
Genome-scale targeted mutagenesis in Brassica napus using a pooled CRISPR library
- Jianjie He1,2,4,
- Kai Zhang1,2,4,
- Shuxiang Yan1,2,
- Mi Tang1,2,
- Weixian Zhou1,2,
- Yongtai Yin1,2,
- Kang Chen1,2,
- Chunyu Zhang3 and
- Maoteng Li1,2
- 1Department of Biotechnology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, China;
- 2Key Laboratory of Molecular Biophysics of the Ministry of Education, Wuhan 430074, China;
- 3National Key Laboratory of Crop Genetic Improvement and College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, China
-
↵4 These authors contributed equally to this work.
Abstract
The recently constructed mutant libraries of diploid crops by the CRISPR-Cas9 system have provided abundant resources for functional genomics and crop breeding. However, because of the genome complexity, it is a big challenge to accomplish large-scale targeted mutagenesis in polyploid plants. Here, we demonstrate the feasibility of using a pooled CRISPR library to achieve genome-scale targeted editing in an allotetraploid crop of Brassica napus. A total of 18,414 sgRNAs were designed to target 10,480 genes of interest, and afterward, 1104 regenerated transgenic plants harboring 1088 sgRNAs were obtained. Editing interrogation results revealed that 93 of the 178 genes were identified as mutated, thus representing an editing efficiency of 52.2%. Furthermore, we have discovered that Cas9-mediated DNA cleavages tend to occur at all the target sites guided by the same individual sgRNA, a novel finding in polyploid plants. Finally, we show the strong capability of reverse genetic screening for various traits with the postgenotyped plants. Several genes, which might dominate the fatty acid profile and seed oil content and have yet to be reported, were unveiled from the forward genetic studies. Our research provides valuable resources for functional genomics, elite crop breeding, and a good reference for high-throughput targeted mutagenesis in other polyploid plants.
Routine strategies for random mutagenesis, including physical (Henry et al. 2015; Li et al. 2017), chemical (Tang et al. 2020; Jiang et al. 2022), and biological insertional methods (Alonso et al. 2003; Li et al. 2019), have been applied to generate plentiful genetic resources for plant functional genomics. However, these methods have inherent drawbacks: the weak correlation between the genotype and phenotype, and the requirement of a considerable amount of labor. Recently, clustered regularly interspaced short palindromic repeat-associated nuclease 9 (CRISPR-Cas9), which mediates targeted editing with the complex of a sgRNA and a Cas9 protein by recognizing the 20-bp target sites (Jinek et al. 2012), has emerged as a versatile tool for genome engineering (Knott and Doudna 2018). This system has been successfully used for the improvement of crops (Chen et al. 2019), domestication of wild plants (Lemmon et al. 2018; Li et al. 2018; Yu et al. 2021), and generation of clonal seeds (Wang et al. 2019). Moreover, high-throughput mutagenesis in diploid crops, such as rice (Lu et al. 2017; Meng et al. 2017; Chen et al. 2022), tomato (Jacobs et al. 2017), soybean (Bai et al. 2020), and maize (Liu et al. 2020), has already been achieved by the CRISPR-Cas9 owing to its easy manipulation (Shalem et al. 2015). However, to our best knowledge, there has yet to be a report about creating a mutant collection in plants with complex polyploid genomes using targeted mutagenesis.
Rapeseed (Brassica napus, AACC; 2n = 38), a typical polyploid and hybridized from Brassica rapa (AA; 2n = 20) and Brassica oleracea (CC; 2n = 18) ∼7500 years ago, is a vital oil crop with sequenced reference genomes (Chalhoub et al. 2014; Song et al. 2020). Thus far, several genes have been mutated to generate the desired traits in this crop by using the CRISPR-Cas9 system (He et al. 2021). For instance, self-incompatible rapeseed could be obtained by editing the BnS6-Smi2 gene (Dou et al. 2021). Knocking out the BnTT8 could generate mutants with yellow seeds, which lead to increased seed oil content (SOC) and the alteration of fatty acid composition (Zhai et al. 2020). In addition, rapeseed architecture could be improved by mutating two homoeologs of the BnMAX1 gene (Zheng et al. 2020). In our group, editing BnLPAT2 and BnLPAT5 genes led to wrinkled mature seeds and decreased SOC (Zhang et al. 2022). Despite the promising results, high-throughput screening of rapeseed genomes using CRISPR-Cas9 or other CRISPR-based genome editing systems has yet to be reported. Here we describe in detail a rapeseed mutant collection established with a pooled CRISPR library consisting of 18,414 sgRNAs that target 10,480 genes. Then, we show the editing efficiency and the strong capability of reverse and forward genetic screening of this transgenic collection.
Results
sgRNA design and characteristics of the selected target sites
First, 26 queries (Supplemental Fig. S1A) related to the reproductive organs were used to search within the TAIR website, and 2662 Arabidopsis thaliana genes were obtained. The gene GA4, of great interest, which participates in the gibberellic acid biosynthetic pathway, was also picked up. Additionally, 391 A. thaliana genes homologous to 808 highly expressed B. napus genes characterized in our transcriptomes of seeds under six developmental stages were also selected (Supplemental Fig. S1A). As a result, 3054 A. thaliana genes were obtained, corresponding to 11,873 B. napus homologies from the Darmor-bzh reference genome (Chalhoub et al. 2014). The 11,873 rapeseed genes were then divided into 2647 homoeologous groups, and each group corresponded to one A. thaliana gene (Supplemental Table S1). These B. napus genes, related to developing reproductive organs and crop yield, were used for CRISPR-Cas9-mediated targeted mutagenesis. For sgRNA design, the 20-bp target sites on the coding sequences (CDSs) of the chosen 11,873 B. napus genes, followed by the 3-nucleotide (nt) protospacer adjacent motif (PAM) of NGG, were picked out based on the following principles: first, select the ones covering as more homoeologs per group as possible; second, choose the ones closest to the start codons; and, third, three target sites per gene locus are taken if it is possible (Fig. 1A).
