
The impact of SWI/SNF and NuRD inactivation on gene expression is tightly coupled with levels of RNA polymerase II occupancy at promoters
- Sachin Pundhir1,2,3,4,
- Jinyu Su1,2,3,4,
- Marta Tapia1,2,3,
- Anne Meldgaard Hansen1,2,3,
- James Seymour Haile1,2,3,
- Klaus Hansen1,2,3 and
- Bo Torben Porse1,2,3
- 1The Finsen Laboratory, Copenhagen University Hospital–Rigshospitalet, DK2200 Copenhagen, Denmark;
- 2Biotech Research and Innovation Center, Faculty of Health Sciences, University of Copenhagen, DK2200 Copenhagen, Denmark;
- 3Novo Nordisk Foundation Center for Stem Cell Biology, DanStem, Faculty of Health Sciences, University of Copenhagen, DK2200 Copenhagen, Denmark
-
↵4 These authors contributed equally to this work.
Abstract
SWI/SNF and NuRD are protein complexes that antagonistically regulate DNA accessibility. However, repression of their activities often leads to unanticipated changes in target gene expression (paradoxical), highlighting our incomplete understanding of their activities. Here we show that SWI/SNF and NuRD are in a tug-of-war to regulate PRC2 occupancy at lowly expressed and bivalent genes in mouse embryonic stem cells (mESCs). In contrast, at promoters of average or highly expressed genes, SWI/SNF and NuRD antagonistically modulate RNA polymerase II (Pol II) release kinetics, arguably owing to accompanying alterations in H3.3 and H2A.Z levels at promoter-flanking nucleosomes, leading to paradoxical changes in gene expression. Owing to this mechanism, the relative activities of the two remodelers potentiate gene promoters toward Pol II–dependent open or PRC2-dependent closed chromatin states. Our results highlight RNA Pol II occupancy as the key parameter in determining the direction of gene expression changes in response to SWI/SNF and NuRD inactivation at gene promoters in mESCs.
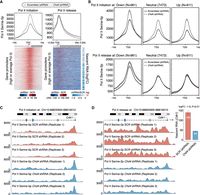
Chromatin remodeling is a fundamental process that ensures the proper presentation of DNA to transcription factors. It is catalyzed by ATP-dependent chromatin remodeling complexes such as the SWI/SNF and NuRD remodelers, both of which bind to numerous common target gene promoters as revealed by the chromatin occupancy profiles of their ATPase subunits (BRG1–SWI/SNF and CHD4–NuRD) in mouse embryonic stem cells (mESCs) (Jiang and Pugh 2009; Yildirim et al. 2011; Morris et al. 2014; Kadoch and Crabtree 2015; Clapier et al. 2017; Brahma and Henikoff 2019; Kubik et al. 2019). At gene promoters, SWI/SNF reorganizes nucleosomes to increase DNA accessibility, whereas NuRD rewraps DNA around nucleosomes to promote a more closed chromatin state (Fig. 1A; Yildirim et al. 2011; Morris et al. 2014; Kadoch et al. 2017; Bornelov et al. 2018; Hodges et al. 2018).
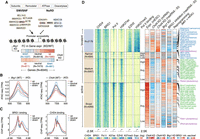
RNA polymerase II (Pol II)–bound genes respond opposite to that of PRC2-bound genes upon perturbation of SWI/SNF or NuRD activities. (A) The composition of the SWI/SNF and NuRD remodeling complexes. BRG1 (SWI/SNF) and CHD4 (NuRD) are the ATPase subunits of these complexes. SWI/SNF supports open whereas NuRD supports closed chromatin states at gene promoters. Also shown are the SWI/SNF and NuRD target genes in mESCs (N = 9345) binned into groups of 25 each and rank-ordered based on the Pol II binding levels at their promoters. For each bin, the median gene expression fold changes following Chd4 or Brg1 knockdown (KD) are shown. (B) Changes in promoter accessibility at genes that are up-regulated, neutral, or down-regulated following KD of Brg1 (left) or Chd4 (right). Standard error is represented by a shaded area around the lines. (C) BRG1 and CHD4 binding signals at the promoter of genes that are up-regulated, neutral, or down-regulated following KD of Brg1 (left) or Chd4 (right). (D) Occupancy levels of CHD4, BRG1, Pol II, EZH2, and histone modifications (H3K27ac and H3K27me3) at active (N = 7166) and bivalent (N = 2179) gene promoters. Active promoters are further subclassified based on their widths: narrow, ≤500 bp; medium, 500–1000 bp; broad, >1000 bp. Genes are binned into groups of 25 each and rank-ordered based on the Pol II binding levels at their promoters. For each bin, the median gene expression fold changes following Chd4 or Brg1 KD or during MEF-to-mESC reprogramming. Also shown are the changes in absolute (spike-in normalized) nascent RNA levels upon BRG1 inhibitor treatment (BRM014 for 3 h) and Chd4 KD. Genes corresponding to the KEGG pathways regulated by high/average Pol II–bound and low Pol II–bound/bivalent genes are marked on the heatmap (Supplemental Fig. S3A).
The antagonistic activities of SWI/SNF and NuRD are finely tuned at gene promoters, which, when altered, lead to moderate but wide-spread changes in gene expression (Yildirim et al. 2011; Bornelov et al. 2018). Importantly, these changes in expression are sufficient to negatively affect self-renewal and pluripotency properties of mESCs (Kaji et al. 2006; Ho et al. 2009). At bivalent promoters, SWI/SNF-associated increases in DNA accessibility and gene expression are linked to its role in directly evicting Polycomb repressive complexes 1 and 2 (PRC1 and PRC2) (de Dieuleveult et al. 2016; Kadoch et al. 2017; Stanton et al. 2017; Bracken et al. 2019). This is in contrast to NuRD, whose activity promotes PRC2 binding at bivalent genes and is accompanied by a decrease in DNA accessibility and in gene expression (Yildirim et al. 2011; Reynolds et al. 2012; Sparmann et al. 2013; de Dieuleveult et al. 2016; Bracken et al. 2019). Thus canonically, SWI/SNF-induced increases in DNA accessibility have a positive effect on gene expression, whereas NuRD-mediated DNA compaction has a negative effect on gene expression. However, the vast majority of SWI/SNF and NuRD target genes are not bivalent and often show paradoxical expression changes (i.e., DNA accessibility and expression changes in opposite directions) in response to perturbations of SWI/SNF and NuRD components (Yildirim et al. 2011; de Dieuleveult et al. 2016; Bornelov et al. 2018). The predominantly overlapping binding patterns of SWI/SNF and NuRD remodelers (Morris et al. 2014), as well as their canonical and paradoxical impact on gene expression, raise two fundamental questions: (1) are accessible DNA at promoters per se necessarily correlated to active transcription, and if not, what features distinguish canonical and paradoxical response genes; and (2) how do the two key chromatin remodeling complexes coordinate their antagonistic activity in order to regulate the expression of their common target genes?
