
Challenges and considerations for reproducibility of STARR-seq assays
- Maitreya Das1,2,3,
- Ayaan Hossain3,4,
- Deepro Banerjee3,4,
- Craig Alan Praul3 and
- Santhosh Girirajan1,2,3,4,5
- 1Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 2Molecular and Cellular Integrative Biosciences Graduate Program, Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 3Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 4Bioinformatics and Genomics Graduate Program, Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 5Department of Anthropology, Pennsylvania State University, University Park, Pennsylvania 16802, USA
Abstract
High-throughput methods such as RNA-seq, ChIP-seq, and ATAC-seq have well-established guidelines, commercial kits, and analysis pipelines that enable consistency and wider adoption for understanding genome function and regulation. STARR-seq, a popular assay for directly quantifying the activities of thousands of enhancer sequences simultaneously, has seen limited standardization across studies. The assay is long, with more than 250 steps, and frequent customization of the protocol and variations in bioinformatics methods raise concerns for reproducibility of STARR-seq studies. Here, we assess each step of the protocol and analysis pipelines from published sources and in-house assays, and identify critical steps and quality control (QC) checkpoints necessary for reproducibility of the assay. We also provide guidelines for experimental design, protocol scaling, customization, and analysis pipelines for better adoption of the assay. These resources will allow better optimization of STARR-seq for specific research needs, enable comparisons and integration across studies, and improve the reproducibility of results.
Enhancers are cis-acting DNA elements that regulate gene expression (Banerji et al. 1981). The ability of enhancers to recruit transcription factors through specific binding motifs to regulate the expression of target genes in a cell, tissue, and developmental stage-specific manner makes them critical components of gene regulatory networks (Shlyueva et al. 2014). Although enhancer–reporter assays (Banerji et al. 1981) and comparative genomics (Pennacchio et al. 2006) enabled initial discoveries, enhancers are usually mapped within nucleosome-free open chromatin regions that are hypersensitive to DNase I (Gross and Garrard 1988) or accessible to transposase (Buenrostro et al. 2013) and to sequences bound by specific transcription factors (Visel et al. 2009) or histone modifications (Heintzman et al. 2007). Such assays that are used to detect putative enhancer regions throughout the genome include DNase-seq (Boyle et al. 2008), ATAC-seq (Buenrostro et al. 2013), and ChIP-seq (Robertson et al. 2007). However, these methods are limited to only providing the location of candidate enhancers and do not assess their functional activity.
Self-transcribing active regulatory region sequencing (STARR-seq), like other episomal massively parallel reporter assays (MPRAs) (Melnikov et al. 2012; Patwardhan et al. 2012), directly quantifies enhancer activity by relying on transcription factors within a host cell system, thereby removing any chromatin-associated biases (Arnold et al. 2013). STARR-seq takes advantage of the property of enhancers to act bidirectionally, and therefore, candidate enhancer fragments cloned downstream from a minimal promoter sequence transcribe themselves in a host cell. A comparison between the final read count of self-transcribed fragments with the initial number of transfected or transduced fragments provides a quantifiable measure of enhancer activity. By assessing candidate enhancer libraries through massively parallel sequencing, researchers have built genome-wide enhancer activity maps (Liu et al. 2017b); functionally validated enhancers identified by other methods such as ChIP-seq (Barakat et al. 2018), ATAC-seq (Wang et al. 2018; Hansen and Hodges 2022), and FAIRE-seq (Chaudhri et al. 2020; Glaser et al. 2021); tested the impact of noncoding variants within or near enhancer sequences (Liu et al. 2017a; Kalita et al. 2018; Schöne et al. 2018; Zhang et al. 2018); and assessed changes in enhancer activity owing to external factors such as drug or hormone treatment (Shlyueva et al. 2014; Johnson et al. 2018). Furthermore, Peng et al. (2020) identified a subset of enhancers in mouse embryonic stem cells (ESCs) that showed activity with STARR-seq but was not associated with active chromatin marks, suggesting that functional assessment using STARR-seq can reveal novel chromatin-masked enhancers in specific cellular contexts. Thus, STARR-seq has emerged as a state-of-the-art method for functional interrogation of the noncoding genome.
Although STARR-seq is powerful and versatile, there are several challenges associated with successfully carrying out this assay. First, the experimental protocol is laborious with more than 250 steps, involving construction of a STARR-seq plasmid library, delivery of the library into a host through methods such transfection or transduction, sequencing of delivered (input) and self-transcribed (output) fragments to sufficient read depth, and bioinformatic analysis to identify peaks at enhancer sites (Neumayr et al. 2019). Several of these steps also require preliminary experiments, for example, optimizing library construction to achieve sufficient complexity, testing transfection efficiency, mitigating host-specific limitations, and assessing target coverage for reproducible discovery of enhancer location and activity. Second, variation across studies and a lack of benchmarking for selecting sequencing and data analysis parameters such as optimal read depth, choice of peak caller, cut-off scores for characterizing enhancer activity, and methods for data validation complicate comparisons across studies. Third, most published studies do not provide sufficient details for researchers to adapt or scale their protocols for specific needs such as modifying target library size and choosing enhancer fragment length. Furthermore, the lack of quality control (QC) details for critical intermediate steps raises significant concerns for the replicability and reproducibility of results from STARR-seq assays.
Several studies have addressed challenges associated with reproducibility in biology, including reports on best practice guidelines for various genomic analysis pipelines, such as RNA-seq (Conesa et al. 2016), ChIP-seq (Landt et al. 2012), and ATAC-seq (Yan et al. 2020), as well as the Reproducibility Project: Cancer Biology (Center of Open Sciences and Science Exchange). For example, Errington et al. (2021) attempted 193 experiments from published reports but were able to replicate only 50 owing to a failure of previous studies to report the descriptive and inferential statistics required to assess effect sizes, as well as a lack of sufficient information on study design. Several of their experiments required modifications to the original protocols, with results showing significant deviations from previous findings. These studies illustrate the difficulties in successfully repeating published functional genomics experiments.
Here, we discuss four major aspects of a STARR-seq assay, including (1) pre-experimental assay design, (2) plasmid library preparation, (3) enhancer screening, and (4) data analysis and reporting. We also delineate several features specific to STARR-seq in the context of general reproducibility of biological experiments (Freedman et al. 2015). We show how each of these features varies across previous studies, and we score them based on details provided in the original publications. Furthermore, through a series of carefully designed STARR-seq experiments (Supplemental Text; Supplemental Protocol), we identify challenges, rate-limiting conditions, and critical checkpoints. We note that a limitation of this study is that all biological results, interpretations, and conclusions drawn from these data sets were excluded because they fall outside the scope of this Perspective. We list these limitations in the Supplemental Materials. Finally, we reanalyze multiple published STARR-seq data sets along with our own data to illustrate limitations of available analysis pipelines. We provide recommendations for study design, library construction, sequencing, and data analysis to help future researchers conduct robust and reproducible STARR-seq assays.