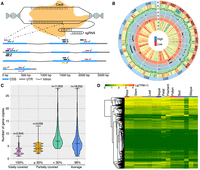
sgRNA design and characteristics of the selected 20-bp target sites. (A) The schematic diagram illustrates the three principles of sgRNA design. Each small bar near the corresponding target gene represents a 20-bp target site, and the identical ones are marked in the same full color. Ticks alongside the target sites indicate the selection, whereas crosses are on the opposite. This example displays the four homoeologs of the BnGPAT4 gene. (B) Circos plot visualizes the integrated information for the 18,414 selected target sites and the corresponding 10,480 target genes. (Tracks 1–3) Expression levels of the target genes in seeds 42 d after flowering (DAF; 1), 30-DAF seeds (2), and 17-DAF seeds (3), detected in our transcriptome data. (Tracks 4–6) Distribution of quantitative trait loci (QTLs) related to thousand-seed weight (TSW; 4), silique length (5), and seed oil content (6). (Tracks 7,8) Density distribution of the target sites (7) and the target genes (8). (Track 9) Nineteen chromosomes of the reference genome. (C) Density distribution of the target sites across the number of homoeologs. The widest part of the violin plot stands for the highest density of target sites; the center line is the median; and the top and bottom lines represent the entire data range (one group containing 65 homoeologs was removed from this analysis). An example for interpretation of this plot is as follows: Each of the 3543 target sites covers all homoeologs from the same homoeologous group and so on for the rest. (D) Expression features of the representative target genes (n = 7791) in different tissues of the ZS11 cultivar. Among the 7791 genes, 6220 (79.8%) were identified as expressed (with TPM ≥ 1) in at least one of the 16 tissues; however, expression of the last 1571 genes (20.2%) was not detected in any tissue.
In total, 18,414 sgRNAs targeting 10,480 genes were designed, representing an average of 2.82 sgRNAs per gene locus (Supplemental Table S2). Further analysis showed that the target sites were distributed on all 19 chromosomes of the rapeseed genome, with the highest density found in A03 (Fig. 1B). We also discovered that one sgRNA could simultaneously target up to nine homoeologs (Fig. 1C). In addition, some target genes were found located in the quantitative trait locus (QTL) intervals related to thousand-seed weight (TSW), silique length, and SOC, suggesting the ability to validate the results from forward genetics (Fig. 1B). In addition to the expression during seed development, 79.8% of the analyzed 7791 target genes were found to express in other tissues of the ZS11 cultivar, indicating that various abnormal traits would be seen in the transgenic collection (Fig. 1B,D).
Quality evaluation of the pooled CRISPR library
The pooled synthesized target recognition sequences were ligated into the expression cassettes of the CRISPR-Cas9 (Supplemental Fig. S1B). To evaluate the quality, total sgRNAs amplified from the pooled plasmids were sequenced by high-throughput sequencing (HTS). These results showed that 89.74% of the sgRNA (16,228) read counts fell into the range between one and 1000. In contrast, only 0.43% were detected with 5000 or more, suggesting a good representation of the pooled plasmids (with an average read count of 432 and a standard deviation of 728) (Supplemental Fig. S2A,B). Further analysis showed that 96.7% of the sgRNA reads were accurate, indicating high accuracy efficiency (Supplemental Fig. S2C). Moreover, 98.2% of the designed sgRNAs and 99.8% of the target genes were detected in the HTS data (Supplemental Fig. S2C). Together, these results indicated that the constructed pooled plasmids were of high quality.
Next, we transformed the pooled plasmids into competent Agrobacterium cells GV3101 by electroporation, and all colonies grown on the selected LB plates were harvested. To evaluate the quality of the pooled Agrobacterium, we randomly selected 355 single colonies and sequenced the sgRNAs (Supplemental Fig. S2D; Supplemental Dataset S1). The results revealed that 52.1% of the colonies (185) harbored individual sgRNAs, representing 171 correct unique ones and an accuracy efficiency of 95.7%, and most of the mutations were 1-nt changes (Supplemental Fig. S2D). We hypothesized that the high percentage (47.9%) of the colonies harboring multiple sgRNAs was caused by the high concentration of the transformed plasmid library. To confirm this speculation, we electroporated the competent Agrobacterium cells with the threefold diluted pooled plasmids and randomly selected 82 single colonies for evaluation (Supplemental Fig. S3A; Supplemental Dataset S2). The sequencing results revealed that 92.7% of the colonies (76) contained single sgRNAs, representing 74 unique ones and a correct efficiency of 97.4%, thus verifying the hypothesis. These results indicated the pooled agrobacteria library's high accuracy, efficiency, and good representation.
Identification of sgRNAs in the transgenic rapeseed collection
We next generated a collection of 1104 transgenic plants by Agrobacterium-mediated hypocotyl transformation with the nondiluted library. We then used HTS to sequence the sgRNA amplicons of each regenerated line in the T0 generation (Supplemental Fig. S4A; Supplemental Table S3; Supplemental Dataset S3). These results showed that 84.2% of the transgenics (929) harbored at least one single sgRNA, indicating a high transformation efficiency (Supplemental Fig. S4B). Moreover, among the 929 positive transgenic lines, 88.3% of them (820) contained single sgRNAs; 11.3% of them (103) harbored two to three different sgRNAs; and a small proportion of them (0.6%) was identified with four to five distinct sgRNAs (Supplemental Fig. S4C). Altogether, we detected 1088 sgRNAs covering 1834 genes in the transgenic collection, and 97.2% of them (1058) were identified as accurate (Fig. 2A), consistent with the correct efficiency of the pooled CRISPR library.