Recent studies have shown that SWI/SNF and NuRD can also regulate the levels of RNA polymerase II (Pol II) at gene promoters. Specifically, increased occupancy of the MBD3 subunit of the NuRD complex led to reduced RNA Pol II levels at promoters in mESCs (Yildirim et al. 2011; Bornelov et al. 2018). Similarly, loss of the PWWP2A/B subunit of the NuRD complex decreased Pol II elongation levels at highly expressed genes in mESCs (Zhang et al. 2018). Furthermore, the SWI/SNF subunit ARID1A has been shown to regulate the levels of Pol II promoter-proximal pausing in ovarian clear cell carcinoma (Trizzino et al. 2018). Collectively, these findings suggest that the SWI/SNF and NuRD chromatin remodelers impact various aspects of Pol II behavior at promoters.
In this study, we aimed to understand the molecular mechanisms by which SWI/SNF and NuRD coordinate their remodeling activity to regulate the expression of common target genes.
Results
Perturbation of SWI/SNF and NuRD activities leads to widespread paradoxical changes in gene expression
We analyzed a previously published data set (de Dieuleveult et al. 2016) to determine changes in DNA accessibility and gene expression in response to perturbations of BRG1 (SWI/SNF) and CHD4 (NuRD) activities at all common target promoters (N = 9345; see Methods). As expected, DNA accessibility decreased upon Brg1 knockdown (KD), whereas it increased upon Chd4 KD (Fig. 1B). However, at a subset of promoters, changes in accessibility led to paradoxical changes in gene expression, meaning that gene expression increased despite the expected loss in DNA accessibility upon Brg1 KD (N = 1390) and decreased despite the gain in promoter accessibility upon Chd4 KD (N = 961) (Fig. 1A,B; Supplemental Fig. S1A,D). Importantly, BRG1 and CHD4 bind at similar intensities at all target promoters (as determined by ChIP-seq for TAP-tagged BRG1 and CHD4 using the same antibody), suggesting that the absolute level of remodeler binding cannot explain the observed paradoxical changes in gene expression (Fig. 1C). Instead, we found that the promoters, where the two remodelers' activity-associated changes in promoter accessibility fit with expression changes, are enriched for EZH2 binding (a subunit of the PRC2 complex) (Supplemental Fig. S1A,D; Pasini et al. 2007). In contrast, the promoters for which we observed paradoxical changes in expression in response to perturbations in remodeler activity are enriched for Pol II binding. To rule out secondary effects and internal sequence-depth normalization issues, we also measured changes in nascent RNA levels using spike-in normalized 4sU nascent RNA-seq and observed similar canonical and paradoxical changes in gene expression upon inhibition of BRG1 activity (3-h BRM014 treatment) and upon Chd4 KD (Supplemental Fig. S1B,C,E–G).
Mutually exclusive binding of RNA Pol II and PRC2 at canonical and paradoxical SWI/SNF and NuRD response promoters
Next, we sought to determine the characteristic features distinguishing genes displaying paradoxical and canonical behavior in response to SWI/SNF or NuRD perturbations and, therefore, asked if the mutually exclusive binding of Pol II and EZH2 at gene promoters can explain these divergent responses.
Pol II–bound and EZH2-bound gene promoters generally represent the ends of the gene expression spectrum, that is, active and bivalent in mESCs, and can be classified based on the H3K4me3 and/or H3K27me3 histone modification patterns at their promoters (Supplemental Fig. S2A; Bernstein et al. 2006). We first rank-ordered genes from the active and bivalent classes according to the Pol II occupancy levels at their promoters and measured changes in their expression following KD of Brg1 and Chd4 (Fig. 1D). To account for any potential effect of promoter width on SWI/SNF and NuRD activities (de Dieuleveult et al. 2016), active genes were further subdivided based on this parameter (narrow, medium, and broad). We found that irrespective of promoter width, genes bound by Pol II at average to high levels showed completely opposite changes in their expression (both in mature and nascent RNA levels) in response to Chd4 and Brg1 KD compared with those bound by EZH2 (bivalent) or those displaying low Pol II occupancy at their promoters (Fig. 1D). Especially among those with medium and broad promoters, the most active genes are the ones showing paradoxical gene expression changes following KD of Brg1 or Chd4 (Fig. 1D). Importantly, we observed similar canonical and paradoxical changes in the expression of genes from the four classes (high, average, or low Pol II and bivalent) when mESCs were treated with the BRG1 inhibitor BRM014, as well as upon the re-expression of MBD3 in Mbd3-null mESCs through the addition of tamoxifen (MBD3 along with CHD4 co-occupy most of the active/bivalent gene promoters as part of the NuRD complex) (Supplemental Figs. S1H, S2A; Bornelov et al. 2018; Lurlaro et al. 2021). We provide examples of the canonical and paradoxical changes in expression by four genes: Cdk1, Ercc6l, Fam107b, and Ets1 (Fig. 2A). These genes display comparable levels of BRG1 and CHD4 binding at their promoters and, importantly, show the anticipated changes in promoter accessibility following down-regulation of SWI/SNF and NuRD activity (Yildirim et al. 2011; de Dieuleveult et al. 2016; Gatchalian et al. 2018; Kloet et al. 2018). Although modulation of remodeler activities resulted in canonical expression changes for Fam107b and Ets1 (low Pol II–bound or high EZH2-bound [bivalent], correlating positively with BRG1 occupancy and negatively with CHD4 occupancy), those for Cdk1 and Ercc6l (high or average Pol II–bound) were completely opposite (Fig. 2A). These findings clearly highlight both the canonical and paradoxical impact of SWI/SNF and NuRD remodelers on gene expression.