Different assay goals result in varying experimental designs
The general strategy for STARR-seq consists of cloning a library of fragments selected from either a list of putative enhancers or the entire genome into a STARR-seq plasmid, which is transfected or transduced into a host, such as cultured cells or live tissues. The “input” library and the transcribed “output” fragments from the host are sequenced to high read depth, followed by bioinformatic analysis for quantification of enhancer peaks. In our assessment of published STARR-seq studies, we found that the original protocol (Arnold et al. 2013) was modified in myriad ways to fit various study goals and underlying biological contexts, including altering design features such as target library size, DNA source, fragment length, sequencing platform, choice of STARR-seq vector, and choice of host (Fig. 1A). Reporting of these features are crucial for understanding the rationale behind downstream strategies, such as protocol scaling, choice of QC measures, and selection of optimal parameters for assay performance and data analysis. Here, we discuss STARR-seq experimental design features and how they vary across studies and affect study reproducibility (Table 1), and suggest rationale for selecting different designs (Table 2). We further provide a list of definitions of frequently used terminology in sequencing-based studies in Box 1.
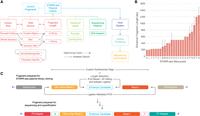
Experimental design features of a STARR-seq experiment. (A) Variations of pre-experimental design features and their impact on library preparation protocol are shown. (B) Fragment length (mean and range are shown) of published STARR-seq libraries reported by deidentified papers A to X (on x-axis) and reported fragment length (on y-axis) are shown. Note that studies W, M, O, P, Q, R, and G reported the range for their fragment lengths indicated by the error bars, and other studies reported the exact length of the fragment. (C) Read architecture schematic of a STARR-seq plasmid library (top) and “input” and “output” sequencing libraries (bottom) are shown.
Reproducibility features for biological assays with a focus on STARR-seq
Rationales for core experimental design factors including library size, fragment length, and DNA source for different biological questions addressed by different adaptations of STARR-seq and MPRA
Definitions of frequently used terminology in sequencing-based studies
Assay reproducibility: Measure of how accurately and consistently assay results can be reobtained to reaffirm previous conclusions.
Bead clean-up: Magnetic DNA or RNA purification beads designed to be selective for DNA or RNA fragments. In DNA purification, the ratio of volume of beads to the volume of sample is selective of the length of DNA being purified. Lower bead volumes fail to bind longer DNA fragments, thereby filtering them out. This is a method for fragment length selection.
Cloning arms or overhangs: Sequences added to adapter-ligated (read1/read2) inserts that are complementary to cloning sites of the STARR-seq vector. Ligation is accomplished using LM_PCR to facilitate cloning of the fragments into the vector.
Deduplication: Bioinformatic process for removing PCR duplicates from sequenced reads.
Demultiplexing: Bioinformatic process by which all high-throughput sequencing reads from pooled libraries are separated into sample-specific reads, based on sample-specific i5/i7 barcodes.
Episomal assay: Assays that transfect one or more DNA sequences into a host cell, whereby the transfected sequences do not integrate within host cell genome.
Experimental replicability: Measure of how accurately an experiment can be repeated with the exact parameters to obtain the same output data.
i5/i7 barcodes or indexes: These are 6- to 8-bp sequences ligated to sequencing libraries to provide a unique identity for each sample when pooled with libraries from other samples. Libraries can use single indexing (either i5 or i7) or dual indexing (both i5 and i7).
Input library: STARR-seq sequencing library constructed either directly from a plasmid library pool or from plasmid DNA extracted from transfected cells. The number of reads from the input is used to normalize the number of “output” reads to quantify enhancer activity.
Library complexity: The total number of unique fragments present in a library.
Library delivery: The process of delivering the prepared STARR-seq library into a host system through processes like transfection or transduction.
Library dynamic range: The ratio of read counts between the most active fragment in the output library to the least active fragment in the same library.
LM_PCR: Ligation-mediated PCR is a type of PCR that uses primers carrying extended sequences (not having complementary sequence on the target strand) and ligates those extended sequences to the region being amplified.
Massively parallel sequencing: High-throughput sequencing techniques that can sequence billions of reads in parallel based on a sequencing library.
MPRA: Massively parallel reporter assays are high-throughput assays that quantify the activity of a test fragment, typically cloned downstream from a minimal promoter, via massively parallel sequencing of barcoded reporter transcripts.
Multiplexing: A process in which multiple libraries are pooled and sequenced on the same lane of the sequencer.
Output library: STARR-seq sequencing library built using self-transcribed reporter transcripts of candidate enhancers extracted from the host cell.
P5/P7 Adapters: Sequences flanking index (i5/i7) barcodes that help hybridize the fragment to the flow cell to enable sequencing.
Peak calling: Automated method by which sequenced STARR-seq reads from the output library are compared with reads from the input library to identify regions with significantly increased read counts or “peaks.”
Read 1/Read 2 adapters: Sequences immediately flanking the fragment of interest that are sequenced as “read 1” and “read 2” on a standard Illumina flow cell. These regions serve as recognition sequences for Illumina sequencing primers.
Sequencing library: Libraries containing fragments of amplicon DNA or cDNA to be sequenced using high-throughput sequencing. Each fragment consists of a core region of interest flanked by read 1 and read 2 adapter sequences, index barcodes, and P5 and P7 flow cell adapters.
STARR-seq plasmid library: Library comprising of candidate enhancer fragments cloned downstream from a minimal promoter in a STARR-seq vector.
STARR-seq: Self-transcribing active regulatory region sequencing is a specialized episomal MPRA that is used to directly measure enhancer activity by comparing output transcripts produced by a candidate enhancer sequence cloned downstream from a minimal promoter to the number of copies of the enhancer fragments used as input before transcription.
UMIs: Unique molecular identifiers are randomly synthesized oligos (unknown sequences) of a fixed length that can be added to fragments. If fragments undergo amplification via PCR, UMIs added to those fragments before PCR are also duplicated during PCR. This enables detection of PCR duplicates, as opposed to identical mRNA self-transcripts.