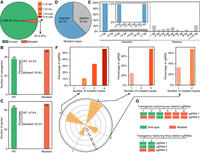
Identification of the 20-nt target recognition sequences and editing efficiency evaluation of the constructed transgenic collection in the T0 generation. (A) The ratio of the correct target recognition sequences to the mutated ones. (del.) Deletion, (ins.) insertion, and (sub.) substitution. (B) Editing efficiency analysis with 86 randomly selected positive transgenic plants. (WT) Wild type. (C) Editing evaluation of the 178 genes in the 86 selected transgenic lines. (D) The proportion of the number of insertions (n = 79) to that of the deletions (n = 47). (E) Percentages of different lengths of inserted and deleted base pairs. (F) Evaluation of editing efficiency of sgRNAs targeting different numbers of homoeologs. The numbers inside the polar plot represent the ratios of mutagenic sgRNAs to the total examined that target the same number of homoeologs; the ones alongside indicate the numbers of homoeologs. The top three bar plots detail the percentages of sgRNAs editing different numbers of homoeologs. For sgRNAs targeting one to four homoeologs: n = 40, n = 41, n = 4, n = 11. (G) Editing identification of the six transgenics harboring two distinct sgRNAs and two transgenic plants with three different sgRNAs. The eight transgenic plants were randomly selected for the examination.
Evaluation of the editing efficiency
We then evaluated the editing efficiency of the positive transgenic collection using the 86 randomly chosen lines of the T0 generation (Supplemental Table S4; Supplemental Dataset S4). These results revealed that 48 out of the 86 plants were identified with editing on the 20-bp target sites, representing an editing efficiency of 55.8% (Fig. 2B). Among the 178 target genes in the 86 examined plants, 93 genes were detected with small insertions or deletions (indels), representing an editing efficiency of 55.2% (Fig. 2C). Genotyping results revealed that 44.1% of the edited genes (41) belonged to monoallelic mutation, and 35 genes belonged to biallelic mutation (14 homozygotes). The last 17 genes were chimeras (Supplemental Fig. S4D). Among them, 32 genes (34.4%) were identified with the genotypes of +1 bp/wild type (WT), and up to four alleles were detected in the chimeric genes (Supplemental Fig. S4E,F). These results highlighted the high effectiveness of the editing. Moreover, sequence variations of six target sites were discovered between the recipient and the reference genomes, unveiling genome variations among distinct cultivars (Supplemental Table S5). We then selected 19 off-target sites for editing examination, possibly targeted by the mutagenic sgRNAs. The results revealed that only one off-target site was detected with editing, suggesting the low off-target effect (Supplemental Table S6; Supplemental Dataset S5).
Examination of the editing events showed that around two-thirds were detected as insertions, with 97.6% characterized as 1-bp insertions; however, the deleted sequence's length did not show a specific preference (Fig. 2D,E). Using the events of 1-bp insertion, we found that most (85.7%) of the cleavage sites by Cas9 were 3-bp upstream of the PAM sequences, which was in line with the previous study (Supplemental Fig. S4G; Jinek et al. 2012). Further analysis using the mutagenic sgRNAs targeting different numbers of homoeologs showed that Cas9-mediated double-stranded breaks tended to occur in all target sites guided by the same sgRNAs, which was an intriguing novel finding in the polyploid plants. The underlying mechanism remained elusive (Fig. 2F). We then analyzed the editing events in the transgenics with multiple sgRNAs (Fig. 2G). The results revealed that three of the six transgenic plants containing two distinct sgRNAs were detected with mutations mediated by both sgRNAs. However, only one mutagenic sgRNA was seen in the two plants containing three different sgRNAs.
Because multiple sgRNAs were separated into distinct single sgRNAs in the successive generation of the Agrobacterium colonies (Supplemental Fig. S3B), we speculated that various sgRNAs harbored by a single plant were from different Agrobacterium cells containing distinct single sgRNAs. To confirm this, we infected the rapeseed with the mixed Agrobacterium solution containing two distinct sgRNAs in equal concentration, and six out of the 19 regenerated lines harbored the two sgRNAs, thereby substantiating this hypothesis (Supplemental Fig. S3C,E). Furthermore, we showed that the mutations mediated by multiple sgRNAs in one transgenic line could be stably inherited from the T0-to-T1 generations (Supplemental Fig. S5). These results suggested that higher-order mutagenesis could be realized by transforming the recipient materials with a pooled Agrobacterium solution containing distinct multi-sgRNA plasmids.
Reverse genetic screening with the postgenotyped plants
To examine the capability of reverse genetic screening, we interrogated various traits of the postgenotyped positive transgenic plants. Phenotyping results of the seed-related traits showed that mutating specific genes could lead to changes in seed area, the aspect ratio of seed, and TSW (Fig. 3A). For instance, mutating the BnLAP6 gene could result in a significant decrease in seed area, and editing the BnEMB3147 gene could distort the typical shape of the seeds. Furthermore, substantial changes in root lengths and fatty acid profiles were uncovered in specific mutants, as shown in Figure 3, B and C. Noticeably, compared with the WT, knocking out the four homoeologs of the BnKANT3 gene might lead to an increased percentage of linoleic acid (C18:2) and to longer roots at the seed germination stage of 6 d after imbibition, which had not yet been reported (Fig. 3B,C). Nonetheless, further experiments are required to consolidate these results and illuminate the underlying molecular mechanisms.