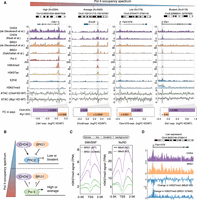
Mutually exclusive binding of RNA Pol II and PRC2 explains the paradox in the antagonistic activities of NuRD and SWI/SNF at gene promoters. (A) Genome browser view for four representative genes (Cdk1, Ercc6l, Fam107b, and Ets1) that are bound by both CHD4 and BRG1 at their promoters. DNA accessibility of all four gene promoters is increased following Chd4 KD (light green) and is decreased following Brg1 KD. Also shown are gene expression fold changes following Chd4 or Brg1 KD (bottom), the orientation of which are opposite for high/average Pol II–bound genes and low Pol II–bound/bivalent genes. (B) A model to explain the differential activity of NuRD and SWI/SNF at Pol II–bound and PRC2-bound genes. (C) Changes in H3K27me3 modification at lowly expressed and bivalent gene promoters (PRC2-bound) following loss of BRG1 activity (left) and KO of Mbd3 (right). (D) Genome browser view for Fam107b (lowly expressed gene) where the loss in BRG1 activity leads to an increase in H3K27me3 modification. The opposite is observed following KO of Mbd3.
Functionally, high or average Pol II–bound genes (paradoxical response) are associated with distinct Gene Ontology categories compared with low Pol II–bound or EZH2-bound genes (canonical response) (Supplemental Fig. S3A). Specifically, paradoxical-response genes become up-regulated during mouse embryonic fibroblast (MEF)-to-mESC reprogramming and are enriched in cell cycle and DNA replication pathways, that is, GO terms that are highly active in mESCs (Fig. 1D; Supplemental Fig. S3A; Agrawal et al. 2014; Liu et al. 2019; Michowski et al. 2020). In contrast, canonical-response genes are involved in WNT signaling and in regulation of pluripotency pathways important for mESCs to exit self-renewal and commit to differentiation and, in agreement, are down-regulated during MEF-to-mESC reprogramming (Fig. 1D; Davidson et al. 2012). Taken together, these results suggest that the modality of SWI/SNF and NuRD activities at gene promoters is dependent on the gene activity level.
SWI/SNF and NuRD antagonistically modulate PRC2 occupancy at low Pol II–occupancy and bivalent gene promoters
Next, we wanted to understand the underlying mechanisms for the differential impact of SWI/SNF and NuRD on bivalent/low Pol II–bound genes and genes showing more pronounced Pol II occupancy at their promoters in mESCs (Fig. 2B; Supplemental Fig. S3B). To determine if the observed phenomena could be extended to differentiated cells, we also defined four classes of promoters (high, average, and low Pol II and bivalent) in MEFs by analyzing available ChIP-seq data (for classification thresholds, see Methods) (Supplemental Fig. S2B; Chronis et al. 2017). First, we examined the role of the two remodelers in regulating PRC2 activity at low Pol II–bound and bivalent genes (Fig. 2B). Specifically, we determined H3K27me3 levels at these promoters in mESCs expressing a catalytic dead version of BRG1G784E/+ (ATPase-dead mutant Gly784Glu, G784E) or in Mbd3 knockout (KO) mESCs, and compared it to WT controls (Reynolds et al. 2012; Stanton et al. 2017). Similar to the roles of these remodelers at bivalent genes, we observed an increase in PRC2 as well as in PRC1 (RING1B) activity at low Pol II–bound genes, concomitant with a decrease in promoter accessibility and gene expression following loss of BRG1 activity (Fig. 2C,D; Supplemental Fig. S3C,D). In contrast, PRC2 activity decreased at these genes, accompanied by an increase in promoter accessibility and expression in Mbd3 KO mESCs (Fig. 2C,D). MBD3 colocalizes with CHD4 at gene promoters in mESCs (Supplemental Figs. S2A, S3E; Bornelov et al. 2018; Burgold et al. 2019), and the gene expression profile of Chd4 KD mESCs (de Dieuleveult et al. 2016) positively correlates with that of Mbd3 KD mESCs (Supplemental Fig. S3F; Yildirim et al. 2011). In further support of a positive role of CHD4 in regulating PRC2 occupancy, high CHD4 occupancy levels relative to BRG1 at gene promoters positively associate with high H3K27me3 levels and low gene expression in both mESCs and MEFs, as well as during MEF-to-mESC reprogramming (as measured by ChIP-seq) (Supplemental Fig. S4A–D). In summary, we conclude that SWI/SNF and NuRD antagonize each other to regulate PRC2 activity and gene expression at lowly expressed and bivalent genes in mESCs in a manner compatible with their impact on DNA accessibility.
SWI/SNF and NuRD antagonistically modulate Pol II release kinetics at average- and high Pol II–occupancy gene promoters
Having analyzed the canonical impact of SWI/SNF and NuRD at bivalent- and low Pol II–occupancy gene promoters, we next addressed the paradoxical impact of these remodelers at average- or high Pol II–occupancy gene promoters (Fig. 2B). The paradoxical increase in expression following Brg1 KD may potentially be explained by the concomitant increase in Pol II levels at gene promoters, and vice versa for Chd4 KD. However, in contrast, we observed a decrease in promoter Pol II levels following functional inactivation of BRG1 (as assessed in BRG1G784E/+ [ATPase-dead mutant Gly784Glu, G784E] and Brg1 KD mESCs), as well as the opposite in response to the loss of the NuRD components CHD4 and MBD3 (Fig. 3A,D; Supplemental Fig. S5A,B). We observed similar changes in promoter proximal nascent RNA at promoters, which complements the ChIP-seq-based measure of Pol II levels in mESCs (Fig. 3B,E; Bornelov et al. 2018; Hainer et al. 2015). In fact, Pol II occupancy positively associates with SWI/SNF activity and negatively with NuRD activity at gene promoters in both mESCs and MEFs (Fig. 3G; Supplemental Fig. S5E).
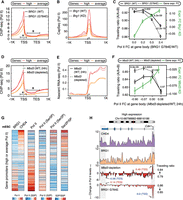
Differential effects of SWI/SNF and NuRD on RNA Pol II release kinetics at promoters of average/high Pol II–bound genes. (A,B) Changes in Pol II occupancy (A) and nascent RNA levels (B) at promoters (N = 6387) upon loss of BRG1 activity. Standard error is represented by a shaded area around the lines. (C) Genes with average/high Pol II promoter occupancy in mESCs (N = 6387) are binned into four groups (1597 genes each) based on the changes in the levels of Pol II released into the gene body upon loss of BRG1 activity (x-axis). For each bin, the changes in traveling ratio (solid vs. dashed black lines) and gene expression (green line) upon loss of BRG1 activity are shown (Wilcoxon rank-sum test, two-sided). (D–F) Same as A–C but following Mbd3 loss. (G) Genes (N = 6387) are binned into groups of 25 each based on the CHD4 and BRG1 binding ratio at their promoters. For each group, the median occupancy levels of initiating (Ser5p) and elongating (Ser2p) Pol II forms are shown. (H) Genome browser view for Cdk1, where loss of BRG1 activity leads to a decrease in Pol II promoter occupancy. The opposite is observed following Mbd3 depletion.
