyond only considering tree topology and mutation placement, one could incorporate cancer progression models that incorporate measurements of clonal prevalences to improve predictions (Beerenwinkel et al. 2015). Second, one could use CloMu's low-dimensional representations of clones and mutations to predict measured properties of cancer clones or mutations, such as response to treatment or other patient outcomes. Third, one could extend CloMu's model to support somatic mutations beyond SNVs such as copy-number aberrations (CNAs) and their interplay with SNVs, including mutation loss. Fourth, one could improve CloMu's computational efficiency on data sets with large numbers of mutations per patient and vast numbers of possible trees per patient, especially when including mutations beyond SNVs such as CNAs. We note that to keep runtimes manageable, our present analyses restricted the number of SNVs per patient on real data sets. Fifth, the effects of germline predispositions can be captured by encoding them in the same way as mutations, that is, including an additional predisposition-specific “mutation” at the root node followed by the rest of the original tree. Similarly, one could encode therapeutics as occurring on all clones at the same time in the same way as mutations are encoded. Encoding such events in the input trees will enable the model to learn their downstream effects on future mutations. Sixth, we only evaluated multimutation effects on simulated data, which required a large number n ≈ 500 of patients and large effect sizes. Our simulations showed that it is feasible to detect complex patterns such as two mutations s and t only causing a third mutation r when paired together. Additionally, because of limited cohort sizes, we collapsed mutations in the real data to the gene level and only retained frequently recurrent genes, preventing the detection of mutation-specific patterns or the effects of rare genes. To perform more detailed analyses on real data in future work, we require much larger data sizes than currently available, which we expect to see in the future.
Methods
Independent clonal evolution problem
As previously stated, we take as input a set of possible tumor phylogeny trees 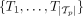 for each patient p among a cohort of n patients with m total mutations. Additionally, we have assumed independent clonal evolution. Although clonal cooperation has been described
in the literature (Tabassum and Polyak 2015; Kuipers et al. 2021), the resulting problem under our assumption still enables meaningful analyses even for cases with violations to this assumption
(Supplemental Material B.3.6). Our function,
for each patient p among a cohort of n patients with m total mutations. Additionally, we have assumed independent clonal evolution. Although clonal cooperation has been described
in the literature (Tabassum and Polyak 2015; Kuipers et al. 2021), the resulting problem under our assumption still enables meaningful analyses even for cases with violations to this assumption
(Supplemental Material B.3.6). Our function, 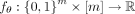 , outputs the logarithm of the rate at which the mutation occurs on the clone (Fig. 1A). As such, the probability of mutation s occurring on the clone c in the next δ time units is
, outputs the logarithm of the rate at which the mutation occurs on the clone (Fig. 1A). As such, the probability of mutation s occurring on the clone c in the next δ time units is 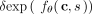 for small δ. Our trees are generated by iteratively building upon partial trees starting with the tree T(0) with only a single normal clone
for small δ. Our trees are generated by iteratively building upon partial trees starting with the tree T(0) with only a single normal clone 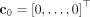 . Given a tree T(k) with clones
. Given a tree T(k) with clones 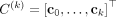 , the conditional probability
, the conditional probability 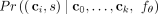 of the next mutation s to occur on clone ci among clones C(k), denoted by (ci, s), equals
of the next mutation s to occur on clone ci among clones C(k), denoted by (ci, s), equals
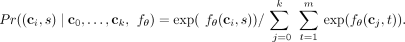 (1)
This yields a new tree T(k+1) with a new clone ck+1 with ci as its parent. Equivalently, we may represent the conditional probability density function with a softmax as P =
(1)
This yields a new tree T(k+1) with a new clone ck+1 with ci as its parent. Equivalently, we may represent the conditional probability density function with a softmax as P = 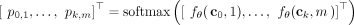 where
where 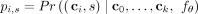 (see Fig. 1B). Importantly, we assume mutations are irreversible as clones can only be modified by adding additional mutations. Optionally,
the infinite site assumption can be enforced by setting the probabilities p0,s, …, pk,s of a new mutation s to zero if mutation s already exists in one of the clones c0, …, ck. We terminate the tree generation process when we reach a prespecified number ℓ ≤ m of mutations.