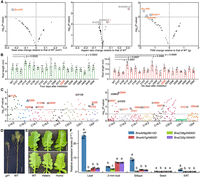
Phenotyping of the selected positive transgenic lines after interrogating the editing. (A) Phenotyping of seed-related traits. Each dot represents a transgenic line, and the corresponding edited gene is alongside. (TSW) Thousand-seed weight. (B) Phenotyping of root length at the stages of 4 and 6 d after seed imbibition. P-values less than 0.01 are shown. The ID of the plant with four edited homoeologs of the BnKNAT3 gene is marked in red. (C) Phenotyping of the fatty acid composition in self-pollinated (by bagging; left panel) and open-pollinated (naturally mature; right panel) seeds. Each dot represents a transgenic plant, and the mutant IDs are marked. (ODP) Oleic desaturation proportion. The asterisk indicates the ID of the plant with four edited homoeologs of the BnKNAT3 gene. Seeds used for the phenotyping in A–C were from plants of the T0 generation. (D) A representative photo displays the dwarf phenotype of the gif1 mutant in the T1 generation. Scale bar, 15 cm. (E) Representative photo shows the glossy phenotype of the examined kcs6.A09 homozygous mutant in the T1 generation. (Hetero) Heterozygote, (Homo) homozygote. Scale bar, 2 cm. (F) Relative expression levels of the four homoeologs of the BnKCS6 gene in the indicated five tissues of J9709 WT. (SAT) Shoot apical tissue. Different letters represent significant differences at P < 0.05 by two-way ANOVA with Tukey's honest significant difference (HSD). In A–C, the Student's t-test was used for the statistical analysis.
Two mutants of the T1 generation showed visualized dwarf and glossy phenotypes, respectively (Fig. 3D,E). For further detailed analyses, we selected those mutants with target genes of biallelic mutations and/or no exogenous T-DNA fragments (Supplemental Fig. S6; Supplemental Dataset S6). These results revealed that knocking out of the four homoeologs of the BnGIF1 gene led to the dwarf plants (Fig. 3D) and a decrease of seed number per silique and silique length (Supplemental Fig. S7A,B) but an increased percentage of linolenic acid (C18:3) (Supplemental Fig. S7C). In addition, compared with the J9709 WT, the gif1 mutants of rapeseed are leafier, with smaller flowers of narrow petals and abnormal inflorescences (Supplemental Fig. S8A); the trait of narrow petals is in line with that of the A. thaliana gif1 mutants (Kim and Kende 2004). The glossy mutant was identified with the homozygous mutated gene of BnCER6.A09, which is one copy of the BnCER6 gene located on Chromosome A09 and involved in the biosynthesis of very long chain fatty acids (VLCFs) (Fig. 3E; Supplemental Fig. S6; Jenks et al. 1995). Compared with the WT, RT-qPCR results showed that the expression level of this gene was significantly lower in the leaf and 2-mm bud tissues of the homozygote. In contrast, no significant changes in the expression level and the phenotype were found in the heterozygote (Supplemental Fig. S7D). We then examined the relative expression levels of the four homoeologs of the BnCER6 gene in the five tissues of J9709 WT (Fig. 3F). The results showed that the expression level of BnCER6.A09 was significantly higher than that of the other copies in leaf and silique tissues, thus explaining the reason why knocking out only one copy of the BnCER6.A09 could lead to the glossy phenotype. Together, these results showed that the constructed mutant collection was powerful in reverse genetic screening.
Forward genetic screening with the positive transgenic collection
To illustrate the forward genetic screening, self-pollinated seeds from plants of the T0 generation were used for the study. We phenotyped the seeds’ oil content and fatty acid composition using 23 ungenotyped positive transgenic lines to screen for the potential mutants (Fig. 4A,B; Supplemental Fig. S9). These plants harbored sgRNAs targeting two or more homoeologs, as they were likely to contain the edited targets. According to the phenotyping results, nine lines with significant changes in the stable parameter of oleic desaturation proportion (ODP) and one transgenic line with an increase of SOC were used for the editing interrogation (Fig. 4A–C; Supplemental Table S7; Supplemental Dataset S7). These results showed that seven of 10 plants contained the mutated target genes. At the same time, no editing events were identified in the target sites of the last three plants (Fig. 4C).
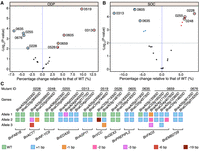
Illustration of the forward genetic screening. (A,B) ODP parameter (A) and SOC (B) of the 23 randomly chosen positive transgenic lines with sgRNAs targeting two or more homoeologs. (ODP) Oleic desaturation proportion, (SOC) seed oil content. Each dot represents a plant, and the IDs of the transgenic plants used for genotyping are marked alongside. The red indicates an increase, and the blue represents a decrease. In A and B, the Student's t-test was used for the statistical analysis. (C) Editing interrogation of the target genes in the 10 selected plants that show significant changes in the fatty acid profiles. The seeds used for the forward genetics were from the plants of the T0 generation.
Specifically, editing two homoeologs of the BnAGP11 gene might lead not only to a significant decrease of C18:1 and an increase of C18:2 (Supplemental Fig. S9) but also to delayed emergence of true leaves during seed germination (Fig. 4C; Supplemental Fig. S8B). Although no editing events were found in line 0255, the transgenic plant had wrinkled yellow leaves, suggesting other mutations, such as insertional mutagenesis underlying the phenotype (Fig. 4C; Supplemental Fig. S8C). Noteworthily, the sharp decrease of C18:3 by thoroughly knocking out the six homoeologs of the BnFAD3 gene might consolidate these findings, but further experiments are required to verify these results. In addition, other visible abnormal traits, such as abnormal petals and short filaments, were also seen in the field, providing valuable resources for future studies (Supplemental Fig. S8D–J). Above all, our generated transgenic collection is efficient for performing forward genetic screenings.
BnEDA32 and BnFAB1B.C01 might dominate the fatty acid composition and SOC
Two mutants with significantly altered fatty acid composition and SOC were used for further analysis. The results showed that thoroughly knocking out four homoeologs of the BnEDA32 gene (mutant ID: 0313) might result in a significant decrease of SOC by 36.7% and a reduction of oleic acid (C18:1) from 68.5% to 56.4% but an increase of ODP by 12.8%, an increase of C18:2 by 48.5%, and an increase of C18:3 from 6.5% to 10.1%, which had not yet been reported (Fig. 5A,B). In addition, RT-qPCR results showed that two homoeologs (BnaAnng21770D and BnaA04g04000D) of the BnEDA32 gene were significantly expressed relative to the others in the siliques 14 d after flowering (DAF) and in 26-DAF seeds, suggesting the main contribution to fatty acid composition and SOC from both genes (Fig. 5E).