Target library size
The versatility of STARR-seq allows for enhancer screening across both whole genomes and targeted regions. Studies screening for enhancer activity across numerous targets typically require multiple reactions for all library preparation steps, as well as high cloning and transformation efficiencies to achieve adequate representation of all fragments compared with studies with fewer targets. For example, a library generated by Johnson et al. (2018) to assess the effect of glucocorticoids on genome-wide enhancer activity contained more than 560 million unique fragments. The investigators performed 60 reactions of sequencing adapter ligations and 72 transformations and grew the pooled libraries in 18-L LB broth. In contrast, Klein et al. (2020) synthesized a targeted library of 2440 unique fragments along with customized adapter and cloning sequences in three reactions to compare context-specific differences among seven different MPRA designs, including two variations of STARR-seq. Cloning and transformation of each tested design were performed in duplicates, and each reaction was grown in 100 mL of LB broth. Although adequately scaled protocols exist for whole-genome STARR-seq in Drosophila melanogaster and humans (Arnold et al. 2013; Neumayr et al. 2019), scaling guidelines for focused libraries are not well reported because their sizes can vary from more than 7 million unique fragments (Wang et al. 2018) to a few hundred (Vockley et al. 2015) or thousand fragments (Klein et al. 2020). We provide a rationale for custom designing STARR-seq assays in Table 2 and scaling guidelines based on library size, fragment length, and read coverage in Supplemental Table S1.
DNA source
A strong advantage of STARR-seq is its ability to screen random fragments of DNA from any source for enhancer activity. To this effect, DNA can be sourced from commercially available DNA repositories (Liu et al. 2017b), specific populations carrying noncoding mutations or SNPs to be assayed (Liu et al. 2017a), cultured bacterial artificial chromosomes (BACs) (Arnold et al. 2013), or custom oligo pools (Kalita et al. 2018). Different sources have their own advantages and drawbacks that are important to consider for efficient library building (Table 2). For example, custom synthesized oligo pools are generally designed to be of uniform length and may include sequencing and cloning adapters as part of the design. These libraries may not require fragment length selection and adapter ligation and are also devoid of any representation bias caused by fragments of nonuniform lengths. However, we note that synthesized fragments may also have sequence-specific biases owing to factors such as repetitiveness, single-stranded DNA structures, and mappability issues associated with the underlying sequence (Halper et al. 2020). Furthermore, most STARR-seq studies using synthesized oligos have a fragment synthesis-based length restriction of 230 bp, although recent advances can allow up to 300 bp. Therefore, current limitations for custom-made fragments include synthesis-based length restrictions, higher costs, and a smaller library size as opposed to larger focused or whole-genome libraries using genomic DNA. Of note, genomic DNA derived from a host cell is recommended for focused studies that aim to assess enhancers captured through chromatin-based techniques such as ATAC-STARR-seq (Wang et al. 2018), ChIP-STARR-seq (Barakat et al. 2018), and FAIRE-STARR-seq (Chaudhri et al. 2020). Using the host cell DNA for these methods enables selection of cell type–specific candidate enhancers, which can then be functionally validated with STARR-seq upon redelivery into the host. Although in theory any DNA fragment can be tested in any cell line, this recommendation can be advantageous to test cell type–specific effects.
Fragment length
The length of each candidate enhancer fragment that is compatible with the assay goal, library size, and DNA source (Fig. 1A) determines critical experimental parameters such as the extension temperature of ligation-mediated PCR (LM_PCR), sample volume-to-bead ratio for bead clean-up, insert-to-vector ligation ratio, and fragment molarity for cloning reactions. Most STARR-seq studies either use ∼500-bp fragments sourced from sheared whole-genome DNA or DNA isolated from BACs or use ≤230-bp oligo pools (Fig. 1B). However, an “ideal” fragment length is dependent on the assay goal, because different fragment lengths have different benefits and limitations (Table 2). Fragments >500 bp may be more economical and better suited for genome-wide screens or enhancer discovery within larger targets. Notably, larger fragments may not have the resolution to detect the activity of individual enhancers but are useful to identify the compounding effect of multiple closely located enhancers or the influence of flanking sequences on transcription factor binding (Schöne et al. 2018; Klein et al. 2020). In contrast, focused studies using shorter fragments allow for fine mapping the enhancer effects of individual TF binding sites but may not uncover synergistic effects detectable by longer fragments. For example, Klein et al. (2020) compared the effects of fragment length on enhancer activity by extending the flanking genomic sequences of the same candidate enhancer sites to create 192-bp, 354-bp, and 678-bp fragments using hierarchical multiplex pairwise assembly. The investigators found less concordance in enhancer activity for the same candidate enhancers when fragments of different lengths were compared, with higher correlations between smaller fragments than larger fragments. Schöne et al. (2018) showed that the flanking sequences have an effect on DNA shape that affects TF binding, thus impacting enhancer activity. Hansen and Hodges (2022) reported that differently sized fragments around the same genomic region can identify distinct active regions. However, despite these observations, there is still a lack of sufficient understanding of the role played by variations in fragment length on enhancer activity within specific contexts. Library preparation from sheared DNA requires accurate size-selection methods as fragments of nonuniform length are more prone to uneven representation in the final library owing to PCR and cloning biases. In fact, Neumayr et al. (2019) suggest keeping the fragment lengths within ∼300 bp of each other to avoid extreme biases. If the assay requires variable fragment lengths, then similarly sized fragments can be batched together in reactions and pooled before enhancer screening.
Choice of vector and basal promoter
The choice of STARR-seq vector and basal promoter primarily depends on the host type, the class of candidate enhancers being assayed, and the biological question. The original STARR-seq vector included a backbone sequence derived from vectors previously used for reporter assays or MPRA and featured a species-specific minimal or core promoter, that is, a promoter that binds a set of general transcription factors and RNA polymerase II to initiate transcription (Haberle and Stark 2018). Although most studies have continued to use a similarly designed vector, the backbone has been modified to fit different biological contexts. For example, Inoue et al. (2017) developed LentiMPRA, which included candidates cloned upstream of the minimal promoter on a lentiviral construct. This allows the enhancer–promoter sequence to get integrated into the host genome, thus providing a genomic context to the assay. Lambert et al. (2021) adapted a recombinant adeno-associated virus (AAV) vector to enable library delivery into mouse retina and brain. Muerdter et al. (2018) observed that enhancer signals from the original human STARR-seq minimal promoter, Super Core Promoter 1 (SCP1), were not consistent owing to interference from the origin of replication (ORI) promoter that was present on the same vector. The investigators mitigated this issue by redesigning the vector to carry just the ORI promoter and showed a more robust assay. Sahu et al. (2022) used a minimal δ1-crystallin gene (Sasaki) promoter for their experiments across multiple libraries. They also used a CpG-free vector backbone containing the Lucia reporter gene driven by eukaryotic translation elongation factor 1 alpha 1 (EEF1A1) promoter to assess the effect of CpG methylation on enhancer activity. Klein et al. (2020) compared seven different MPRA and STARR-seq designs and concluded that LentiMPRA and the ORI vector showed the highest consistencies in activity across replicates. Thus, the vector can be designed based on the assay goal and modified to be compatible with the host genome.