(see Fig. 1B). Importantly, we assume mutations are irreversible as clones can only be modified by adding additional mutations. Optionally,
the infinite site assumption can be enforced by setting the probabilities p0,s, …, pk,s of a new mutation s to zero if mutation s already exists in one of the clones c0, …, ck. We terminate the tree generation process when we reach a prespecified number ℓ ≤ m of mutations.
As stated in the Independent Clonal Evolution problem, our goal is maximizing the probability of observing the input data. Thus, we must describe exactly how to define
the probability 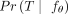 of any tree T. However, there are multiple possible ways of generating each tree. For example, consider the tree T with ℓ = 2 mutations having the normal clone c0 as the root with two children: a clone with mutation 1 and a clone with mutation 2. There are two ways to generate T, either mutation 1 or mutation 2 could occur first on c0 followed by the other mutation, namely, (c0, 1), (c0, 2) or (c0, 2), (c0, 1). We refer to a specific way of generating a tree T with ℓ mutations as a tree generating process G, which is an ordered list
of any tree T. However, there are multiple possible ways of generating each tree. For example, consider the tree T with ℓ = 2 mutations having the normal clone c0 as the root with two children: a clone with mutation 1 and a clone with mutation 2. There are two ways to generate T, either mutation 1 or mutation 2 could occur first on c0 followed by the other mutation, namely, (c0, 1), (c0, 2) or (c0, 2), (c0, 1). We refer to a specific way of generating a tree T with ℓ mutations as a tree generating process G, which is an ordered list 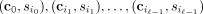 , where each pair indicates the source clone and the mutation that was added to it. Using the independent clonal evolution
assumption, we can now express the probability of a tree generating process
, where each pair indicates the source clone and the mutation that was added to it. Using the independent clonal evolution
assumption, we can now express the probability of a tree generating process 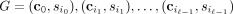 as
as
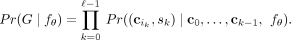 (2)
We denote the set of all tree generating processes that yield a tree T by
(2)
We denote the set of all tree generating processes that yield a tree T by 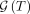 such that
such that 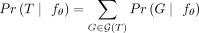 . As tumors are separate evolutionary processes, we have independence across patients and can therefore express the probability
of our data as
. As tumors are separate evolutionary processes, we have independence across patients and can therefore express the probability
of our data as
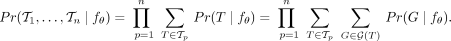 (3)
We note that HINTRA (Khakabimamaghani et al. 2019) and TreeMHN (Luo et al. 2023) solve the Independent Clonal Evolution problem with different types of models (Table 2). The function
(3)
We note that HINTRA (Khakabimamaghani et al. 2019) and TreeMHN (Luo et al. 2023) solve the Independent Clonal Evolution problem with different types of models (Table 2). The function 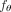 used by HINTRA explicitly enumerates all 2m × m clone–mutation pairs. The advantage of HINTRA's model is its flexibility to fit any pattern of clone-to-mutation interactions,
but this comes at the expense of severely restricting its scalability to a small number of mutations. On the other hand, TreeMHN
defines
used by HINTRA explicitly enumerates all 2m × m clone–mutation pairs. The advantage of HINTRA's model is its flexibility to fit any pattern of clone-to-mutation interactions,
but this comes at the expense of severely restricting its scalability to a small number of mutations. On the other hand, TreeMHN
defines 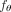 using a linear model of additive rate coefficients for all m × m pairs of mutations. As we showed in the section “Benchmarking on simulated data,” this scales well but does not capture nonadditive
clone-to-mutation interactions nor shared properties of mutations such as two mutations similarly affecting the probability
of other mutations. As discussed in the section “CloMu: low-parameter neural network trained via reinforcement learning,”
in our method CloMu, the model
using a linear model of additive rate coefficients for all m × m pairs of mutations. As we showed in the section “Benchmarking on simulated data,” this scales well but does not capture nonadditive
clone-to-mutation interactions nor shared properties of mutations such as two mutations similarly affecting the probability
of other mutations. As discussed in the section “CloMu: low-parameter neural network trained via reinforcement learning,”
in our method CloMu, the model 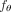 is a two-layer neural network with L = 5 ≪ m hidden neurons (Fig. 1C). The small number of parameters and the ability to model nonadditive clone-to-mutation interactions lead CloMu to achieve
flexibility like HINTRA while also being more resistant to overfitting than either TreeMHN or HINTRA on data sets with a large
number of mutations. Beyond the model type, there are other subtle differences. Rather than considering the set
is a two-layer neural network with L = 5 ≪ m hidden neurons (Fig. 1C). The small number of parameters and the ability to model nonadditive clone-to-mutation interactions lead CloMu to achieve
flexibility like HINTRA while also being more resistant to overfitting than either TreeMHN or HINTRA on data sets with a large
number of mutations. Beyond the model type, there are other subtle differences. Rather than considering the set 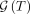 of all tree generative processes of a tree T, HINTRA considers a single tree generative process G to compute the probability of T.