We hypothesized that if reduced Pol II occupancy upon SWI/SNF inactivation is accompanied by an increase of Pol II being released into the gene body, then it could explain the paradoxical increase in gene expression, and vice-versa in case of perturbation of NuRD activity. We therefore measured changes in Pol II occupancy levels at promoter versus gene body following loss of BRG1 (traveling ratio: Pol II [promoter]/Pol II [promoter + gene body]; see Methods). Indeed, we observed a decrease in the traveling ratio following BRG1 loss (Fig. 3C; Supplemental Fig. S5C), which is most prominent at the group of genes that displayed the highest increase in Pol II levels at the gene body and, consequently, in mRNA levels (Fig. 3C). In contrast, we observed an increase in the traveling ratio in response to Mbd3 loss (Fig. 3F; Supplemental Fig. S5D), which is most prominent at the group of genes showing the steepest decline in Pol II levels at the gene body and, consequently, in mRNA levels (Fig. 3F).
A key function of maintaining a high Pol II fraction at the transcription start site (TSS) relative to the gene body is to prevent the formation of a repressive chromatin architecture at promoters by competing with nucleosomes for occupancy (Gilchrist et al. 2010; Teves et al. 2014; Erickson et al. 2018). Thus, it is plausible that SWI/SNF maintains a high level of promoter-proximal Pol II, which in turn results in a relatively open chromatin state at promoters, whereas NuRD may oppose Pol II buildup at promoters by increasing its constant release into the gene body, thus maintaining a relatively closed chromatin state at promoters. To examine this possibility, we rank-ordered paradoxical response genes by their CHD4/BRG1 occupancy ratio at promoters and compared the levels of Pol II engaged in initiation (characterized by serine5 phosphorylation of its C-terminal domain; Pol II–ser5p) and productive transcription (characterized by serine2 phosphorylation of its C-terminal domain; Pol II–ser2p) (Fig. 3G). Indeed, among paradoxically affected genes, promoters with relative high levels of BRG1 are characterized by higher levels of Pol II in the promoter-proximal initiation state (Pol II–ser5p). In contrast, high CHD4 occupancy is positively associated with high levels of Pol II engaged in productive transcription (Pol II–ser2p) (Fig. 3G,H; Supplemental Fig. S5F). Similar observations were made during MEF-to-mESC reprogramming (Supplemental Fig. S5G). These findings are consistent with a model in which SWI/SNF stabilizes promoter-proximal Pol II, thereby explaining why its loss leads to an increase in active transcription.
Next, we wanted to further understand how changes in the promoter distribution of Pol II–ser2P and Pol II–ser5P following changes in SWI/SNF occupancy could explain the paradoxical impact of these remodelers on gene expression. We therefore performed ChIP-seq with reference exogenous genome (ChIP-Rx) (Orlando et al. 2014) to accurately measure changes in the levels of initiating (Pol II–ser5p) and elongating (Pol II–ser2p) forms of Pol II at gene promoters in mESCs following 3-h inhibition of the ATPase activity of BRG1 using the BRM014 inhibitor (Lurlaro et al. 2021). In agreement with our earlier observation, both globally as well as for the representative gene Cdk1, Pol II initiation levels decreased at gene promoters following BRM014 treatment. This decrease was accompanied by an increase in Pol II release from the promoters and into the gene bodies (Fig. 4A,C,D; Supplemental Fig. S6A,B,E,F). The observed increase in Pol II release is further supported by a predominant increase in nascent transcription of these genes upon BRG1 inhibition (Fig. 4A). Moreover, Pol II initiation levels decrease irrespective of gene expression changes (down-regulated, neutral, or up-regulated) upon BRG1 inhibitor treatment (Fig. 4B). In contrast, the increases in Pol II release levels downstream from the gene promoters were restricted to up-regulated genes (Fig. 4B).
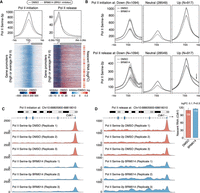
SWI/SNF modulate the rates of Pol II release into the gene body to paradoxically affect the expression of genes having average/high Pol II promoter occupancy levels. (A) Changes in Pol II initiation (left) and Pol II release (right) levels at gene promoters (N = 6387) in mESCs upon inhibiting BRG1 activity by BRM014. Also shown are the changes in the nascent RNA levels of these genes upon BRG1 inhibition (far right). (B) Changes in Pol II initiation (top) and Pol II release (bottom) at genes stratified based on changes in their expression upon inhibition of BRG1 activity by BRM014 in mESCs. (C,D) Genome browser view for Cdk1, where inhibition of BRG1 activity using BRM014 leads to a decrease in Pol II initiation at the promoter (C) and an increase in Pol II release into the gene body (D).
Similarly, we performed ChIP-Rx experiments on Pol II–ser5p and Pol II–ser2p in Chd4 KD mESCs (Supplemental Fig. S6I). In complete contrast to our observation for BRG1 inhibition, we observed an increase in Pol II initiation levels accompanied by a decrease in Pol II release into the gene body as well as in nascent transcription in Chd4 KD mESCs (Fig. 5A; Supplemental Fig. S6C,D,G,H). Furthermore, the loss of Pol II at gene bodies following Chd4 KD is specific to down-regulated genes (Fig. 5B), exemplified by Cdk1 (Fig. 5C,D). Taken together, these results suggest that whereas SWI/SNF shifts the relative balance of Pol II occupancy toward the TSS to maintain promoter accessibility, NuRD shifts it toward the gene body to reduce promoter accessibility. Thus, our results explain the paradoxical impact of these remodelers on the expression of average- and high Pol II–occupancy gene promoters.
NuRD modulate the rates of Pol II release into the gene body to paradoxically affect the expression of genes having average/high Pol II promoter occupancy levels. (A) Changes in Pol II initiation (left) and Pol II release (right) levels at gene promoters (N = 6387) in mESCs upon KD of Chd4 versus a scrambled control. Also shown are the changes in the nascent RNA levels of these genes upon Chd4 KD (far right). (B) Changes in Pol II initiation (top) and Pol II release (bottom) at genes stratified based on changes in their expression in mESCs upon KD of Chd4 versus a scrambled control. (C,D) Genome browser view for Cdk1, where KD of Chd4 leads to an increase in Pol II initiation at the promoter (C) and a decrease in Pol II release into the gene body (D).