Studies have also shown that core promoters are not only specific to a particular species but also specific to different cofactors (Haberle et al. 2019), genomic environments (Hong and Cohen 2022), and enhancers targeting genes of particular function (Zabidi et al. 2015). For example, Zabidi et al. (2015) performed seven whole-genome STARR-seq assays in different Drosophila-derived cell lines using different core promoters and found two distinct promoter classes, housekeeping and developmental, based on promoter–enhancer specificities. Jores et al. (2021) showed that the composition of core promoter elements and the presence of distinct TF binding sites determined the basal strength of the promoter, as well as its specificity to different enhancers. In fact, the low strength of basal promoters adds limitations to measuring the repressive functions of enhancers (Tewhey et al. 2016). Recent MPRA studies have also reported automated methods for the design and synthesis of nonrepetitive promoter sequences (Hossain et al. 2020), and proposed models to predict transcription initiation rates for different promoters (LaFleur et al. 2022). Martinez-Ara et al. (2022) tested pairwise enhancer–promoter combinations in mouse ESCs and reported that enhancer–promoter pairs targeting housekeeping genes show more consistent activity than pairs targeting other genes. Bergman et al. (2022) also tested pairwise combinations of 1000 enhancers with 1000 promoters in human K562 cells using ExP-STARR-seq. However, they found that promoters were activated uniformly by most enhancers, with enhancer–promoter pairs for housekeeping genes only showing subtle differences compared with other tested combinations. Barakat et al. (2018) tested their ChIP-STARR-seq libraries with multiple promoters, including SCP1, cytomegalovirus (CMV), and AAV on human ESCs, and they did not find significant differences between the promoters. Although core promoters may have specificity to certain enhancers, there is still ambiguity in our understanding of promoter–enhancer interactions. Therefore, variable effects of a basal promoter must be considered when designing MPRA or STARR-seq assays (Mulvey et al. 2021).
Control fragments
The plasmid library can also be designed to include control sequences to validate STARR-seq results. Commonly used control sequences include fragments previously validated to be active (positive control) or inactive (negative control) by MPRA or classical reporter assays (Klein et al. 2020), or “scrambled” sequences can be used as negative controls (Martinez-Ara et al. 2022). Although currently available whole-genome STARR-seq data sets may serve as a repository for shortlisting control sequences, factors such as cell type and enhancer–promoter specificities make it difficult to identify controls specific to a cell type or minimal promoter. To this end, Neumayr et al. (2019) suggest running a small-scale focused STARR-seq screen before an experiment to identify potential control regions. Alternatively, studies have also used regions predicted to be inactive, such as coding sequences of genes (Arnold et al. 2013) or CTCF-binding regions (Vanhille et al. 2015), as a proxy for negative controls.
Sequencing methodology
The final set of considerations for library design include the choice of sequencing platform and the compatible adapters and kits required for library preparation. Almost all STARR-seq libraries use Illumina-compatible designs, in which the putative enhancer fragment is flanked by two adapters consisting of read 1 and read 2, followed by unique index sequences or barcodes (i5 and i7) and P5 and P7 sequences to facilitate the sequencing-by-synthesis method (Fig. 1C). The two adapters flanking the candidate fragment serve as “constant” sequences for primer annealing during library amplification, as well as primer recognition sites during Illumina sequencing. Cloning overhang arms are designed based on the selected STARR-seq vector and are ligated adjacent to the adapters through LM_PCR and are used for library cloning. The overhangs are subsequently replaced by the index barcodes i5/i7 and adapters P5/P7 to facilitate sequencing (Fig. 1C; Supplemental Fig. S1A–E). These adapter sequences are especially important for designing oligo libraries, LM_PCR primers, and blocking oligos during hybridization and capture of focused libraries.
Roadmap to successful library construction
With all the design details in place, the construction of a STARR-seq plasmid library includes several optimization and QC steps with each step contributing to varying levels of assay reproducibility. Here, we highlight key elements of the protocol that strongly influence the outcome and offer clarity and explanations behind various optimizations. The features that impact library building include insert preparation, vector linearization, library cloning, and library amplification (Fig. 2A). Additionally, focused libraries also require a library enrichment step to capture the fragments of interest. This enrichment can be performed before insert preparation using ATAC-seq (Wang et al. 2018), ChIP-seq (Barakat et al. 2018), FAIRE-seq (Chaudhri et al. 2020), fragments extracted from specific BACs, or fragments synthesized as custom oligo pools. Alternatively, library enrichment can also be performed after insert preparation using techniques like CapSTARR-seq, which uses special microarrays (Vanhille et al. 2015), or by using custom hybridization and capture probes (Liu et al. 2017a).
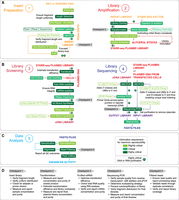
Roadmap to a successful STARR-seq assay. Schematic illustrates five major sections of STARR-seq: (A) Insert preparation and library amplification, (B) library screening and library sequencing, (C) data analysis. Individual steps in each section are categorized as slightly critical (light green), critical (green), or highly critical (dark green) based on the importance of reporting the methodological detail and intermediate results for a reproducible assay. (D) Assay checkpoints. Each section also has a checkpoint, which may serve as stopping points in the protocol to perform validation of previous steps before moving to the next step. Key QC measures that may be performed at these checkpoints are also provided in the bottom panel. Of note, panel B, section 4, “Library Sequencing,” illustrates a methodology for adding unique molecular identifiers (UMIs) and unique dual indexes. A more detailed schematic along with sequence information is provided in Supplemental Figure S1C–E, Supplemental Table S2, and Supplemental Protocol. Alternative methods for UMI addition have also been reported in several studies.
