
Targeted, programmable, and precise tandem duplication in the mammalian genome
- Yaoge Jiao1,4,
- Min Li1,4,
- Xingyu He1,4,
- Yanhong Wang1,4,
- Junwei Song1,
- Yun Hu1,
- Li Li1,
- Lifang Zhou1,
- Lurong Jiang1,
- Junyan Qu2,
- Lifang Xie3,
- Qiang Chen1 and
- Shaohua Yao1
- 1Department of Biotherapy, Cancer Center and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China;
- 2Center of Infectious Disease, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China;
- 3Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, Sichuan 610065, China
-
↵4 These authors contributed equally to this work.
Abstract
Tandem duplications are frequent structural variations of the genome and play important roles in genetic disease and cancer. However, interpreting the phenotypic consequences of tandem duplications remains challenging, in part owing to the lack of genetic tools to model such variations. Here, we developed a strategy, tandem duplication via prime editing (TD-PE), to create targeted, programmable, and precise tandem duplication in the mammalian genome. In this strategy, we design a pair of in trans prime editing guide RNAs (pegRNAs) for each targeted tandem duplication, which encode the same edits but prime the single-stranded DNA (ssDNA) extension in opposite directions. The reverse transcriptase (RT) template of each extension is designed homologous to the target region of the other single guide RNA (sgRNA) to promote the reannealing of the edited DNA strands and the duplication of the fragment in between. We showed that TD-PE produced robust and precise in situ tandem duplications of genomic fragments ranging from ∼50 bp to ∼10 kb, with a maximal efficiency up to 28.33%. By fine-tuning the pegRNAs, we achieved simultaneous targeted duplication and fragment insertion. Finally, we successfully produced multiple disease-relevant tandem duplications, showing the general utility of TD-PE in genetic research.
Tandem duplication (TD) of genomic regions containing entire or partial genes serves as a primary driving force for genetic evolution and also plays an important role in the development of human disorders including genetic disease and cancer (Kottaridis et al. 2001; Jones et al. 2008; Roy et al. 2015; Khater et al. 2019). Many of these duplications are >100 bp in size, which causes structural variations in the genome, resulting in expansion of specific genes, dysregulation of gene expression, or acquisition of novel gene functions (Fan et al. 2008; Tyo et al. 2009; Lan and Pritchard 2016; Teufel et al. 2019). Despite their importance, modeling TDs is difficult, and the extent that TDs contribute to pathological changes remains poorly understood in a large variety of events.
The recently developed gene editing techniques, especially the ones based on the CRISPR-Cas9 system, are readily reprogrammable for genome manipulating purpose, providing powerful tools for basic biomedical research and clinical translation (Cong et al. 2013; Nelson et al. 2016; Eyquem et al. 2017; Rees and Liu 2018; Schene et al. 2020). In particular, the prime editing (PE) tool enables targeted edition of the small-sized genetic fragment in a precise and versatile manner without causing robust levels of double-strand breaks (DSBs), thereby minimizing unintended insertions and deletions (indels) (Anzalone et al. 2019). Basically, a PE system contains two components, a Cas9 nickase and reverse transcriptase (RT) fusion protein and a prime editing guide RNA (pegRNA). Under the guidance of pegRNA, RT writes specific information at the 3′ end of the nick site of the nontarget strand (NTS), which is then integrated into the genome by endogenous DNA repair or replication mechanisms. Currently, PE shows great potential in a wide range of applications, including small fragment indels (Anzalone et al. 2019), single-nucleotide conversions (Anzalone et al. 2019; Adikusuma et al. 2021; Zhuang et al. 2022), and large fragment deletions (Anzalone et al. 2022; Choi et al. 2022; Jiang et al. 2022; Tao et al. 2022a,b), but not gene duplication. Here, we designed a novel editing strategy, named tandem duplication via prime editing (TD-PE), to precisely create TD in the mammalian genome, with the length of the duplication ranging from tens to thousands of base pairs.