of all tree generative processes of a tree T, HINTRA considers a single tree generative process G to compute the probability of T.
Prediction tasks
We gave a brief description of the prediction task in the Results. These prediction tasks are elaborated on and defined more
precisely here. Importantly, these prediction tasks correspond to postprocessing the model 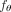 learned only once for each data set
learned only once for each data set  .
.
Reducing phylogeny uncertainty can be accomplished by selecting trees 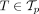 with a high value of
with a high value of 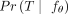 for each patient p (Fig. 1D). To select a single tree per patient, one can simply choose the tree T that maximizes
for each patient p (Fig. 1D). To select a single tree per patient, one can simply choose the tree T that maximizes 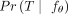 .
.
For determining the fitness of mutations, we consider the set {c1, …, cm} of clones in which each clone cs consists of only mutation s. Intuitively, the fitness of a clone cs and, consequently, mutation s is proportional to the rate at which a mutation would occur on the clone (Fig. 1E). As such, normalizing this over all clones c1, …, cm yields the expression of mutation fitness Fs for mutations s:
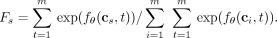 (4)
As an example, if the fitness Fs of mutation s is 10 times larger than the fitness Ft of mutation t, this implies mutations occur on the clone cs at a rate that is 10 times higher than the rate at which new mutations occur on the clone ct. Although the fitness value is defined only in terms of clones with a single mutation, we typically expect the fitness value
to generalize such that on all clones, having mutation s rather than t will increase the rate of new mutations occurring. Additionally, our fitness definition assumes that an increasing rate of
mutations on a clone is associated with clonal expansion. However, there exist some exceptions to this assumption. For instance,
mutations that interfere with DNA mismatch repair mechanisms may increase the rate of mutations, and thus, our definition
may overestimate their fitness. That is, unless there is specific evidence of nonlinear effects of combinations of mutations
in the data set, our regularized neural network model would naturally predict the same linear effect of a mutation on the
rates of all other mutations for all clones. In cases with such nonlinear effects, assigning a single fitness value to a mutation
will not capture these effects. An important note is that our fitness predictions for a mutation s are entirely learned based on the occurrence of additional mutations in clones with mutation s in the phylogeny. As such, if mutation s is always terminal, our model would not be able to conclude s to be high fitness. This is an intrinsic limitation of only using tree data but could be addressed by incorporating additional
information such as clonal prevalence values.