Although these steps typically follow kit-based protocols, there are limited scaling guidelines for differently sized libraries. To ensure replicability of the scaled protocols, details such as the number of reactions performed, sample concentration and purity, details of the purification method used for intermediate product stages, use of control reactions, and the experimental parameters must be reported. Additionally, reporting details of validations for intermediate steps such as length verification of fragments following length selection, as well as adapter and cloning arm ligation steps, provide checkpoints for researchers attempting similarly designed experiments. It is often difficult to understand whether the library being constructed is successful or not without sequencing the library, which is both time-consuming and expensive, and it is also hard to detect and pinpoint experimental errors owing to the length of the protocol. Therefore, having validation checkpoints at intermediate stages helps address these problems. While performing STARR-seq, we shortlisted five such checkpoints in the protocol that require product assessment and validation before proceeding to the next step (Fig. 2A–D). We also categorized intermediate assay steps as “highly critical,” “critical,” and “slightly critical” based on the requirements of data reporting to replicate those steps (Fig. 2A–D).
The final determinant of library coverage is the efficiency of molecular cloning and bacterial transformation. Cloning strategies such as In-fusion HD, Gibson assembly, and NEBuilder HiFi DNA assembly allow for fast and one-step reactions that use complimentary overhang sequences on the inserts and the vector. Although these methods have comparable accuracy and efficiency, the chosen method needs to be optimized before cloning the final library pool. The critical parameters to consider, optimize, and report include (1) purity and concentration of the insert and the vector, (2) insert-to-vector ratio by mass or moles and the maximum molarity per reaction supported by the cloning enzyme, (3) the number of cloning reactions required and pooled, (4) details of negative control used for cloning (typically using a linearized vector-only reaction), (5) competent cell strain and volume per reaction, (6) the amount of cloned product transformed per reaction and the total number of reactions performed and pooled, (7) details of positive (such as supercoiled plasmids like pUC19) and negative (such as unligated, linearized vector) controls used for transformation, (8) the volume of culture used to grow the library, and (9) the number of unique colonies observed in a dilution series for estimating transformation efficiency and library complexity. For example, Barakat et al. (2018) used genomic fragments obtained from chromatin immunoprecipitation to perform multiple PCR reactions and generate inserts for cloning via Gibson assembly. Each reaction was performed in duplicate and pooled before transformation in DH10β cells. The transformed product was grown in SOC medium, serially diluted, plated, and grown in LB to obtain between 8 and 31 million unique colonies. In contrast, Kalita et al. (2018) used synthesized oligos for two rounds of nested PCRs to obtain the inserts for cloning using the infusion HD strategy, followed by multiple transformation reactions. For each of these steps, the investigators provided the exact volumes for each reaction, elution steps, the number of reactions used, and the number of colonies obtained from the dilution series, enabling accurate replication of these experiments.
Before final library transformation, pilot transformations may also be performed using a positive control plasmid and the cloned library to estimate the transformation efficiency by calculating the colony forming units per microgram of plasmid (CFU/μg). This step helps estimate the number of transformations required to achieve the required complexity (Supplemental Table S1). The final complexity of the plasmid library is typically assessed by evaluating sequencing read representation of the target regions. For example, Johnson et al. (2018) performed MiSeq and used the preseq tool (Daley and Smith 2013) to assess library complexity for their whole-genome library. The verified STARR-seq plasmid library is then transfected or transduced into the host for screening. For sequencing, candidate fragments need to be reamplified from the plasmid pool and fitted with complete P5 and P7 adapters and index barcodes replacing the cloning arms (Fig. 1C; Supplemental Fig. S1C–E). To compare self-transcribed transcripts (output) with fragments present in the plasmid library (input), two sets of sequencing libraries (output and input) are prepared. Although there is a consensus on strategy for building the output library, the input library preparation varies across studies. For example, some studies built the input library by directly amplifying the plasmid pool in a second round of LM_PCR, independent of library transfection (Muerdter et al. 2018; Neumayr et al. 2019). Other studies have isolated the input fragment DNA from a fraction of the transfected cells (Arnold et al. 2013; Peng et al. 2020) or extracted DNA and RNA from the same cells to account for fragment loss during transfection (Klein et al. 2020). The latter strategy may be advantageous in discriminating inactive fragments from fragments lost during library delivery for host–genome integrating libraries such as in LentiMPRA (Inoue et al. 2017). This is especially useful for hosts with lower transduction efficiency causing high fragment loss.
Enhancer screening using STARR-seq
Enhancer screening involves preparation of the STARR-seq “output” library followed by deep sequencing of the input and output libraries in parallel. The output library is derived from STARR-seq transcripts generated by functional enhancers when delivered into a host, chosen based on the desired cellular environment. For studies using cultured cells, the choice of host cell line determines multiple transfection parameters such as (1) transfection methodology, (2) amount of plasmid per transfection, (3) the total number of cells transfected per replicate, and (4) incubation time of transfected cells. These details can vary across studies and determine downstream experimental features for screening. For example, for focused STARR-seq, Wang et al. (2018) electroporated 120–130 million lymphoblastoid cell lines (LCLs) per replicate at 5 µg per 1 million cells and used five replicates for their library comprising of more than 7 million unique fragments. Kalita et al. (2018) also electroporated LCLs but used 3 µg per 7.5 million cells per replicate in nine replicates for their library of 75,501 unique fragments. For whole-genome STARR-seq, Sahu et al. (2022) transfected 1 µg of their library per 1 million cells and used two replicates comprising 35–500 million cells per replicate in multiple cell lines. In contrast, Lee et al. (2020) electroporated 8 µg of their whole-genome library per 1 million cells and used two replicates of 700 million to 1 billion cells per replicate in multiple cell lines. Therefore, it is important to consider the exact cell line–specific transfection method and its nuances. For instance, transfection through electroporation is more efficient for harder-to-transfect cells such as ESCs or LCLs and involves shorter incubation time of about 6 h. However, this process requires higher amounts of plasmid and more cells per library owing to the loss of cells during the electroporation process. Methods using lipofectamine may be recommended for various mammalian cell lines such as HEK293T, as they are more efficient and gentler on the cells, but this procedure involves longer incubation times of 12–24 h. Furthermore, higher transfection efficiency reduces the need for multiple transfections for each biological replicate. Another important consideration for selecting the cell line is to test whether there is a need for kinase inhibitors to prevent interferon response from the host. Muerdter et al. (2018) showed that cell lines such as HeLa S3 trigger a type I interferon (INF-1) response upon library transfection, which disrupts enhancer signals and leads to false results. The investigators suggest the use of kinase inhibitors BX-795 hydrochloride to inhibit TANK binding kinase 1 (TBK1) and imidazolo-oxindole PKR inhibitor C16 to inhibit protein kinase R (PKR) to mitigate the situation (Muerdter et al. 2018). Our assessment of recent studies showed enrichment of interferon gene motifs within enhancer peaks, illustrating the significance of interferon response on specific cell types (Johnson et al. 2018; Sahu et al. 2022). Although STARR-seq and most MPRAs have predominantly been performed on cultured cells, some studies have also used in vivo or ex vivo systems such as mouse retina explants (White et al. 2013), cortex (Shen et al. 2016), and postnatal mouse brain (Lambert et al. 2021). Here, considerations include the mode of library delivery such as injections or electroporation (Montana et al. 2011) and transduction efficiency for AAV-based libraries.