Results
Design of TD-PE strategy
In the TD-PE strategy, we designed a pair of in trans pegRNAs with opposite directions to facilitate the ssDNA extension outward from the aimed duplication (Fig. 1). Our hypothesis was based on the assumption that producing double nicks located 3′ away from each other while installing a paired homologous arm (HA) to each nick by paired pegRNA–mediated PE would trigger the reannealing of the sequences around the nicks. We anticipated that such a mechanism would harness the endogenous DNA repair mechanisms to produce TD of sequences between the nicks (Fig. 1).
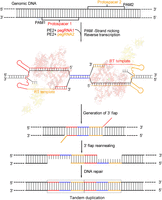
Overview of editing strategies for TD-PE. Schematic diagram showing the design and the putative editing process of TD-PE strategy. The TD-PE strategy contains a pair of in trans pegRNAs, pegRNA1 and pegRNA2, which are shown in red and orange, respectively. Each pegRNA contains an RT-template that is homologous to the sequence of the 5′ end or 3′ end of the fragment to be duplicated. These homologous regions are marked by colors that are the same as their corresponding pegRNAs. A blue line indicates the fragment between the homologous region. The prime edited ssDNA strands reannealed to form an intermediate that contains partial tandem duplication (TD), which was then repaired by endogenous DNA repair mechanisms to accomplish the TD.
TD-PE creates small fragment TD at endogenous genomic locus
To test this hypothesis, we designed two pairs of pegRNAs aiming to produce a 50-bp TD in the FANCF locus and a 57-bp TD in the EMX1 locus, respectively. Each pegRNA in the pair contained an RT-template that was homologous to the 3′ residue sequences of the opposite nick (Fig. 1). To differentiate these RT-templates from other types, we nominated the RT-template as an HA. Cotransfection of the resulting pegRNAs along with PE2 plasmids into HEK293T cells revealed that they did produce considerable levels of TDs. As shown in Figure 2, A and B, the analysis of the amplicons flanking the targeted regions via agarose gel detected bands with a size corresponding to the TD-containing fragment specifically in the edited cells. Sanger sequencing of the amplicons confirmed double peaks exactly occurring at the positions of the second nicks, where the theoretical duplication occurred (Supplemental Fig. S1). Moreover, in the sequencing chromatograph, we observed that fragments with single but lower peaks occurred at the end of the double-peak region, with lengths matching each duplicated region and sequences corresponding to each end of the amplicons (Supplemental Fig. S1). Analyses of the chromatographs by CRISPR ID software (Dehairs et al. 2016) predicted that there were two sequences in each of them, with one being the wild-type sequence and the other one being the targeted TD.

TD-PE generates a small fragment TD at the endogenous genomic locus. (A) PCR strategy for detecting the presence and frequencies of TDs. For each TD, a pair of primers flanking the duplicated fragment was designed. Successful TDs would generate bands with a larger size. (B) Agarose gel electrophoresis analysis of the presence of targeted TD on indicated loci. (C) HTS analysis of the frequencies of TD. The fragments containing TD in B were amplified by the HTS primers listed in Supplemental Table S5. Then the products were purified and subjected to HTS to analyze editing efficiency and product purity. Values and error bars reflect the mean ± SD of n = 3 independent biological replicates. (D) HTS analysis of the relative ratio of accurate and undesired editing outcomes of TD-PE. Values and error bars reflect the mean ± SD of n = 3 independent biological replicates. (E) HTS sequences showing the editing result of 2 × TD editing by TD-PE at the FANCF locus (reads with frequencies ≥0.02% were shown). The duplicated sequences are placed on separate lines and are indicated by blue curved arrows. Deletion, insertion, and base substitution are indicated by a black dash, a red box, or green letters, respectively. The insertion sequence of the red box was consistent with its 5′ upstream sequence. Note that sequences of ≥3 × TDs were displayed in Supplemental Figure S2. (F) Schematic diagram showing the pattern of insertion or deletion within undesired TDs, with the majority occurring at the boundaries of the duplications.