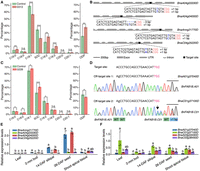
Significant alteration of the fatty acid profiles in two knockout mutants. (A) Fatty acid profile of the self-pollinated seeds in line 0313 of the T0 generation and the control background. (ODP) Oleic desaturation proportion, (SOC) seed oil content. (B) Detailed information for the editing interrogation of the four homoeologs of the BnEDA32 gene in the T0 generation. The PAM sequences are marked in red; the inserted nucleotides, in blue; and the deleted nucleotides, in blue dashes. The gene names are displayed on the right side of the structures. (C) Fatty acid profile of the self-pollinated seeds in the transgenic line 0228 of the T0 generation and the control background. (D) Editing interrogation in the two off-target sites of line 0228 in the T0 generation. The mismatched nucleotides are marked in lowercase, and the PAM sequences are marked in red and indicated by the black lines above the sequencing chromatograms. The arrow indicates the insertion site of the BnFAB1B.C01 gene. (E) Relative expression levels of the four homoeologs of the BnEDA32 gene in the five tissues of J9709 WT. (F) Relative expression levels of the four homoeologs of the BnFAB1B gene in the five tissues of J9709 WT. In A and C, the Student's t-test was used for statistical analysis: (n.s.) not significant, (*) P < 0.01, (**) P < 0.001, and (***) P < 0.0001. In E and F, different letters represent significant differences at P < 0.05 by two-way ANOVA with Tukey's HSD. (A,C,E,F) Error bars represent SDs.
Line 0228 showed a significant increase of SOC by 26.8% and an alteration of the fatty acid profile, such as increased C18:1 (from 64.6% to 67.7%), decreased C18:2 (from 19.8% to 16.7%), and a change in the ODP parameter by 3.6%. Although no editing events were found in the target genes of this plant, one off-target site, located in the CDS of the BnFAB1B.C01 gene, was identified with a 1-bp insertion (Fig. 5C,D). The edited off-target gene is homoeologous to the target ones. The expression level of this gene is significantly higher than the other three homoeologs in the leaf, 2-mm bud, 14-DAF silique, and 26-DAF seed tissues of the WT, thereby suggesting that mutating only this homoeolog might be sufficient to alter the fatty acid composition significantly and may increase the SOC (Fig. 5F). These results provide helpful information for breeding elite B. napus cultivars with a high percentage of C18:1 and SOC.
Discussion
In this study, we accomplished genome-scale targeted editing in the polyploid crop B. napus. We first built a pooled CRISPR library comprising 18,414 sgRNAs targeting 10,480 genes and then unveiled its high quality. Afterward, we generated a transgenic rapeseed collection containing 1104 plants and identified the sgRNAs harbored by each line by HTS-based amplicon sequencing. Editing interrogation results revealed a mutation efficiency of 55.8% in the positive transgenic plants and 52.2% in the target genes. We also discovered that Cas9-mediated DNA double-stranded breaks tended to happen in all target sites guided by the same sgRNAs, a novel finding in polyploid plants. Finally, we showed the ability to perform reverse and forward genetic screenings with the constructed transgenic collection.
Compared with the pooled CRISPR libraries constructed in rice, which have an accuracy efficiency of ∼93% in the plasmids (Lu et al. 2017; Meng et al. 2017) and 79.1% in the transgenic positive rice plants (Meng et al. 2017), our established one is of higher accuracy efficiency (∼97%) in the plasmids and the transgenic positive rapeseed plants. Although the evaluated editing efficiency is higher in the transgenic rice plants (78.1%–86.5%) (Lu et al. 2017), our result is comparable to those (14.4%–68%) assessed by knocking out the individual genes in B. napus (Zhang et al. 2019; Zhai et al. 2020). In addition, compared with the large-scale functional genomic screenings using iFOX hunting (Ling et al. 2018) and EMS-mediated mutagenesis (Tang et al. 2020), our generated transgenic collection showed a markedly stronger correlation between the phenotypes and genotypes, easy screening, and stable inheritance during the functional genomic studies.
The mechanism(s) underlying the CRISPR-Cas9-mediated cut tendency to all target sites in rapeseed guided by the single sgRNA remains elusive. Our results showed that average on-target efficacy scores of total mutagenic sgRNAs were higher than those nonmutagenic ones under the human and mouse genome backgrounds, accounting for those targeting four or more homoeologs (Supplemental Fig. 10A,B). This might be caused by the small collection of nonmutagenic sgRNAs targeting three or more genes. In addition, the 13th nucleotide of the mutagenic sgRNAs targeting four or more gene loci strongly prefers guanine. On the contrary, no such biases were found in total or other mutagenic sgRNAs targeting two or fewer homoeologs with our data (Supplemental Fig. 10C,D). Nevertheless, quite a few nonmutagenic sgRNAs have high on-target efficacy scores comparable to those of the mutagenic ones, suggesting the different scoring mechanisms between B. napus and the mammal backgrounds. Using logistic regression or deep learning, our generated data would help improve the sgRNA on-target efficacy score prediction under the rapeseed genome background.
Editing specific target genes not identified as expressed in the transcriptome data might lead to abnormal phenotypes, as one example shown in our study (Supplemental Fig. S11). Public transcriptome data and our RT-qPCR results showed that the two target genes of line 0635 were not detected with any expression (Supplemental Fig. S11A,B). However, even though more experiments are required to consolidate these results, thorough editing of these two genes possibly has led to the significant perturbation of plant height observed in the T1 generation (Supplemental Fig. S11C,D). In addition, a substantial alteration of fatty acid composition was also found in this mutant in the T0 generation (Supplemental Fig. S9). The results suggested that the expression of some critical genes might be transient and difficult to capture. Our results thus provide novel insights into the roles of genes identified as nonexpressed.