The factors that determine the extent and scalability of enhancer screening include library size and the number of biological replicates. The library size determines the multiplicity of reactions required per biological replicate. Here, the term “reactions” indicates smaller experimental setups that are pooled for a single large experiment, and “replicates” typically refers to additional experiments conducted for validation and reproducibility. Although reactions may be pooled at intermediate steps of the protocol, replicates may only be pooled post hoc during peak calling. The impact of the number of replicates on final output has been assessed previously. For example, Hansen and Hodges (2022) found that an increase in the number of replicates increased enhancer calls in their ATAC-STARR-seq assays. However, the investigators also caution that increasing replicates may also increase the number of false-positive calls. Thus, it is important to consider the number of replicates, especially for studies that pool reads from replicates for peak calling.
Following library delivery, the next series of steps involve total RNA isolation, mRNA isolation, and reverse transcription of STARR-seq transcript-specific mRNA using a reporter-specific primer for first-strand synthesis to build a cDNA library. A majority of studies have used well-described kits for these steps and follow standardized scaling guidelines. However, in order to replicate these steps, investigators should report reaction parameters like sample concentrations, amount of starting material for each step, replicate information, and sample pooling details to ensure replicability of the steps. Additional contributors to reproducibility here include intermediate sample validations and QC assessments including RNA integrity (RIN) analysis and RNA purification using TURBO DNase and RNAClean XP beads, all of which increase library quality. The cDNA library is then amplified using junction PCR primers to remove plasmid artifacts followed by ligation of sequencing adapters and index barcodes using LM_PCR, similar to the input sequencing library (Fig. 2B). An important consideration here is to prevent overamplification of the library, which leads to sequencing bias, by adjusting the number of amplification cycles during the final PCR step. Sequencing libraries may be visualized on an agarose gel for a characteristic “smear” at the expected fragment length as opposed to a tight band (Neumayr et al. 2019), unless the library is built using synthesized fragments of uniform length.
Although preparing the screening library is simpler compared with the plasmid library, certain anomalies may occur that hamper library quality and replicability. For example, batch effects during library preparation can be minimized by constructing biological replicates independently and on different days. For example, Klein et al. (2020) transfected each replicate on separate days and prepared the libraries independently of each other. Another common occurrence in STARR-seq data is the presence of PCR duplicates owing to the use of PCR-based sequencing library preparation methods. Self-transcribed mRNA may resemble these PCR duplicates, leading to data anomalies. Previous studies have typically removed PCR duplicates (deduplication) by filtering reads originating from a single fragment of DNA using computational techniques such as the “MarkDuplicates” function in Picard (Peng et al. 2020). However, Picard may not be an ideal tool as it may also remove STARR-seq transcripts, thus resulting in reduced enhancer signals and high false negatives (Liu et al. 2017b). This tool can be particularly challenging for libraries assaying variants of the same enhancer as it removes fragments of identical length with the same start and end coordinates. Therefore, for accurate removal of PCR duplicates, Kalita et al. (2018) added unique molecular identifiers (UMIs) during the reverse transcription step, such that PCR duplicates possess the same UMI sequence, unlike the self-transcribed mRNA. There are now several published variations of the UMI method. For instance, Neumayr et al. (2019) suggested adding UMIs following an extra second-strand synthesis step for cDNA in a single-cycle PCR reaction. Any UMI-based STARR-seq experiment can use relevant UMI-based read deduplication algorithms such as UMI-tools (Smith et al. 2017), Picard's “UmiAwareMarkDuplicatesWithMateCigar” function (http://broadinstitute.github.io/picard), or the calib library (Orabi et al. 2019).
The final step is sequencing of the input and output libraries. Libraries are usually pooled and subsampled before sequencing. Although these are standardized procedures, small variations in these steps can largely impact data reproducibility. For example, while running multiple STARR-seq samples on the same lane of an Illumina-based sequencer, a phenomenon called “index hopping” may occur, in which index barcodes are assigned to the wrong libraries, leading to inaccurate sequencing data (Kircher et al. 2012). To mitigate this, each library can be assigned two indexes or unique dual indexes (UDI) to detect these incidents and increase the accuracy of demultiplexing individual libraries from a library pool (Figs. 1C, 2B; MacConaill et al. 2018).
STARR-seq analysis and reporting
Despite strictly following experimental design and protocols for reproducing an assay, a lack of data analysis guidelines can result in inconsistent findings. The main features that contribute to reproducibility here include sequencing depth, read processing, and read QC, as well as choice of peak caller, cut-offs used for analysis, data QC, and validations. Although there are well-described guidelines and data standards set for sequencing techniques such as RNA-seq (Conesa et al. 2016), ChIP-seq (Landt et al. 2012), Hi-C (Lajoie et al. 2015), and ATAC-seq (Yan et al. 2020), data analysis for STARR-seq is not standardized.
Sequence depth, coverage, and library complexity
A major factor for any high-throughput study is to attain sufficient coverage of the sequenced library. For example, the Sequence QC project (SEQC) showed that read depth and choice of analysis pipelines are key aspects for reproducibility of RNA-seq experiments (Su et al. 2014). Read depth and coverage requirements for techniques including ChIP-seq (Landt et al. 2012) and RNA-seq (Tarazona et al. 2011; Sims et al. 2014; Conesa et al. 2016) have been thoroughly discussed, and multiple data standards have been set by consortia such as ENCODE (The ENCODE Project Consortium 2007). On the other hand, STARR-seq read depth requirements are vague. Even calculating library coverage of STARR-seq data using available RNA-seq guidelines results in inaccurate estimates and faulty inferences of current studies.