To obtain a more precise understanding of the existence of any other types of TDs and their frequencies, as well as to evaluate the accuracy of the edited products, we analyzed those amplicons with high-throughput sequencing (HTS). In addition to two copies of the target region (2 × TD), the HTS also detected three or more copies (Fig. 2C; Supplemental Figs. S2, S3). Taking FANCF locus as an example (50-bp TD), four types of TDs were detected by HTS, containing 2×, 3×, 4×, or 6× copies of the targeted region, respectively. The efficiencies of these TDs gradually decreased as the duplication copies increased (28.33% for 2 × TD, 9.66% for 3 × TD, 2.19% for 4 × TD, and 0.04% for 6 × TD). The same phenomenon was also observed in the EMX1 locus (TD 57 bp; 16.80% for 2 × TD, 2.75% for 3 × TD, 0.09% for 4 × TD, and 0.03% for 5 × TD) (Fig. 2C).
The HTS analysis revealed that there were no significant indels in the amplicons without duplication (0.1% in EMX1 locus and not detectable in FANCF locus) (Fig. 2C). However, in the amplicons with duplication, undesired editing outcomes were detected, although they only accounted for a relatively small proportion. These undesired TDs contained insertions or deletions (Fig. 2D). In the FANCF locus, the frequency of accurate TDs (including 2 × to 6 × TDs) was ∼40.18%, and the one of undesired TDs was ∼3.96%. The portions of undesired outcomes varied with the copies of TDs (2 × TDs, 3.57% undesired edit; 3 × TD, 0.38%; 4 × TD, 0.01%; 6 × TD, not detectable). In the EMX1 locus, the frequency of accurate TDs was ∼16.70%, and the one of undesired TDs was 0.10% (only detected in 2 × TD) (Fig. 2D). Detailed analysis of the undesired TDs revealed that the majority of deletions and insertions occurred at the boundaries of the duplications (Fig. 2E,F). Nearly all the insertions (∼10 bp in length) had sequences that exactly matched those of their upstream fragments, indicating that these insertions were duplicated from target regions (Supplemental Figs. S4, S5). To test the possibility that these multiple TDs or indels containing variants may result from the artificial effects of PCR, we constructed two plasmids containing the wild-type allele or the allele with a perfect 2 × TD (FANCF TD 50 bp) to imitate the wild-type or the duplicated allele, respectively. We then PCR amplified the target regions by using these verified plasmids as templates. Sequencing the resulting amplicons with HTS did not identify obvious indels or multiple TDs, indicating that these indels or multiple TDs were unlikely to be owing to artificial effects. Taken together, these analyses suggested that our TD-PE strategy produced only mild undesired editing outcomes.
TD-PE creates large TDs
Above results showed the activity of TD-PE in targeted duplications of small fragment. Then we sought to test if TD-PE is also functional in large fragment duplications (>100 bp). We designed three TDs in the AAVS1 locus, with the lengths of target regions ranging from 376 bp to 846 bp. As shown in the gel electrophoresis results (Fig. 3A), we identified candidate bands with a molecular weight that exactly matched the aimed edits, and the frequencies of these edits decreased as the length of target regions increased (8.30% for 376 bp, 2.74% for 532 bp, and 1.14% for 846 bp) (Fig. 3B). We then designed additional 10 large fragment duplications across three different loci (EMX1, FANCF, and HEK3) (Fig. 3B). All 10 TD-PEs generated detectable TDs, which were consistent with the observation in the AAVS1 locus, and the frequencies of these TDs were also gradually decreased with an increase in the lengths of the target regions. It is noteworthy that the sequences of all visible bands were confirmed by Sanger sequencing (Supplemental Figs. S6–S9), showing that targeted TD-PEs occurred between the flanking regions.