(4)
As an example, if the fitness Fs of mutation s is 10 times larger than the fitness Ft of mutation t, this implies mutations occur on the clone cs at a rate that is 10 times higher than the rate at which new mutations occur on the clone ct. Although the fitness value is defined only in terms of clones with a single mutation, we typically expect the fitness value
to generalize such that on all clones, having mutation s rather than t will increase the rate of new mutations occurring. Additionally, our fitness definition assumes that an increasing rate of
mutations on a clone is associated with clonal expansion. However, there exist some exceptions to this assumption. For instance,
mutations that interfere with DNA mismatch repair mechanisms may increase the rate of mutations, and thus, our definition
may overestimate their fitness. That is, unless there is specific evidence of nonlinear effects of combinations of mutations
in the data set, our regularized neural network model would naturally predict the same linear effect of a mutation on the
rates of all other mutations for all clones. In cases with such nonlinear effects, assigning a single fitness value to a mutation
will not capture these effects. An important note is that our fitness predictions for a mutation s are entirely learned based on the occurrence of additional mutations in clones with mutation s in the phylogeny. As such, if mutation s is always terminal, our model would not be able to conclude s to be high fitness. This is an intrinsic limitation of only using tree data but could be addressed by incorporating additional
information such as clonal prevalence values.
We determine causal relationships between pairs of mutations. By causal relationships, we mean temporal patterns of co-occurrence
and mutual exclusivity on a clonal level in the context of phylogenetic tree. Under our simplified model of clonal evolution,
these patterns are causal. However, the biology of clonal evolution is too complex to know definitively if they are causal
or simply correlative in the true biological system of clonal evolution. We define absolute causality as
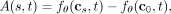 (5)
namely, the log ratio of rates of mutation t occurring on clone cs versus clone c0 (Fig. 1F). Recall that
(5)
namely, the log ratio of rates of mutation t occurring on clone cs versus clone c0 (Fig. 1F). Recall that 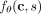 is the logarithm of the rate at which a mutation s occurs on clone c. In practice, some threshold value τ > 0 in the strength of the causal relationship must be used in order to avoid false
positives. Therefore, for absolute causality, we say s causes t if
is the logarithm of the rate at which a mutation s occurs on clone c. In practice, some threshold value τ > 0 in the strength of the causal relationship must be used in order to avoid false
positives. Therefore, for absolute causality, we say s causes t if 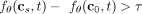 , s inhibits t if
, s inhibits t if 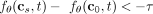 , and there is no causal relationship from s to t otherwise. Note that causality is bidirectional, namely, we can assess causality from t to s by evaluating
, and there is no causal relationship from s to t otherwise. Note that causality is bidirectional, namely, we can assess causality from t to s by evaluating  , amounting to a total of nine possible pairs of causal relationships between any two mutations. One potential issue with
absolute causality is that a high fitness mutation s may increase the rates of all other mutations t ≠ s and thus have a positive causal relationship with all other mutations t. To address this issue, we define relative causality R(s, t) from s to t as
, amounting to a total of nine possible pairs of causal relationships between any two mutations. One potential issue with
absolute causality is that a high fitness mutation s may increase the rates of all other mutations t ≠ s and thus have a positive causal relationship with all other mutations t. To address this issue, we define relative causality R(s, t) from s to t as
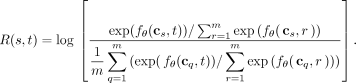 (6)
This measures how much a mutation s increases the relative probability that t will be the next mutation to occur on the clone, relative to the average of all clones cq with one mutation q. As an example, consider three mutations s, t, and r such that R(s, t) = R(r, t) + x. Then the probability that the next mutation is t on a clone that already has s is exp (x) times larger than the probability that the next mutation is t on a clone that already has r. Let 0.1 be the probability that the next mutation on a clone with r is mutation t. Also, let x = log2. An example further showing relative and absolute causality is provided in Supplemental Table S1. Then the probability of the next mutation on a clone with s being mutation t is exp (log2) · 0.1 = 0.2. Similarly to absolute causality, we say s causes t if R(s, t) > τ, s inhibits t if R(s, t) < −τ, and there is no causal relationship from s to t otherwise. In general, one should use absolute causality if one simply wants to know if having mutation s increases the rate of mutation t occurring for any reason including mutation s being high fitness. Alternatively, one should use relative causality if one wants to know if there is a specific relationship
between s and t such that s increases the likelihood of t occurring relative to all other mutations. Additionally, we define multimutation causality to capture the nonlinear effect of a combination of multiple mutations causing some other mutation (Supplemental Material A.2.2).