Whole-genome STARR-seq assays typically have lower coverage owing to technical limitations, although these assays are highly useful for screening entire genomes for strong and distinct enhancer signals. For example, Johnson et al. (2018) obtained 59× coverage of the human genome in their library, whereas Liu et al. (2017b) reported that 74.3% of their library was covered by at least 10 reads. Whole-genome studies, which primarily focus on mapping the entire enhancer landscape of the organism, may not require a high-resolution view of enhancer activity. In contrast, smaller focused libraries such as those quantifying the effect of noncoding mutations require a higher coverage to reduce false-negative signals. For example, Schöne et al. (2018) obtained more than 100× coverage of the glucocorticoid receptor binding sites they assessed for activity. Hence, there needs to be an established minimum sequencing depth requirement for STARR-seq experiments and guidelines for reporting read length, number of sequenced reads per library, and sequencing parameters used to generate reproducible data. Computational tools such as preseq (Daley and Smith 2013) can be used to estimate the number of sequenced reads required to obtain sufficient coverage for all the unique fragments present in a sequencing library, but they require a preliminary “shallow” sequencing run of the STARR-seq plasmid library. Based on the preliminary run, preseq estimates a library complexity curve that can be used to determine the efficacy of deeper sequencing runs. Alternatively, the required number of reads can also be estimated from the library size, required library complexity, and expected dynamic range (i.e., range of enhancer activity) of the library, as shown in Supplemental Table S1.
There is also a large variation in how sequence depth is reported by different studies. For example, some studies have reported the percentage of unique library fragments with at least “N” reads as a measure of sequencing depth, whereas others have reported the average number of reads that aligned to all library fragments. We suggest reporting both these metrics because they each summarize sequencing depth distinctly; the former presents an atomistic view of read coverage of the fragment, whereas the latter indicates library coverage as a whole.
Read processing and QC
After completion of sequencing, raw reads generated from the sequencer are first demultiplexed, and the correct sample labels are assigned. Reads are then mapped to a reference genome and filtered to remove unmapped reads, off-target reads, PCR duplicates, and reads with low mapping quality based on the assigned mapping quality (MAPQ) scores. Read filtering results in the loss of a large percentage of reads and reduces the overall coverage of the library. Loss of coverage will reduce the strength of enhancer signals and may result in incorrect quantification of target regions. For example, Johnson et al. (2018) reported up to a 40% loss of their reads after QC filtering. To compare the read loss statistics and QC metrics across studies, we analyzed multiple published STARR-seq data sets in addition to our own using a custom analysis pipeline (Supplemental Methods). In our assay, which included a modified sequencing library design to include UMIs and UDIs (Fig. 3A; Supplemental Fig. S1C–E; Supplemental Table S2), we filtered out ∼75% of all reads after QC (Fig. 3B; Supplemental Methods). Because the reanalyzed data sets did not contain UMIs, precluding the use of our deduplication strategy, we instead modified our pipeline to use Picard (http://broadinstitute.github.io/picard) to remove duplicate reads. We observed that the percentage of reads lost after read QC for focused STARR-seq libraries ranged between 70% and 80%, whereas whole-genome libraries lost approximately half as much (Supplemental Fig. S2). We also observed that the percentage of reads lost owing to deduplication was significantly lower in our study compared with the reanalyzed data. It is possible that Picard's deduplication strategy inadvertently eliminated self-transcribing STARR-seq fragments along with PCR duplicates. Therefore, it is important not only to consider read loss after sequencing when designing the experiment but also to report read QC steps such as filtering parameters and cut-offs used, on public repositories such as GitHub to enable replication of the analysis.
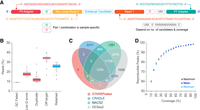
Read architecture, read depth loss, and peak calling comparisons. (A) Schematic illustrates read architecture of final STARR-seq sequencing library (input and output) before sequencing. (B) Box plots show the percentage of STARR-seq reads retained and those removed owing to various QC filters during analysis of in-house STARR-seq assays. (C) Overlap of STARR-seq peaks identified from the same experiment by four different callers. (D) Percentage of peaks that were reproduced from in-house STARR-seq data set after down-sampling to various coverage thresholds. Analysis details are provided in Supplemental Methods.
After ensuring read quality, studies often evaluate data quality and reproducibility by calculating correlation coefficients for read counts between library replicates. Some studies also calculate correlation of fold change between read counts of RNA from output compared with DNA from input libraries, across sample replicates to check for consistency of STARR-seq activity. Our analysis of published data sets and our own data showed strong correlations between library replicates (Supplemental Fig. S3A–C; Supplemental Methods) similar to correlations (Pearson correlation coefficients >0.8) reported by other STARR-seq studies (Supplemental Fig. S4). Correlation values can vary across studies depending on (1) the type of correlation metric (Pearson or Spearman's) and type of correlation measured (input vs. input, output vs. output, or output/input vs. output/input), (2) normalization of reads (RPM, RPKM, or not normalized), and (3) the read scaling factor used (log scale or not scaled). Additionally, data quality can be assessed for batch effects using principal component analysis (PCA) for all library replicates, in which clustering reflects library consistency. For instance, PCA of our libraries showed strong clustering of library replicates and were consistent with the reanalyzed data sets from published studies (Supplemental Fig. S5A–C; Supplemental Methods).
Enhancer calling
Following data QC assessment, the next step involves comparing the input and output libraries to quantify enhancer activity. Here, read counts for candidate regions are compared between the output and input libraries to determine a “peak” of concentrated reads that are indicative of enhancer activity. Previously, peak calling was made using either the MACS2 algorithm, which was originally developed for ChIP-seq analysis (Arnold et al. 2013), or the DESeq2 algorithm (Love et al. 2014), or enhancer activity was detected by calculating statistically significant fold-change in reads per region between the input and output libraries. Recently, STARR-seq-specific peak calling algorithms such as STARRPeaker (Lee et al. 2020) and CRADLE (Kim et al. 2021) have also been developed. The primary difference between the peak calling algorithms lies in the probabilistic read density distribution used to estimate expected STARR-seq read density across the regions of interest. MACS2 and CRADLE use a Poisson distribution to model region-wide read density, whereas DESeq2 and STARRPeaker use negative binomial distribution. Additionally, CRADLE and STARRPeaker use regression models to account for sequence-based biases before modeling the read density distribution. We also note that each of these callers have distinct utilities based on the data obtained and the assay design. For example, CRADLE may be better suited for studies with low correlation and irregular clustering across replicates, as it takes variance between replicates into account before merging reads for peak calling.