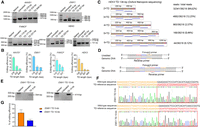
TD-PE generates large fragment TDs at endogenous genomic loci. (A) Agarose gel electrophoresis analysis of the presence of targeted TD on indicated loci. Red stars represent multiple copies of TDs. Sanger sequencing results of targeted TDs were shown in Supplemental Figures S7–S10. (B) Quantification of the efficiencies of the indicated TDs. Values and error bars reflect the mean ± SD of n = 3 independent biological replicates. (C) Different types of TDs in HEK3 TD 136 bp were analyzed by Oxford Nanopore sequencing. Each blue line in the output represents one single copy of the duplications. (D) PCR strategy for detecting the presence of targeted large fragment TDs. A pair of primers were designed for testing kilobase-scale TDs (>5 kb), with forward primers flanking the downstream side of the amplified regions and reverse primers flanking the upstream side of the amplified regions. These primers would specifically amplify the boundary of targeted TDs. The length of the blue fragment was equal to the length that needs to be duplicated minus the red and yellow. (E) Agarose gel electrophoresis analysis of the presence of large fragment TDs in EXM1 loci. Primers used for the detection are listed in Supplemental Table S3. The amplicons with expected sizes are marked by arrows. (F) Sanger sequencing results of PCR products in E. The sequences corresponding to each end of the TD boundary were marked by yellow and red boxes, respectively. (G) Quantifying the frequencies of targeted TDs (EMX1 TD 5 kb/10 kb) by absolute quantitative PCR. The standard curves are shown in Supplemental Figure S24B. The junction fragments of each TD were used to calculate the duplicated allele, and the flanking fragment was used to calculate total allele. Values and error bars reflect the mean ± SD of n = 3 independent biological replicates.
To gain a deeper insight into the entire editing outcomes of these larger TDs, we performed Oxford Nanopore Technologies (ONT) sequencing. We extracted the DNA fragments with molecular weights equivalent to or larger than each 2 × TD for Nanopore sequencing. The sequencing results confirmed the presence of 2 × TDs and also identified multiple TDs in all 10 events examined. The ratios of different copies of TDs were similar to those observed in smaller TDs, with 2 × TDs being the most abundant. For example, at the HEK3 TD 116-bp site, ∼84.6% events were 2 × TD (12.2% for 3 × TD, 2.63% for 4 × TD, 0.44% for 5 × TD, and 0.13% for 6 × TD) (Fig. 3C; Supplemental Fig. S10). Because ONT sequencing is less accurate than HTS and because smaller TDs showed that most undesired edits occur at the junctions of TDs, we next sought to perform HTS to characterize the editing accuracy of these larger TDs. We amplified these junctions of those larger TDs for HTS analysis. Similar to the observation in smaller TDs, the analysis revealed that only a small fraction of TDs contained undesired insertions or deletions (Supplemental Figs. S11–S21). Sequences derived from the sgRNA scaffold were identified in the insertions, which was consistent with previous observations showing that the sgRNA scaffold can be transcribed (Anzalone et al. 2019) and integrated into the genome (Jiang et al. 2020).
To further confirm the presence of the targeted TDs and to examine whether those TDs could be stably passed to progeny cells, we cultured the edited cells for monoclonal analysis. As shown in Supplemental Figure S22 (HEK3 TD 136 bp), we did detect colonies that harbored targeted TDs, albeit the frequency of them was lower than that observed immediately after the editing. This is likely because of the editing conferring a proliferation or survival disadvantage.
In addition, to examine whether TD-PE was also active in other cells, we chose the HeLa cell line for further testing because it had been shown to support PE in previous studies (Anzalone et al. 2019, 2022; Tao et al. 2022b). Two TD-PE sites (HEK3 TD 136 bp and EMX1 TD 116 bp) that showed robust efficiency in HEK293T cells were selected for the test. As shown in Supplemental Figure S23, both sites generated efficiently targeted TDs, indicating that the TD-PE strategy is active in a broad range of cell types.
Next, we tested if TD-PE could achieve TDs at a kilobase scale by designing ∼5-kb and ∼10-kb duplications in the EMX1 locus. Because the length of the resulting TDs was >10 kb, which was difficult to be detected through full-length amplification, we designed a PCR protocol to specifically amplify the junctions of the TDs by using forward primers targeting their 3′ regions and reverse primers targeting their 5′ regions. Therefore, these paired primers should not amplify the wild-type sequences (Fig. 3D). As shown in Figure 3E, we did detect positive bands with aimed length specifically in samples treated with TD-PE. Sanger sequencing these positive bands identified sequences corresponding to the junction of TDs (Fig. 3F). To determine the editing efficiencies of those kilobase-scale TDs, we performed absolute quantitative PCR analysis. In the analysis, we used amplicons spanning the TD junction to indicate the TD alleles and used those within the flanking region to indicate total alleles (Supplemental Fig. S24). Through the analysis, we found that the efficiencies of TD 5 kb and TD 10 kb were ∼6.14% and 2.62%, respectively (Fig. 3G). We then performed HTS to analyze the accuracy of the editing by sequencing the junction amplicons. The sequencing identified relatively lower ratios of undesired edits compared with smaller TDs (0.07% for TD 5 kb, and 0.13% for TD 10 kb) (Supplemental Fig. S25). Taken together, these results suggest that TD-PE is also effective in producing targeted TDs of large fragments.