Our results also indicate that the generation of abnormal phenotypes is not necessary to edit all of the homoeologs in polyploid plants, suggesting the evolutionary divergence of these homoeologs. Hence, our study provides valuable resources for gene functional evolution analysis. In addition, although further experiments are needed to consolidate the results, our findings imply that overexpressing BnaAnng21770D and BnaA04g04000D of the BnEDA32 gene or knocking out BnaC01g37100D of the BnFAB1B gene might result in the increase of SOC in rapeseed, which is valuable information for breeding elite B. napus cultivar with higher SOC and has yet to be reported. Moreover, because CRISPR-based genome editing in the hexaploid wheat has been successfully achieved (Li et al. 2022), our research provides a good reference for genome-scale targeted mutagenesis in this crop and other plants with polyploid genomes.
Nonetheless, our study has several drawbacks. First, the collection of the generated transgenic plants is small, and the recipient material is not an elite rapeseed cultivar. This problem can be solved by obtaining tremendous transgenic lines using the ZS11 or other elite cultivars as the recipient. Second, even though genome variations between the reference cultivar of Darmor-bzh and the recipient cultivar of J9709 were detected in a small ratio (3.3%), sequencing and assembly of the recipient genome are necessary for future study. Third, this study did not show an appropriate distribution of phenotypes from negative controls. This drawback can be overcome using the positive transgenic plants containing the random nontargeting sgRNAs. Last but not least, because of the strict limit of the PAM sequence of NGG, no target sites for CRISPR-Cas9 are available in 11.7% of the 11,873 selected genes. This obstacle can be easily solved using the near PAMless genome editor with SpRY protein (Walton et al. 2020), which has been successfully used in plants recently (Ren et al. 2021).
In summary, we showed the feasibility of genome-scale targeted mutagenesis in B. napus with complex polyploid genomes. Our works provide valuable resources for functional genomics and rapeseed breeding and a good reference for high-throughput genome screening in other polyploid plants.
Methods
Screening for target genes
Before the screening, a gene index table of A. thaliana genes and their homologies in B. napus was retrieved from the BnVIR online database (Yang et al. 2022). Then 26 queries (Supplemental Fig. S1A) related to the reproductive organs were used to search for the A. thaliana genes on the TAIR website (https://www.arabidopsis.org); as a result, a total of 2662 genes were obtained. The gene GA4, involved in the gibberellic acid biosynthetic pathway, was also of great interest and was selected. Furthermore, 808 highly expressed rapeseed genes (with an average FPKM ≥ 125) were obtained by analyzing our transcriptomes of seeds (from the high and low oil content materials) under six developing stages (10 DAF, 17 DAF, 24 DAF, 30 DAF, 36 DAF, 42 DAF). These 808 rapeseed genes correspond to 391 A. thaliana homologies. In total, 3054 A. thaliana genes were obtained, homologous to 11,873 B. napus homologies picked out with the gene index table, representing 2647 groups of homoeologs. The selected A. thaliana genes and the B. napus target ones are provided in Supplemental Table S1.
sgRNA design
For sgRNA design, the 20-bp target sites on the CDS of each gene, followed by the PAM of NGG, were picked up using the online tool of GPP sgRNA Designer (Doench et al. 2014). First, split CDSs of the 10,480 gene models were extracted using the AGAT tool (https://github.com/NBISweden/AGAT) with the input of the GFF3 and the genome FASTA files, followed by checking the sequence length ≥ 30 bp. For those lengths ≤ 30 bp, an in-house Python script was used to locate the NGG motif and obtain the upstream 20-bp target sites. For those lengths ≥ 30 bp, the target sites were retrieved with the GPP sgRNA Designer using the human reference genome (Human GRCh38, NCBI), following the parameter of CRISPRko and Hsu (2013) tracrRNA. All possible target sites were collected, regardless of the on-target efficacy scores. The total target sites were aligned against the rapeseed genome to exclude those with <3-nt mismatch with the off-target sites. The target sites containing fewer than four thymidines were also ruled out. Afterward, the target sites were picked out as follows. The target sites covering as many homoeologs as possible in each group were selected in priority. The following principle for selecting target sites is according to the distances between them and the start codons, and the closest ones were chosen using an in-house Python script. Ideally, three target sites covering each gene locus were selected for the guaranteed editing with high possibility; however, owing to the PAM limit of NGG, fewer than three target sites were available for some of the homoeologs. The primary data, chosen target sites, and the corresponding target genes are provided in Supplemental Table S2 and Supplemental Dataset S8.
Analysis of the target genes
Expression data of the 10,480 target genes in 17-DAF, 30-DAF, and 42-DAF seeds were obtained from our transcriptome data, and the values in tracks 1–3 of the Circos plot were displayed as log10(FPKM + 1). Data for the QTL related to SOC, silique length, and TSW were obtained from our previously published papers (Chao et al. 2017; Zhao et al. 2019). The expression levels of the representative 7791 target genes in other tissues of the ZS11 cultivar were retrieved from the BnTIR online database (Liu et al. 2021). CDSs of the ZS11 and Darmor-bzh cultivars were aligned against each other with the NCBI BLASTN (2.9.0) (Altschul et al. 1997) program with a search cutoff e-value of 1 × 10−10 to determine the IDs of identical genes between the two genomes. The BBH method was used to analyze the BLAST results to construct a gene index table between the ZS11 and Darmor-bzh gene models as described previously (Zhang et al. 2021). As a result, 7791 ZS11 genes identical to those of the target genes were picked out with the gene index table. The 7791 gene IDs of the ZS11 cultivar were used as queries to search within the BnTIR (now BnIR; http://yanglab.hzau.edu.cn/BnIR/expression_zs11) to obtain the expression data.