A role for H2A.Z and H3.3 histone variants in NuRD- and SWI/SNF-mediated changes in Pol II release kinetics at target promoters
SWI/SNF and NuRD predominantly target nucleosomes flanking the TSS at gene promoters (de Dieuleveult et al. 2016), and canonical H3 and H2A components of these nucleosomes are frequently replaced by specialized H3.3 and H2A.Z histone variants, respectively. Our analysis revealed a strong positive association of BRG1 occupancy with the levels of H3.3-substituted nucleosomes and of CHD4 occupancy with the levels of H2A-Z-substituted nucleosomes at promoters both in mESCs and MEFs (Fig. 6A), as well as during MEF-to-mESC reprogramming (Supplemental Fig. S5G).
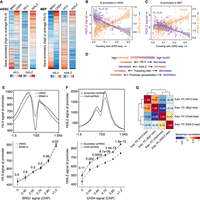
Role of H2A.Z and H3.3 in SWI/SNF- and NuRD-mediated changes in Pol II release kinetics. (A) Genes with average/high Pol II promoter occupancy in mESCs (N = 6387; left) and MEFs (N = 7234; right) are binned into groups of 25 each based on the CHD4 and BRG1 binding ratio at their promoters. For each group, the median CHD4, BRG1, H3.3, and H2A.Z ChIP-seq signals at promoters are shown. (B) Genes (N = 6387) are binned into groups of 25 each based on their Pol II traveling ratio. For each bin, shown are the median H2A.Z and H3.3 levels (Loess regression lines are included). (C) Genes with average/high Pol II promoter occupancy in MEFs (N = 7234) are binned into groups of 25 each based on their Pol II traveling ratio. For each bin, shown are the median H2A.Z and H3.3 levels (Loess regression lines are included). (D) Schematic diagram showing the relationship between SWI/SNF and NuRD occupancy levels with Pol II traveling ratio, DNA accessibility, and H3.3 and H2A.Z levels at gene promoters. (E) Changes in H3.3 levels at promoters in mESCs upon inhibiting BRG1 activity using BRM014 (top). Genes (N = 6387) are binned based on BRG1 occupancy levels in mESCs. For each bin, the H3.3 levels in WT and BRG1 inhibitor-treated mESCs are shown (bottom). (F) Same as E but showing the changes in H2A.Z levels in Chd4 KD mESCs compared with a scrambled control. (G) Hierarchical clustering (based on Spearman's rank correlation) of the gene expression profiles of Chd4 KD, Brg1 KD, H2A.Z-depleted, and H3.3-depleted mESCs. P-values: (***) <0.001, (*) <0.05.
Functionally, TSS-flanking canonical nucleosomes constitute a high-energy barrier for Pol II release into the gene body (Aoi et al. 2020); however, this barrier is reduced at H2A.Z, containing nucleosomes (Santisteban et al. 2011; Weber et al. 2014). Indeed, we observed a gradual decline in traveling ratio (less Pol II at promoter relative to at the gene body) and in promotor accessibility as the levels of H2A.Z increase at active promoters in mESCs and MEFs (Fig. 6B,C, purple lines; Supplemental Fig. S7A–C). In contrast H3.3 has been implicated, via its function as a nucleosomal cofactor for EP300-mediated deposition of H3K27ac, in maintaining high Pol II levels at promoters (Ray-Gallet et al. 2011; Boija et al. 2017; Martire et al. 2019). In agreement, we observed a gradual increase in the traveling ratio and in promoter accessibility as the levels of H3.3 increase at active promoters in both mESCs and MEFs (Fig. 6B,C, orange lines; Supplemental Fig. S7A–C).
These observations beg the question if the inverse association of the two histone variants with Pol II occupancy levels at promoters and their specific enrichment at TSS-flanking nucleosomes can reveal insights into the mechanisms by which the two chromatin remodelers modulate Pol II release kinetics at gene promoters (Fig. 6D). We therefore performed H3.3 and H2A.Z ChIP-Rx experiments (Orlando et al. 2014) in mESCs treated with the BRG1 inhibitor (BRM014) for 3 h and in DMSO controls. In support of a positive relationship between SWI/SNF activity and H3.3 deposition at nucleosomes, we observed reduced H3.3 levels upon inhibition of BRG1 activity (Fig. 6E, top; Supplemental Fig. S7D,F). Importantly, promoters with high BRG1 occupancy show a steeper decline in H3.3 levels following BRG1 inhibitor treatment (Fig. 6E, bottom). Arguing for a selective role of BRG1 activity in regulating H3.3 levels, we observed no changes in H2A.Z levels upon BRG1 inhibition (Supplemental Fig. S7D,F). In contrast, ChIP-Rx experiments in Chd4 KD mESCs revealed a loss in H2A.Z but not in H3.3 levels at gene promoters (Fig. 6F, top; Supplemental Fig. S7E,G). Furthermore, H2A.Z loss was most predominant at promoters having the highest CHD4 occupancy levels (Fig. 6F, bottom). Lastly, the gene expression profiles of BRG1- and H3.3-depleted mESCs are positively correlated (Fig. 6G; Gehre et al. 2020), whereas the gene expression profiles of Chd4 KD mESCs positively correlate with that of H2A.Z-depleted ES cells, suggesting convergent responses on gene expression (Fig. 6G; Subramanian et al. 2013). Collectively, these findings argue for a model in which the antagonistic activity of the SWI/SNF and NuRD remodelers in modulating Pol II release kinetics is facilitated by the selective deposition of H3.3 and H2A.Z histone variants at TSS flanking nucleosomes.
Lesions in SWI/SNF confer paradoxical gene expression changes in a cancer setting
Finally, we wanted to address the extent to which the paradoxical changes in gene expression following perturbation of NuRD and SWI/SNF activity extended to a disease setting. To do so, we performed a pan-cancer gene expression–based regression analysis (see Methods) comparing patients harboring nonsynonymous and damaging mutations in SMARCA4 (encoding BRG1) or CHD4 to patients devoid of these mutations (Fig. 7A; for the number of patients in each group, see Supplemental Table S2). We find that lesions in SMARCA4 are associated with increased gene expression, which is more evident for highly expressed genes (Fig. 7B). Similarly, CHD4 mutations are associated with a tendency toward decreased gene expression levels; however, this is only evident for the most highly expressed genes. Collectively, these analyses suggest that perturbation of SWI/SNF activity drives paradoxical gene expression changes even in a cancer setting and, consequently, that these changes may be of functional importance in this context.