Improving the flexibility of TD-PE by coupling Cas9 variant and fine-tuning pegRNA
The action mode of above TD-PE strategy suggested that its application is restricted by protospacer adjacent motif (PAM) sequences and the nick position of Cas9. To expand the application of TD-PE, we substituted TD-PE with Cas9-NG, a variant recognizing NG PAM that has been shown to be effective in PE (Kweon et al. 2021). As shown in Supplemental Figure S26, the Cas9-NG variant was also effective in TD-PE target sites. We then sought to fine-tune the design of pegRNAs to insert additional sequences to relax the requirement of the nick position. As shown in Figure 4A, 2- to 10-nt fragments were added in between the HA and PBS of pegRNAs, so that they could prolong the original duplications. We chose AAVS1 and HEK3 sites to test this design and found that it could support the targeted duplications, as evidenced by PCR and Sanger sequencing analysis (Fig. 4B,C; Supplemental Figs. S27, S28).
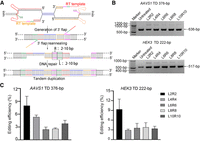
Improving the flexibility of TD-PE by fine-tuning pegRNA design. (A) Schematic diagram showing the design of TD-PE with additional insertions. Insertions with different lengths (2–10 nt; shown in bold pink and green) were added between the original RT-template and the PBS so as to prolong the original duplications and relax the requirement of the nick position. (B) Agarose gel electrophoresis analysis of the presence of TD at the HEK3 and AAVS1 loci. (C) Quantification of the efficiency of targeted TD in B. Values and error bars reflect mean ± SD of n = 3 independent biological replicates.
Applications of TD-PE in disease-relevant TDs
Based on these improvements, we next applied TD-PE to modeling disease-related TDs. We designed four TDs spreading on RYR1, NCF2, ENG, and SATB2 loci, respectively (RYR1:VCV000831179.2; NCF2:VCV000832066.2; SATB2:VCV001066449.1; ENG: VCV001068859.2) (http://www.ncbi.nlm.nih.gov/clinvar/). According to sequence context of the regions to be duplicated, four paired pegRNAs were designed for these TDs, with NCF2 and RYR1 pegRNAs using NGG PAMs and with SATB2 and ENG pegRNAs using NGA PAMs (Fig. 5A; Supplemental Fig. S29). The RT-templates of those pegRNAs were fine-tuned by inserting additional sequences so as to exactly genocopy the disease-related TDs. As shown in Figure 5B, we detected obvious TDs in HEK293T cells at four specific loci by analyzing the amplicons of the target regions using gel electrophoresis, with averaged editing efficiencies ranging from 2.34% to 16.52% (Fig. 5C). To confirm the success of TDs, those TD-containing amplicons were further verified by Sanger sequencing, which confirmed the presence of duplications (Supplemental Fig. S30).
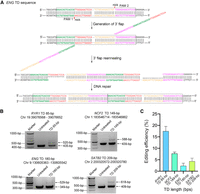
Applications of TD-PE in the investigation of disease-relevant TDs. (A) PegRNAs design at the ENG site and the putative editing process of TD-PE strategy. (B) Agarose gel electrophoresis analysis of the presence of TD at four disease-related loci. (C) Quantification of the efficiency of targeted TD in B. Values and error bars reflect mean ± SD of n = 3 independent biological replicates.
Discussion
In summary, we developed a novel PE strategy, TD-PE, to produce TDs of large DNA fragment in the mammalian genome by using a pair of in trans pegRNAs with opposite directions, which induced in situ ssDNA extensions outward from the aimed region. The extended paired ssDNAs and their boundary sequences were designed complementary with each other, so as to facilitate the homology searching process and promote the outcomes of DNA repair toward targeted TD. We found that TD-PE produced robust and precise in situ TDs of large DNA fragments in endogenous genomic loci, with the length of duplication ranging from ∼50 bp to ∼10 kb. We generated multiple disease-related TDs by using fine-tuned TD-PE, showing its potential as a powerful tool in genetic research.