To assess reproducibility of enhancer calls, we used four methods on our STARR-seq data and assessed overlap of peaks between callers (Fig. 3C; Supplemental Methods). Although peaks called by STARRPeaker overlapped with all other peak callers, the number of peaks varied across different callers, suggesting higher experimental noise than enhancer signal for a subset of enhancers. Akin to other massively parallel sequencing experiments, signal-to-noise ratio in STARR-seq can be measured by evaluating the ratio between the mean and the standard deviation of enhancer activity values across multiple replicates. Furthermore, STARR-seq data might also be confounded by DNA sequence-related biases that cannot be removed solely by increasing the number of replicates (Kim et al. 2021). Thus, we recommend using STARR-seq-specific callers such as STARRPeaker and CRADLE that can identify and eliminate sequence-associated biases.
To investigate the effect of read coverage on peak calling, we subsampled our reads and used STARRPeaker to call peaks to delineate the percentage of peaks retained at different levels of sequencing coverage (Fig. 3D; Supplemental Methods). We observed a steady increase in peaks with increase in coverage up to 280×, showing the importance of read depth for STARR-seq. For example, we found that only 60% of the peaks remained at ∼28× coverage and 90% at 112× coverage, indicating the need to benchmark these parameters. After peak calling, the next step involves peak validation and assessment of control regions. Comparing peaks at candidate regions with control regions ensures robust detection of enhancer activity. For example, in our study, we compared the peaks at candidate enhancer regions to those within exons and noticed significantly reduced activity across the exonic regions (Supplemental Fig. S6), in line with our assumption that coding regions of genes on an average display lower activity than candidate enhancer sites (Supplemental Methods).
Another component of STARR-seq data analysis is to determine the sequence features of the regions designated as peaks. Traditionally, motif enrichment analysis (MEA) tools such as HOMER (Heinz et al. 2010) or MEME (Bailey and Elkan 1994) have been used to detect enriched binding sites of known TF motifs within a set of active enhancer peaks. However, machine learning–based classification models can also serve a similar purpose. For example, Sahu et al. (2022) used logistic regression to predict enhancer activity based on the presence of TF binding motifs. Furthermore, deep learning methods, such as the convolutional neural networks that predict enhancer activity by automatically learning the underlying sequence features, can be powerful (de Almeida et al. 2022). Insights derived from these models add biological contexts to the identified peaks.
Conclusion
Successful functional genomic studies take years to design and perform, and each step goes through repeated iterations in multiple replicates to optimize parameters for meaningful outcomes. Even steps involving established kits and protocols need to be scaled according to the study design and often require customizations and intermediate step validations. Although each of these factors contribute to overall assay reproducibility, most published studies focus solely on the final outcome and tend to underexplain the methods, optimizations, and intermediate validations, resulting in large gaps of knowledge for researchers attempting to reproduce the results or tailor the study for their own biological questions (Box 2; Fig. 4A–G). In fact, a comparison of human whole-genome STARR-seq data sets across studies showed variations (Supplemental Fig. S7; Supplemental Methods). Therefore, standardization of protocols and data reporting guidelines would benefit researchers and significantly improve the quality of the conducted assays. Here, we highlighted the different challenges in performing STARR-seq, a particularly long and difficult assay with huge potential to identify detailed enhancer landscapes and validate enhancer function. We emphasize the importance of reporting details related to biological context, underlying hypothesis, and experimental design, as well as protocol and bioinformatic analysis parameters, to ensure replicability of each step and outcome of the assay.
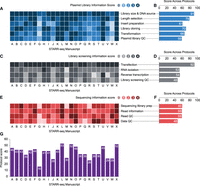
Reproducibility in current STARR-seq studies. (A,C,E) Heatmap shows feature scores for deidentified studies A–X for assay design and plasmid library preparation (A), library screening (C), and sequencing information (E). (B,D,F) Cumulative scores for each feature across studies illustrating reporting trends and highlighting features that are typically well explained or underexplained. (G) Cumulative scores for each assay (A–X) for all features enabling direct comparisons of reproducibility. We observed that critical design factors, such as library scale and source and fragment length, or experimental features, such as transfection, scored three or more across 87.5%–100% of publications, with studies missing only minor details and explanations. In contrast, features such as library screening QC, data QC, and data transparency scored three or more across 16.6%–37.5% of publications.
Reproducibility across STARR-seq studies
To assess and quantify reproducibility for STARR-seq based studies, we systematically compared 24 studies (Arnold et al. 2013; Vanhille et al. 2015; Vockley et al. 2015; Verfaillie et al. 2016; Liu et al. 2017a,b; Barakat et al. 2018; Brandt et al. 2018; Johnson et al. 2018; Kalita et al. 2018; Klein et al. 2018; Muerdter et al. 2018; Schöne et al. 2018; Wang et al. 2018; Zhang et al. 2018; Chaudhri et al. 2020; Klein et al. 2020; Lee et al. 2020; Peng et al. 2020; Van Ouwerkerk et al. 2020; van Weerd et al. 2020; Glaser et al. 2021; Selvarajan et al. 2021; Sahu et al. 2022) that performed STARR-seq experiments on mammalian or Drosophila cells (Fig. 4A–G). We further identified 15 features critical for assay success and assessed their potential for reproducibility across the published studies. We scored each feature from zero to four based on the reporting of methodological details, design rationale, and QC measures, with zero indicating no detail and four indicating reporting of complete details for each feature that would allow for replication of the assay. The scoring was performed independently by four individuals and aggregated. A detailed scoring rubric for each feature is provided in Supplemental Table S3, A through C. Our approach provides a method for quantifying the extent of reproducibility of published studies. In particular, we observed that investigators typically mentioned steps such as read filtering, mapping tools, peak callers, and QC experiments but failed to provide associated intermediate data or parameters and cut-off thresholds used for their analyses, thereby reducing replicability of the steps. Furthermore, critical intermediate steps for sequencing such as library multiplexing and pooling that may be performed at shared sequencing cores are often overlooked, leading to additional disparities in data quality. Therefore, standardization of these assay features and implementation of uniform data reporting guidelines will strongly improve reproducibility of STARR-seq assays.
Data access
All sequence data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA879724. Detailed descriptions of analysis pipelines used are provided in Supplemental Methods. All source code is provided as an additional Supplemental Code file and at GitHub (https://github.com/deeprob/starrseq_reproducibility).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Drs. Matthew Jensen, Yasuhiro Kyono, Istvan Albert, Aswathy Sebastian, Howard Salis, Ross Hardison, Corrine Smolen, and the Penn State Genomics Core Facility for technical support for this project. This work was supported by the National Institutes of Health grants R01-GM121907 (National Institute of General Medical Sciences) and R21-NS122398 (National Institute of Neurological Disorders and Stroke), and resources from the Huck Institutes of the Life Sciences to S.G.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277204.122.
- Received August 14, 2022.
- Accepted March 15, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