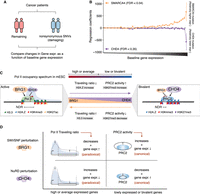
The impact of SMARCA4 and CHD4 lesions in cancer on gene expression. (A) Schematic illustration of the approach used to compare gene expression differences between patients having mutations in SMARCA4 or CHD4 to those of the remaining patients from 11 different cancer cohorts. (B) Mean regression coefficients for genes (N = 19,592 binned into 50 groups of 392 genes each and sorted by their expression levels) across 11 cancer cohorts along with standard error bars. Regression coefficients reflect a relatively higher (positive) or lower (negative) expression level of genes in patients having SMARCA4 or CHD4 mutations in comparison to those of the remaining patients. (C) A model explaining the antagonistic activities of SWI/SNF and NuRD chromatin remodelers at gene promoters during steady-state and MEF-to-mESC reprogramming. (D) Summary of the canonical and paradoxical impact of SWI/SNF and NuRD activities on gene expression following their perturbation.
Discussion
Here we have undertaken an integrative approach using more than 60 previously published and newly generated data sets to understand the functional consequences of the chromatin remodeling activities of NuRD and SWI/SNF at gene promoters. Positive changes in DNA accessibility at promoter regions are normally believed to be positively associated with gene expression; thus, SWI/SNF is generally considered a transcriptional activator and NuRD a repressor. However, functional ablation of key protein subunits of these complexes are associated with both canonical and paradoxical changes in gene expression in the context of their remodeling activities, which have remained unresolved (Yildirim et al. 2011; de Dieuleveult et al. 2016; Bornelov et al. 2018). Here we have shown that NuRD, indeed, opposes, whereas SWI/SNF supports an open chromatin state at target gene promoters. However, we found that the antagonistic nucleosome reorganization activities of the two remodelers are conferred by their distinct modes of action at RNA Pol II–bound active genes versus PRC2-bound repressed/bivalent genes. We explain this difference in the form of a unified model in which remodeler behavior is dependent on the position of the genes along the spectrum of Pol II occupancy at the promoters (Fig. 7C). Thus, a key tenet of the model is that the two remodelers have an opposing impact on Pol II buildup at promoters and, as a consequence, are in a constant tug-of-war in the context of Pol II distribution at genes.
In a steady-state situation, as in the context of proliferating mESCs, genes with a high SWI/SNF-to-NuRD ratio are characterized by high Pol II promoter occupancy (high Pol II occupancy at promoter vs. gene body), high promoter accessibility, and high mRNA production (absolute levels of Pol II increases, both at promoters and at gene bodies). The opposite is true for genes with a low SWI/SNF-to-NuRD ratio. Thus, in a steady-state situation, the SWI/SNF-to-NuRD ratio is reflected in the Pol II occupancy spectrum and the mRNA output of these genes, which is in line with the canonical impact of these two antagonistic chromatin remodelers (Fig. 7C). The same is true for SWI/SNF- and NuRD-mediated changes during differentiation/reprogramming, that is, processes involving major chromatin changes in order to facilitate the rewiring of the transcriptional landscape of the cell. Here, alterations in gene expression are also driven by the marked changes in DNA accessibility and Pol II occupancy levels. In contrast, the paradoxical changes in mRNA levels that we observed following perturbation of SWI/SNF or NuRD activities are centered on highly expressed genes, and we were able to show that these were owing to alterations in the delicate balance of Pol II states. Specifically, the relative decrease in the ratio of Pol II at the promoter versus the gene body that occurs following interference with SWI/SNF activity resulted in higher mRNA levels. Ablation of NuRD activity, on the other hand, increased the ratio of Pol II at the promoter versus the gene body and resulted in lower mRNA levels (Fig. 7D).
At the other end of the promoter Pol II occupancy spectrum, that is, at promoters devoid of or with low levels of Pol II, the impact of SWI/SNF and NuRD on PRC2 (eviction and recruitment, respectively) (Yildirim et al. 2011; Reynolds et al. 2012; Sparmann et al. 2013; Kadoch and Crabtree 2015; Stanton et al. 2017) drives the response to a shift in the balance between these modelers (Fig. 7C). At these canonical response promoters, an increase in SWI/SNF activity will reduce PRC2 levels and drive genes toward activation (i.e., increased transcription and increased mRNA levels) and vice versa for a shift toward increased NuRD activity. Perturbation in SWI/SNF activity at these promoters increases PRC2 activity, leading to reduced DNA accessibility and mRNA production, and vice versa upon perturbation of NuRD activity (Fig. 7D). In agreement, KO of either NuRD or PRC2 components results in differentiation defects in mESCs (Hu and Wade 2012). Thus, in this context, DNA accessibility correlates positively with the expression of genes with no or low Pol II promoter occupancy, as measured by mRNA production. In summary, these findings highlight the crucial role of the ratio between SWI/SNF and NuRD activities in fine-tuning gene expression as a function of Pol II promoter occupancy. Consistent with the functional importance of these observations, the loss of function of these complexes in mESCs is associated with self-renewal and differentiation defects (Kaji et al. 2006; Ho et al. 2009; Hu and Wade 2012).
Our data show that both remodelers regulate Pol II levels by modulating its release into the gene body, which is reflected in altered Pol II occupancy levels at gene promoters (Ehrensberger et al. 2013; Shao and Zeitlinger 2017; Core and Adelman 2019). Similar to our observation following reduction of SWI/SNF activity, loss of Mediator complex activity also leads to reduced Pol II levels at the gene promoters accompanied by an increase in the rate of Pol II released into the gene body (Jaeger et al. 2020). This may suggest a more general mechanism by which gene regulatory factors operate to regulate gene expression. Here we provide evidence for a model in which SWI/SNF and NuRD alter the energy barrier for commitment to productive transcription by modulating the use of specific histone variants. Specifically, we find that CHD4-bound promoters harbor H2A.Z-containing downstream nucleosomes, which have been reported to reduce the energy barrier for commitment to productive elongation (Santisteban et al. 2011; Weber et al. 2014). These data are also consistent with an earlier report showing that the conditional loss of CHD4 in mouse cerebellum leads to a decrease of H2A.Z occupancy at active promoters (Yang et al. 2016). In contrast, we provide evidence for a role of SWI/SNF in promoting the incorporation of H3.3 variant-containing nucleosomes. Consistent with the findings of our work, the H3.3 variant was recently shown to act as a cofactor of EP300, which is known to promote Pol II binding to promoters (Ray-Gallet et al. 2011; Boija et al. 2017; Martire et al. 2019), and its loss results in reduced BRG1 occupancy at gene promoters (Gehre et al. 2020).