Pooled CRISPR library construction
The 18,414 20-nt target recognition sequences with adapters (full length < 100 nt) and their complementary ones were synthesized by an array-based method using 5′-DMT prevention of polymerization of the nucleoside, following the parameters of 1 fmol per oligonucleotide (oligo) and 12,000 oligos per DNA chip (GeneBiogist). The synthesized pooled single-stranded oligos were then eluted into a 2-mL centrifuge tube, followed by the agarose gel electrophoresis quality check. The pooled double-stranded oligos were formed with a 10-μL solution of 4.5-μL forward primer, 4.5-μL reverse primer, and 1-μL eluted single-stranded oligos under the following conditions: 5 min at 95°C, a descent from 94°C to 73°C (−1°C/min, 22 min in total), 30 min at 72°C, a drop from 71°C to 26°C (−1°C/min, 46 min in total), 2 h at 4°C, 5 min at 95°C, and then cooling down naturally. The pooled double-stranded oligos were then ligated into the linearized CRISPR-Cas9 expression vector DP-05 with the T4 DNA ligase under a 10-μL solution of 4-μL linearized DP-05 vectors (12.7 ng/μL), 1-μL annealed double-stranded oligos, 1-μL T4 buffer (10×), 1-μL T4 DNA ligase, and 3-μL ddH2O. During the ligation, plasmid-safe exonuclease was used to prevent self-ligation. Afterward, all ligated plasmids were transformed into the competent Escherichia coli cells of Dh5α and grown on the LB plates with kanamycin (0.1%). All colonies grown on the selection plates were collected, and the plasmids were extracted with a FastPure plasmid mini kit (Vazyme) according to the manufacturer's instructions. sgRNA amplicons of the plasmid library were amplified by a 100-μL PCR solution of 10-μL buffer (10×), 5-μL forward primer, 5-μL reverse primer, 1-μg DNA, and 80-μL ddH2O in the following conditions: 3 min at 95°C; 35 cycles of 30 sec at 94°C, 30 sec at 58°C, and 30 sec at 72°C; and final extension of 10 min at 72°C. The generated pooled amplicons were subject to HTS (Illumina HiSeq strategy) to evaluate the constructed plasmids’ quality.
Electroporation of Agrobacterium
The pooled plasmids were then transformed into the competent Agrobacterium cells of GV3101. The first transformation experiment was conducted using a multiporator (Eppendorf) with a solution of 5-μL pooled plasmids (305 ng/μL) and 100-μL competent cells (OD600 = 2.8) under an electroporation condition of 2500 V for 5 msec. A total of 355 single colonies were randomly picked out to check the quality of this pooled library. The second transformation was performed with a solution of 5-μL threefold diluted pooled plasmids (90 ng/μL) and 100-μL competent Agrobacterium cells (OD600 = 2.8) to estimate the percentage of single sgRNAs. A total of 82 single colonies were randomly selected to check the quality of the diluted library. Then the transformed Agrobacterium were grown on screened LB plates with the addition of kanamycin (0.1%), gentamicin (0.1%), and rifampicin (0.1%). All colonies were harvested and added with the sterile glycerol in a ratio of 7:3. These Agrobacterium solutions were stored under −80°C in refrigerators to transform rapeseed hypocotyls in future studies.
Rapeseed transformation
Agrobacterium-mediated hypocotyl transformation and tissue culture experiments were performed to generate transgenic seedlings as previously described (Zhang et al. 2020), and the rapeseed cultivar J9709 was used as the recipient material. In brief, hypocotyls were infected by the pooled Agrobacterium solutions for 10 min and cocultivated for 48 h. Subsequent experiments, including induction of calluses, shoots, and roots, were performed to obtain the regenerated B. napus plants. Then the rooted plantlets were transplanted into the pots with nutrient soil in the greenhouse (25°C/18°C, 16-h/8-h day/night) for 2 wk and transferred to the field afterward. All transgenic lines were grown in Wuhan, 2020–2022, and most lines’ self-pollinated and open-pollinated seeds were harvested and stored under −20°C in refrigerators.
sgRNA identification
Before identifying the sgRNAs, the genomic DNA of the 1104 transgenic seedlings of the T0 generation was extracted from leaf tissues with the NuClean plant genomic DNA kits (CWBio) following the manufacturer's protocol and stored in 96-well plates. HTS was used to identify each transgenic plant's sgRNA(s) as described previously (Lu et al. 2017), with minor modifications. In brief, 20 PCR primers, which are tailed with 6-nt barcodes and used to amplify the sgRNA sequences, were designed and verified by the OligoAnalyzer tool (https://sg.idtdna.com/calc/analyzer). Among them, eight primers were used to discriminate the rows of each 96-well plate, whereas the rest marked the 12 columns. PCR reactions with a 20-μL solution of 10-μL Hieff PCR master mix (Yeasen), 1-μL forward primers, 1-μL reverse primers, 7-μL ddH2O, and 1-μL genomic DNA were conducted in the following conditions: 5 min at 94°C; 40 cycles of 15 sec at 94°C, 20 sec at 55°C, and 15 sec at 72°C; and 5 min at 72°C in the final extension. Ten microliters of each reaction from the same 96-well plates was mixed, thus generating 12 pooled solutions. Subsequently, 70-μL of each pooled amplicon was purified using a FastPure gel DNA extraction mini kit (Vazyme) according to the manufacturer's protocol. The 12 pooled amplicons were then subject to high-throughput sequencing (Illumina platforms, PE150/PE 250 strategy). The 20-nt target recognition sequences of the sgRNAs from each transgenic line were identified using a Python script by analyzing the sequencing data with the combinations of the 6-nt barcodes. Primers used for amplifying the sgRNAs are listed in Supplemental Table S8. Primers used for the quality evaluation of sgRNAs and detecting marker-free plants are as follows: GCACAATACATCATTTCTTCTTAGCTTT, CAAGTTGATAACGGACTAGCCTT. sgRNA(s) and the target gene(s) in each plant are given in Supplemental Table S3.