However, it should be noted that the performance of TD-PE strategy requires the coordinated actions of multiple parameters. In addition to the general requirements of standard prime editors, the choice of appropriate PAM and the design of HAs and inserted sequences that match the targeted fragment to be amplified are important for a successful targeted TD, especially in cases in which there are no exact matched PAMs. In such cases, the PAMs should be designed to be located inside the duplication, and additional insertions should be supplemented to the PE to achieve the desired duplications. We have shown that TD-PE allows an additional insertion of up to 14 bp (Fig. 5A,B). However, as the length of the insertion increases, the editing efficiency appears to decrease (Fig. 4B,C). Therefore, we suggest minimizing the insertion, particularly to <10 bp, to improve the editing efficiency. Alternatively, using Cas9 variants with relaxed PAM requirements, such as Cas9-NG and SpRY (Kweon et al. 2021), may be helpful in choosing PAMs that allow shorter insertions.
Methods
Plasmid construction
PE2 plasmid was obtained from Addgene (132775). PE2-NGs were constructed by substituting the PAM interacting (PI) domain of PE2 with the Cas9-NG PI domain amplified from NG-ABEmax (Addgene plasmid 124163) (Kweon et al. 2021). To construct sgRNA plasmids, oligos containing spacer sequences were annealed and inserted into the Bbs1 site of the pU6-sgRNA vector. Oligos used to generate the spacers of sgRNAs were listed in Supplemental Table S1. The pegRNA expression cassettes were generated by installing PBS and RT-template sequences with PCR into sgRNA sequences. The resulting PCR fragments were then cloned into blunt vector (Yeasen) to generate pegRNA expression plasmids. Detailed sequences of pegRNAs are shown in Supplemental Table S2.
Cell culture and transfection
HEK293T cells were cultured in Dulbecco's Modified Eagle Medium (Gibco by Life Technologies), supplemented with 10% (v/v) fetal bovine serum (Life Technologies) and 1% penicillin/streptomycin (Boster Biological Technology). They were seeded on a 96-well plate (Biofil) 12–16 h before transfection, and each well was seeded with 2 × 105 cells. Transfection was conducted using 400 ng of plasmids (276 ng of PE2 plasmid DNA, 62 ng of pegRNA 1 plasmid DNA, and 62 ng of pegRNA 2 plasmid DNA) and 0.7 μL of Transeasy (Forgene) at a confluence of ∼70%–80%, according to the manufacturer's protocol.
Genomic DNA preparation
Genomic DNA (gDNA) was extracted 72 h after transfection. The cells were washed with 1 × PBS solution, and gDNA was extracted by the addition of 30 µL of freshly prepared lysis buffer (10 mM Tris-HCl at pH 7.5, 0.05% SDS, 25 µg/mL Proteinase K [Beyotime]) directly into each well of the 96-well plate. Then the gDNA mixture was incubated for 30 min at 55°C, followed by additional 20 min at 95°C to inactivate the Proteinase K.
TD efficiency analysis
We assessed the efficiency of TD by agarose gel electrophoresis. The PCR amplification of gDNA was performed using Phanta Max superfidelity DNA Polymerase (Vazyme) with the primers listed in Supplemental Table S3. The resulting amplicons were analyzed by agarose gel electrophoresis. Then gel images were analyzed by ImageJ to calculate the efficiency of TD.
For the TD of EMX1 TD 5 kb/10 kb, quantitative PCR was performed using the SYBR Green (Yeasen) approach (Tao et al. 2022a), and the qPCR primers are shown in Supplemental Table S4. The flanking fragment of the target site or the junction fragment of TD obtained by PCR was ligated to the blunt vector (Yeasen) to construct reference plasmids. The standard curves for the reference plasmids were determined by CT values against log-transformed concentrations of serial 10-fold dilutions (2 × 102, 2 × 103, 2 × 104, 2 × 105, 2 × 106, 2 × 107, 2 × 108, and 2 × 109 copies per 1 μL). Absolute copy numbers of the flanking fragment or the fragment of TD in each gDNA were calculated with CT values based on their standard curves. The efficiency of TD was the copies of TD DNA divided by the copies of flanking DNA.