Here, we studied the activity of SWI/SNF and NuRD at gene promoters. However, we note that both SWI/SNF and NuRD also bind at enhancers to regulate gene expression. Therefore, some of the observed activities of the two remodelers at promoters might be influenced by their activity at distal regulatory elements, and future studies along these lines are warranted. Collectively, our findings deliver a unified model for how two antagonistic chromatin remodelers collaborate to fine-tune gene expression across the entire gene expression spectrum.
Methods
BRG1 inhibition
mESCs were treated with 10 μM BRM/BRG1 ATP inhibitor-1, BRM014 (MedChemExpress HY-119374), or an equivalent volume of dimethyl sulfoxide (DMSO) as control for 3 h and directly fixed and subjected to ChIP.
shRNA construction
To generate Chd4 and scrambled shRNA constructs, PLKO.1 plasmid forward and reverse oligos were annealed to generate double-stranded Chd4 and scrambled shRNAs inserts containing AgeI- and EcoRI-compatible overhangs. The inserts were subsequently cloned in PLKO.1 linearized with AgeI and EcoRI:
Chd4 shRNA forward oligo,
5′-CCGGGCTCGAAGATTCAAGCTCTTACTCGAGTAAGAGCTTGAATCTTCGAGCTTTTTG-3′;
Chd4 shRNA reverse oligo,
5′-AATTCAAAAAGCTCGAAGATTCAAGCTCTTACTCGAGTAAGAGCTTGAATCTTCGAGC-3′;
Scramble shRNA forward oligo,
5′-CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTTG-3′; and
Scramble shRNA reverse oligo,
5′-ATTCAAAAACAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTG-3′.
Chromatin immunoprecipitation
ChIP with reference exogenous genome (ChIP-Rx) was performed according to standard protocols. Briefly, proteins and chromatin were cross-linked by the addition of 1% formaldehyde (Thermo Fisher Scientific) for 10 min and subsequently quenched by the addition of 125 mM glycine and three washes with PBS. Cells were lysed in ChIP SDS lysis buffer (50 mM Tris at pH 8.0, 100 mM NaCl, 5 mM EDTA at pH 8.0, 0.5% [w/v] SDS, 50 mM NaF, 20 mM β-glycerophosphate, and protease inhibitor cocktail tablet [Roche]), and chromatin was pelleted by centrifugation at 22,000g for 15 min and resuspended in immunoprecipitation (IP) buffer (a 2:1 mix 2 of ChIP SDS lysis buffer and ChIP dilution buffer containing 100 mM Tris at pH 8.0, 5 mM EDTA at pH 8.0, and 5% Igepal CA630). Chromatin was sheared to 200–500 bp by sonication using either a Bioruptor (Diagenode) or a Covaris (E220) system. To assess Pol II phosphorylation, 400–500 μg fragmented chromatin in IP buffer was immunoprecipitated using either 5 µg of anti-RNA Pol II CTD repeat YSPTSPS (ser5p) antibody (Abcam ab5131) or 5 µg of anti-RNA Pol II CTD repeat YSPTSPS (ser2p) antibody (ab238146). To assess histone variants, 100 μg fragmented chromatin in IP buffer was immunoprecipitated using either 5 µg of antihistone H2A.Z antibody (ab4174, Abcam) or recombinant antihistone H3.3 antibody (Abcam ab176840). Each ChIP was spiked (2%) with sonicated Drosophila chromatin (from S2 cells). For H3.3 and H2A.Z ChIP, 1 μL of antibody against Drosophila histone H2Av (Active Motif 61751) was added as well. Protein G–Sepharose beads (GE Healthcare GE17-0618) were used to pull down antibody-bound chromatin and washed three times with 150 mM NaCl ChIP washing buffer (20 mM Tris at pH 8.0, 150 mM NaCl, 2 mM EDTA at pH 8.0, 0.1% SDS, and 1% Igepal CA630), one time with RIPA ChIP buffer (50 mM Tris at pH 8.0, 150 mM NaCl, 1 mM EDTA at pH 8.0, 0.1% SDS, 1% Igepal CA630, and 0.5% Na deoxycholate), one time with 150 mM NaCl ChIP washing buffer, one time with 500 mM NaCl ChIP washing buffer (20 mM Tris at pH 8.0, 500 mM NaCl, 2 mM EDTA at pH 8.0, 0.1% SDS, and 1% Igepal CA630), and, finally, one time with 150 mM NaCl ChIP washing buffer. Samples were subsequently de-cross-linked by incubating overnight at 65°C in de-cross-linking buffer (1% SDS and 0.1 M NaHCO3). Purified DNA from ChIP-Rx was subjected to library preparation using the NEBNext ultra kit (E7645) according to the manufacturer's instructions and sequenced on the NextSeq 500.
4sU Nascent RNA-seq
Nascent RNA-seq using 4sU was performed as described previously (Rabani et al. 2011; Siersbæk et al. 2014) with minor modifications. In short, mESCs were seeded in 2i medium in 150-mm dishes at 37°C and 5% CO2. Two days later, 500 μM 4-thiouridine (4sU, Sigma-Aldrich T4509) was added. Following 5-min incubation at 37°C, the medium was removed, and the labeling process was quenched by addition of TRIzol (Invitrogen 15596018) to mESCs. Spike-in RNAs (2.4 ng per million cells) were added, and samples were subjected to chloroform extraction and NaCl/isopropanol precipitation/ethanol washing before being resuspended in in DEPC-treated H2O. Two hundred fifty micrograms of extracted RNA was incubated with 500 μg EZ-Link HPDP-biotin (1 μg/μL in 100% dimethylformamide, Thermo Fisher Scientific 21341) to tag 4sU-labeled nascent RNA with biotin in biotinylating buffer (10 mM Tris at pH 7.5, 1 mM EDTA at pH 8.0) with rotation for 2 h at room temperature. RNA was cleaned up by chloroform:isoamyl alcohol extraction and precipitated with NaCl and isopropanol followed by a wash with ethanol and resuspension in DEPC-treated H2O. Next, to isolate 4sU-labled nascent RNA from total RNA, samples were incubated with 100 μL Dynabeads MyOne streptavidin T1 (Invitrogen 65601), washed twice with solution containing 100 mM NaOH and 50 mM NaCl and twice with 100 mM NaCl before use, and incubated for 15 min in streptavidin T1 in binding and washing buffer (5 mM Tris at pH 7.5, 0.5 mM EDTA at pH 8.0, 1 M NaCl, 0.1% Tween 20) while rotating at room temperature. After washing four times with binding and washing buffer, 4sU-labeled nascent RNA was released from biotin-streptavidin beads by elution in 100 mM DTT (Promega V3151) followed by purification with a NucleoSpin RNA kit (Macherey-Nagel 740955.250) according to the manufacturer's instructions.