(6)
This measures how much a mutation s increases the relative probability that t will be the next mutation to occur on the clone, relative to the average of all clones cq with one mutation q. As an example, consider three mutations s, t, and r such that R(s, t) = R(r, t) + x. Then the probability that the next mutation is t on a clone that already has s is exp (x) times larger than the probability that the next mutation is t on a clone that already has r. Let 0.1 be the probability that the next mutation on a clone with r is mutation t. Also, let x = log2. An example further showing relative and absolute causality is provided in Supplemental Table S1. Then the probability of the next mutation on a clone with s being mutation t is exp (log2) · 0.1 = 0.2. Similarly to absolute causality, we say s causes t if R(s, t) > τ, s inhibits t if R(s, t) < −τ, and there is no causal relationship from s to t otherwise. In general, one should use absolute causality if one simply wants to know if having mutation s increases the rate of mutation t occurring for any reason including mutation s being high fitness. Alternatively, one should use relative causality if one wants to know if there is a specific relationship
between s and t such that s increases the likelihood of t occurring relative to all other mutations. Additionally, we define multimutation causality to capture the nonlinear effect of a combination of multiple mutations causing some other mutation (Supplemental Material A.2.2).
There is an extensive body of work on identifying mutations that are mutually exclusive or co-occur (Leiserson et al. 2015; Dao et al. 2017; Kim et al. 2017; Kuipers et al. 2021). Briefly, mutations in genes in the same biological pathway tend to be mutually exclusive as any individual mutation dysregulates
the same pathway (Yeang et al. 2008). On the other hand, some mutations tend to co-occur as their simultaneous presence might be beneficial for cancer progression
(Muzny et al. 2012; Wang et al. 2014; Hill et al. 2015; Ulz et al. 2016). Although many previous works have focused on identifying such mutations at the patient level without directly considering
their effects on subsequent mutations, here we seek to identify interchangeable mutations that have similar impacts on subsequent
mutation evolution. That is, we define two mutations s ≠ t to be interchangeable if it holds that 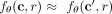 for all mutations
for all mutations 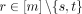 and for any two clones c and c′ that are identical except for c containing mutation s but not t, and vice versa for c′ (Fig. 1G). Note that this definition does not constrain the causal relationship among s and t. If s and t are interchangeable and pairwise inhibitory, this could be a result of them belonging to the same functional pathway. Conversely,
if s and t are interchangeable and co-occur, it could be a result of s and t having an additive effect when simultaneously present. The probability
and for any two clones c and c′ that are identical except for c containing mutation s but not t, and vice versa for c′ (Fig. 1G). Note that this definition does not constrain the causal relationship among s and t. If s and t are interchangeable and pairwise inhibitory, this could be a result of them belonging to the same functional pathway. Conversely,
if s and t are interchangeable and co-occur, it could be a result of s and t having an additive effect when simultaneously present. The probability 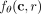 for all r is entirely determined by the values of the hidden neurons of our neural network when c is taken as an input. Consequently, two clones c and c′ behave identically according to our model if their latent representations (hidden neuron values) are identical. Thus, the
clone cs with only mutation s and the clone ct with only mutation t behave identically if they have identical latent representations. Rather than comparing the latent representations on vast
numbers of pairs of clones differing by one mutation, we define the latent representation of a mutation s as the latent representation of the clone cs and predict two mutations to be interchangeable if their latent representations are very close in terms of Euclidean distance.
for all r is entirely determined by the values of the hidden neurons of our neural network when c is taken as an input. Consequently, two clones c and c′ behave identically according to our model if their latent representations (hidden neuron values) are identical. Thus, the
clone cs with only mutation s and the clone ct with only mutation t behave identically if they have identical latent representations. Rather than comparing the latent representations on vast
numbers of pairs of clones differing by one mutation, we define the latent representation of a mutation s as the latent representation of the clone cs and predict two mutations to be interchangeable if their latent representations are very close in terms of Euclidean distance.