Targeted deep sequencing and data analysis
gDNA was extracted 72 h after transfection and used as template. The target region was amplified by high-fidelity DNA polymerase (Phanta Max superfidelity). The PCR reactions consisted of 1 μL of cell lysate and 0.2 μM forward and reverse primers in a final reaction volume of 30 μL. Genomic regions of interest were amplified by PCR with primers flanked with different barcodes (Supplemental Table S5). PCR reactions were performed as follows: 3 min at 95°C; 35 cycles of (15 sec at 95°C, 15 sec at 60°C, and 10 sec at 72°C); and a final extension of 10 min at 72°C. The PCR products were purified with a GeneJET gel extraction kit (Thermo Fisher Scientific) and quantified with NanoDrop (Thermo Fisher Scientific). Samples were sequenced commercially using the Illumina NovaSeq 6000 platform (Personal Biotechnology). The sequencing modes were paired-end, 2 × 250 bp (Fig. 2E; Supplemental Figs. S2–S5), or paired-end, 2 × 150 bp (Supplemental Figs. S11–S21). Raw paired-end reads were merged using fastp software to generate full-length reads (Chen et al. 2018). Adaptor contamination was discarded using AdapterRemoval (Schubert et al. 2016). Alignment of the filtered reads to a reference sequence was performed using Python, and the custom Python script is the same as previously described (Tao et al. 2022b). TD and indel frequencies were quantified as a percentage of total sequencing reads, and the threshold for editing activity was set >0.02%.
ONT library preparation and sequencing
TD sequence amplification and validation
The PCR amplicons in the suspected TDs area were gel-purified with GeneJET gel extraction kit (Thermo Fisher Scientific) and quantified with NanoDrop (Thermo Fisher Scientific).
ONT sequencing and base-calling
One hundred fifty femtomolars of PCR amplicons for each sample were prepared using the ONT ligation sequencing kit (SQK-LSK109) according to the manufacturer's instructions with only minor modifications. The PCR amplicons required end-repair and A-tailing addition for 30 min, followed by native barcode ligation for an additional 30 min. The library was loaded onto a FLO-MIN106 flow cell R9.4.1 and sequenced by MinKNOW version 19.12.5 software (ONT). The resulting data in FAST5 format were base-called to FASTQ format using Guppy (ONT, version 4.0.14, config file: dna_r9.4.1_450 bps_sup.c).
Read alignments and visualization
Base-calling reads (FASTQ) were aligned to reference sequences at different copies of TD using minimap2 (version 2.17-r941, parameter -ax map-ont, ‐‐secondary=no) (Li 2018). Mapped reads were filtered for unique alignments and mapping quality (MAPQ > 1) by SAMtools (Li et al. 2009), and the distribution across different copies of TD in one sample was counted. Integrative Genomics Viewer (IGV) software (Thorvaldsdóttir et al. 2013) was used to visualize the mapped reads on the reference sequences. A custom shell and R scripts provided in Supplemental Code were used to analyze and quantify the ratios of different copies of TDs.
Statistical analyses
GraphPad Prism 8 software was used to analyze the relevant experimental data. All data are presented as the mean ± SD of three independent biological replicates.
Data access
All sequencing data generated in this study, including Sanger, Illumina, and ONT sequencing data, have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA939900. Wild-type and 2× TD amplicons are accessible under accession numbers SRR24206828 and SRR24206829, respectively.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. 81974238 and no. U19A2002) and the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC21018). We thank Prof. Lu Chen for his help in Oxford Nanopore sequencing.
Author contributions: S.Y., Q.C., and Y.J. designed the research. Y.J., M.L., X.H., Y.W., L.L., L.Z., L.J., J.Q., and L.X. performed the experiments. Y.J., L.L., L.X., Y.H., L.Z., and L.J. analyzed the data. S.Y. and Y.J. wrote the manuscript. J.S. performed the Oxford Nanopore library sequencing experiments and analyzed the data. All authors have read and approved the article.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277261.122.
- Received August 29, 2022.
- Accepted April 20, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