Editing interrogation of the target sites
To begin, the CDS of the gene(s) targeted by each sgRNA was used to BLAT in the Genoscope (https://www.genoscope.cns.fr/blat-server/cgi-bin/colza/webBlat) to search for all the homoeologs. Then CDSs of these genes were checked to determine if they contained the target sites targeted by the sgRNA. This step was performed owing to the update of the reference genome (Rousseau-Gueutin et al. 2020). Afterward, specific primers flanking the target site of each gene were designed and used for the PCR amplification of the target sequence. The amplicons were first verified by agarose gel electrophoresis and then subject to Sanger sequencing. The sequencing chromatograms were analyzed using TIDE (Brinkman et al. 2014) or ICE (Conant et al. 2022) to identify the specific editing. Detailed information for the editing efficiency evaluation with the 96 selected target sites, located in 178 target genes and from 86 transgenic lines, is provided in Supplemental Table S4. Sequence variations of the target sites between the reference and recipient genomes and the primers used to amplify the target sequences are listed in Supplemental Table S5. Detailed information for the editing interrogation in the forward genetic screening is listed in Supplemental Table S7.
Phenotyping of the seed-related traits
Seed area and aspect ratio data were obtained by scanning naturally mature open-pollinated seeds with the SC-G automatic seed testing and a thousand-particle weight analyzer (Wseen). Seed weight was determined using an analytical balance (Mettler Toledo) to get the TSW value. Measurement of root lengths was conducted 4 and 6 d after the imbibition of self-pollinated seeds (by bagging) using the germination pouches (Phytotc) with the addition of 15 mL water.
Fatty acid composition and SOC detection were referred to a previous description (Yin et al. 2022), subject to minor modifications. In brief, a total of 10–30 mg of mature, dry seeds was immersed in 3 mL of methanol solution (2.5% H2SO4 and 0.01% BHT), followed by the addition of 0.8-mL methylbenzene and 0.4-mL C17:0 toluene solution (2 mg/mL; set as the internal standard) after grinding the seeds. The mixture was then incubated for 1 h at 98°C, with 3.6 mL of deionized water and 2-mL hexane added afterward. The supernatant was pipetted for GC analysis with flame ionization detection (6850 Network GC system, Agilent Technologies). Helpful in illustrating the activity of FAD2, the parameter ODP was calculated as follows: (%C18:2 + %C18:3)/(%C18:1 + %C18:2 + %C18:3), as described previously (Stoutjesdijk et al. 2002).
Analysis of the off-target effect
Genome-wide identification of the off-target sites with up to 3-bp mismatch of the entire 18,414 target sites was conducted by the stand-alone Cas-OFFinder (Bae et al. 2014). Amplicons of the selected off-target sites were sequenced by the Sanger method, and the editing interrogation was conducted by the online server of TIDE (Brinkman et al. 2014). The primers used to amplify the selected mutants’ off-target sites are listed in Supplemental Table S6.
RT-qPCR experiments
For the reverse transcription (RT) experiment, fresh plant tissues were flash-frozen in liquid nitrogen and ground with the automatic grinder (Jing Xing). Then the total RNA was extracted using the RNAprep pure plant plus kits (Tiangen) according to the manufacturer's protocol. Around 0.1 μg total RNA was added to a solution of oligo-(dT)20VN primers and HiScript III enzyme mix (Vazyme) for RT. Quantitative PCR (qPCR) was performed on a QuantStudio 3 real-time PCR system (Applied Biosystems) with a mixed 20-μL solution of 1-μL cDNA, 0.8-μL primers (10 μM), 8.2-μL ddH2O, and 10-μL SYBR Green master mix (Yeasen). Each experiment was conducted in triplicate, and the gene BnTIP41 was set as the internal reference. Data analysis was performed following the relative quantification method (2−ΔΔCT). Primers used for the RT-qPCR experiments are listed in Supplemental Table S9.
Figure generation
The Circos plot was generated by the Circos package (Krzywinski et al. 2009), and gene structures were visualized with GSDS 2.0 (Hu et al. 2015). The statistical charts were generated by the Python package Matplotlib in this article (Hunter 2007). The figures were edited by Adobe Illustrator to improve their interpretation.
Data access
The HTS output files of the pooled plasmids and reads of the sgRNA amplicons of the 1104 regenerated transgenic plants generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA902177. The Sanger sequencing data generated in this study have been submitted to Figshare (https://doi.org/10.6084/m9.figshare.22215838.v1) and can be accessed in the Supplemental Datasets S1 through S8. The source data and code used for generating the figures are available as Supplemental Codes S1 through S12. The code used for extracting the 20-nt target recognition sequences of the sgRNAs from the amplicon-seq files has been submitted to GitHub (https://github.com/Jianjie-He/Extract-sgRNA) and is available as Supplemental Code S13. The transcriptome data used in this study have been submitted to Figshare (https://doi.org/10.6084/m9.figshare.22639510.v1) and as Supplemental Material. Supplemental figures, summaries of supplemental tables, code, and data sets are public in the Supplemental Information.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was supported by the National Key Research and Development Program of China (2022YFD1200402) and the National Natural Science Foundation of China (32272067 and 32072098).
Author contributions: J.H. performed the experiments and bioinformatic analyses, prepared the figures and tables, and drafted the manuscript. K.Z. conceived the idea and performed some of the experiments. S.Y., M.T., and W.Z. performed parts of the field experiment. Y.Y. and K.C. made helpful suggestions for this study. C.Z. and M.L. supervised the research and acquired funding. All authors have reviewed and edited the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277650.123.
- Received January 3, 2023.
- Accepted April 19, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