A pathway of interchangeable mutations requires that mutations in some set A and some set B are required together in order to increase the likelihood of mutations in some set C (Fig. 1H). Mathematically, if t ∈ A, s ∈ B, and r ∈ C, this implies 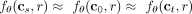 and
and 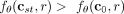 , where cst is the clone that contains only mutations s and t. Note that the case in which each set consists of a single mutation, namely, |A| = |B| = |C| = 1, corresponds to multimutation causality (for discussion, see Supplemental Material A.2.2). For a detailed description on how we infer pathways of interchangeable mutations, see Supplemental Material A.2.1. A final prediction task is the inference of consensus trees, which are a small number of trees that best summarize the diversity
of trees in the cohort (Supplemental Material A.2.3). Finally, we use bootstrapping to assess confidence of CloMu's predictions (Supplemental Material A.3).
, where cst is the clone that contains only mutations s and t. Note that the case in which each set consists of a single mutation, namely, |A| = |B| = |C| = 1, corresponds to multimutation causality (for discussion, see Supplemental Material A.2.2). For a detailed description on how we infer pathways of interchangeable mutations, see Supplemental Material A.2.1. A final prediction task is the inference of consensus trees, which are a small number of trees that best summarize the diversity
of trees in the cohort (Supplemental Material A.2.3). Finally, we use bootstrapping to assess confidence of CloMu's predictions (Supplemental Material A.3).
CloMu: low-parameter neural network trained via reinforcement learning
CloMu uses a two-layer neural network with a small number L of hidden neurons for the function 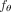 (Fig. 1C). This model has 2Lm + m + L = O(Lm) total parameters. In experiments in this paper, we use L = 5 hidden neurons. As the number of parameters grows linearly in the number of mutations, this model is very resistant to
overfitting. Moreover, we made a slight modification to the neural network to allow for easier regularization, which is used
primarily for the sake of interpretability but also is used to avoid overfitting on some extremely small data sets. This modification
did not affect the scaling of the number parameters. For more details, see Supplemental Material A.1.1.
(Fig. 1C). This model has 2Lm + m + L = O(Lm) total parameters. In experiments in this paper, we use L = 5 hidden neurons. As the number of parameters grows linearly in the number of mutations, this model is very resistant to
overfitting. Moreover, we made a slight modification to the neural network to allow for easier regularization, which is used
primarily for the sake of interpretability but also is used to avoid overfitting on some extremely small data sets. This modification
did not affect the scaling of the number parameters. For more details, see Supplemental Material A.1.1.
The small number L of hidden neurons allows us to form a small L-dimensional representation of any input clone c. Applying this to the set {c1, …, cm} of clones with only one mutation, we obtain latent representations lt = [l1, …, lL] ∈ ℝL for each mutation t ∈ [m] by observing the values of the L hidden neurons given the clone ct. The latent representation encapsulates any effect the mutation could have on some clone according to the model, and thus,
similarity of two latent representations also indicates similarity between the two mutations. In practice, we use the Euclidean
distance 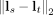 between latent representations to determine the interchangeability of any pair (s, t) of mutations. For the sake of interpretability, we subtract the median value across all mutations from each component of
each latent representation.
between latent representations to determine the interchangeability of any pair (s, t) of mutations. For the sake of interpretability, we subtract the median value across all mutations from each component of
each latent representation.
For the task of modeling evolutionary pathways of interchangeable mutations, it is vital to understand when two mutations are required together in order to cause a third mutation. Having a nonlinear model such as our neural network is thus required for this task. Specifically, we find evolutionary pathways of interchangeable mutations through the following, which is described extensively in Supplemental Material A.2.1. First, we evaluate the probability of the tumor evolving in some evolutionary pathway according to our model. Then, we evaluate the probability of the tumor evolving in that same pathway under a null hypothesis in which mutations have no effect on clonal evolution. Finally, we search for evolutionary pathways in which the probability according to our model vastly exceeds the null hypothesis probability.
The sets  and especially
and especially 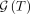 can be very large. For instance, if T is the star tree in which 10 mutations are made on the root node,
can be very large. For instance, if T is the star tree in which 10 mutations are made on the root node, 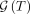 will have 10! = 3,628,800 elements. Explicitly calculating
will have 10! = 3,628,800 elements. Explicitly calculating 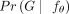 for every
for every 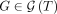 and
and 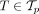 can be infeasible. To avoid doing this, we use reinforcement learning to train the neural network, that is, infer model parameters
θ that maximize the likelihood of the data. Note that the use of reinforcement learning is a standard approach for training
neural networks (François-Lavet et al. 2018). We use policy gradients, adapting the REINFORCE method (Williams 1992), in which the model is given a reward when it generates a tree in the data set. Specifically, the reward is proportional
to how much increasing the probability of that tree would increase the overall log probability of our data set. Typically,
in reinforcement learning, the reward given to the model for a given action sequence is independent of the model itself. In
our case, in order to maximize the probability of observing the data, our reward is a function that depends on the model.
However, to correctly optimize our objective, the rewards must be treated only as numerical values, and the gradient of the
reward must not be taken with respect to the parameters. Although it is less common to have a changing reward function during
training, there exist other examples of this in the reinforcement learning literature. One such example is curiosity driver
exploration in which the intrinsic reward changes during training (Pathak et al. 2017). Another such example is maximum entropy gain exploration, in which entropy is maximized in addition to maximizing total
rewards (Pitis et al. 2020). In CloMu, as well as these papers, the reward changes based on how the probability of states/actions change owing to the
policy changing during training. As is typical, our model is trained via stochastic gradient descent on the typical policy
gradient's loss function. Additionally, we have modified our sampling procedure to increase training speed and accuracy in
the case that there are moderately few trees per patient and consequently adjusted the reward function. For more details,
see Supplemental Material A.1.4.
can be infeasible. To avoid doing this, we use reinforcement learning to train the neural network, that is, infer model parameters
θ that maximize the likelihood of the data. Note that the use of reinforcement learning is a standard approach for training
neural networks (François-Lavet et al. 2018). We use policy gradients, adapting the REINFORCE method (Williams 1992), in which the model is given a reward when it generates a tree in the data set. Specifically, the reward is proportional
to how much increasing the probability of that tree would increase the overall log probability of our data set. Typically,
in reinforcement learning, the reward given to the model for a given action sequence is independent of the model itself. In
our case, in order to maximize the probability of observing the data, our reward is a function that depends on the model.
However, to correctly optimize our objective, the rewards must be treated only as numerical values, and the gradient of the
reward must not be taken with respect to the parameters. Although it is less common to have a changing reward function during
training, there exist other examples of this in the reinforcement learning literature. One such example is curiosity driver
exploration in which the intrinsic reward changes during training (Pathak et al. 2017). Another such example is maximum entropy gain exploration, in which entropy is maximized in addition to maximizing total
rewards (Pitis et al. 2020). In CloMu, as well as these papers, the reward changes based on how the probability of states/actions change owing to the
policy changing during training. As is typical, our model is trained via stochastic gradient descent on the typical policy
gradient's loss function. Additionally, we have modified our sampling procedure to increase training speed and accuracy in
the case that there are moderately few trees per patient and consequently adjusted the reward function. For more details,
see Supplemental Material A.1.4.
We implemented CloMu in Python 3 using the PyTorch library. CloMu is open source and is available at GitHub (https://github.com/elkebir-group/CloMu).
Software availability
The code for CloMu and the used simulated and real data are available at GitHub (https://github.com/elkebir-group/CloMu). These are also available as Supplemental Code and Supplemental Data, respectively.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the Beerenwinkel laboratory and Xiang Ge Luo for providing access to TreeMHN simulation results. This work started as a course project in “CS598MEB: Computational Cancer Genomics.” M.E.-K. was supported by the National Science Foundation (CCF-2046488) as well as funding from the Cancer Center at Illinois.
Author contributions: S.I. and M.E.-K. designed the study. S.I. implemented the method and performed the analyses. S.I. and M.E.-K. interpreted the results and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277672.123.
- Received January 6, 2023.
- Accepted June 9, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