Nascent RNA libraries were prepared using a NEBNext Ultra II directional RNA preparation kit (New England Biolabs E7760S) following the manufacturer's instructions. Briefly, nascent RNA was fragmentated into an average of 200 nt and reverse-transcribed into stranded DNA using random primers, which was followed by addition of sequencing bar-code during PCR amplification. Libraries were paired-end sequenced (40 cycles for each end) on a NextSeq 500 using an Illumina SE75Hi sequencing kit.
Spike-in RNA preparation
Six synthetic RNA spike-in controls (three unlabeled and three 4sU-labeled spike-in RNAs) for validation of labeled RNA enrichment and 4sU nascent RNA-seq global normalization in mESCs are derived from selected RNAs of ERCC RNA spike-in mix (Thermo Fisher Scientific 4456740) (Schwalb et al. 2016). In short, first-strand cDNA was synthesized from ERCC RNAs using a ProtoScript first-strand cDNA synthesis kit (New England Biolabs E6300) with anchored oligo followed by individual amplification with specific primer pairs for the six selected ERCC RNAs (ERCC ID: 00043, 00170, 00136, 00145, 00092, and 00002) using HotStarTaq DNA polymerase (Qiagen 203203) according to the manufacturer's instructions. The six PCR products were purified with QIAquick PCR purification kit (Qiagen 28106) and validated by Sanger sequencing. Six RNA spike-in controls were synthesized with the six ERCC PCR products as templates, respectively, using MEGAscript T7 kit (Invitrogen AM1334) following the manufacturer's instructions, except for the three 4-Thio-UTP-labeled RNA spike-in controls (ERCC ID: 00043, 00136, and 00092), in which 10% of UTP was substituted by 4-Thio-UTP in the RNA synthesis reaction. All synthetic RNAs were cleaned up by chloroform:isoamyl alcohol extraction and precipitated using NaCl and isopropanol followed by wash with ethanol and resuspension in DEPC-treated H2O. Equimolar amounts of the six synthetic RNA spike-in controls (three unlabeled RNAs and three 4sU-labeled RNAs) were mixed to make the final RNA spike-in pool for 4sU nascent RNA normalization in mESCs.
4sU Nascent RNA-seq data analysis
FASTQ reads were trimmed for adapter sequences using Trim Galore! v0.6.4 (Martin 2011). Trimmed reads were mapped to mm10 genome assembly and ERCC spike-in sequences using STAR v2.7.1a (default parameters) (Dobin et al. 2013). Number of reads mapping to Ensembl-defined gene annotations (release 97, mm10) (Cunningham et al. 2022) were determined using featureCounts (allowing multiassignment of reads, and reads mapping to multiple positions are counted as the fraction of number of mapped positions) (Liao et al. 2014). Raw read counts were ERCC spike-in-normalized using EDASeq v2.24.0 (Risso et al. 2011) and RUVSeq v1.24.0 (Risso et al. 2014) and is similar to the analysis performed by Bacon et al. (2020). Specifically, we used RUVSeq to remove unwanted variation in the RNA-seq data by performing factor analysis on ERCC spike-in controls to derive weight factors (using RUVg function), which are then used to normalize the raw read counts for technical variation. Differential gene expression was then performed using edgeR v3.32.1 (Robinson et al. 2010).
Promoter classes in mESCs and MEFs
Gene classes defined based on their activity levels in mESCs and MEFs
We measured Pol II occupancy levels at all active and bivalent promoters in mESCs (N = 9345) and determined the Pol II occupancy thresholds. Next, active genes were grouped based on their promoter Pol II levels as low (first quantile; N = 779), average (second and third quantile; N = 4093), and high (fourth quantile; N = 2294). Similarly, we quantified Pol II occupancy levels at all active and bivalent genes in MEFs (N = 10,351), and active genes were stratified accordingly into low (first quantile; N = 1657), average (second and third quantile; N = 4752), and high Pol II occupancy genes (fourth quantile; N = 2482).
Gene classes defined based on their promoter width in mESCs
We measured the distances between TSS upstream (−1) and downstream (+1) canonical nucleosomes as defined by the width of H3K4me3 peak at promoter (de Dieuleveult et al. 2016) to measure promoter width (Supplemental Fig. S1C). Active gene promoters were subclassified based on their width as narrow, ≤500 bp; medium, 500–1000 bp; and broad, >1000 bp.
Gene classes defined based on changes in the relative occupancy of CHD4 and BRG1 at their promoters during MEF-to-mESC reprogramming
Out of 18,394 genes analyzed in MEFs, we selected 3459 genes for which expression increases or decreases by at least twofold
during reprogramming to mESCs. Next, relevant ChIP-seq signals (CHD4, BRG1, H3K27me3, EZH2, H3.3, H2A.Z, Pol II, Pol II–ser5p,
Pol II–ser2p) at the promoters of these genes, along with their expression in mESCs and MEFs, were quantile normalized using
the normalize.quantiles function in R (R Core Team 2019). Normalized CHD4 and BRG1 occupancy levels at 3459 gene promoters in mESCs and MEFs were used to compare changes in their
occupancy levels during MEF-to-mESC reprogramming (Supplemental Fig. S4C):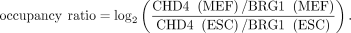
Genes were rank-ordered based on their occupancy ratio, and the top 376 genes in which CHD4 occupancy increases whereas BRG1 occupancy decreases during reprogramming are designated as CHD4-driven. Conversely, the bottom 380 genes in which CHD4 occupancy decreases whereas BRG1 occupancy increases during reprogramming are designated as BRG1-driven. The remaining 2703 genes are designated as a neutral group (Supplemental Fig. S4C).
Quantification and statistical analysis
To determine the significance of all comparisons, the Wilcoxon rank-sum test was used to calculate P-values. All statistical analyses were performed in the R programming environment (R Core Team 2019). Log without any base corresponds to the natural log.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE206497. The source of all the publicly available data analyzed in this study has been listed in Supplemental Table S1. The code to generate figures is available as Supplemental Code, as well as at GitHub (https://github.com/porseLab/Chd4_Brg1).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Jesper Svejstrup, Chirag Nepal, Adrija Kalvisa, Faizaan Mohammad, and Kristian Helin for critical comments on this manuscript and Joachim Weischenfeldt for suggestions regarding patient data analysis. This work was supported through a grant from the Novo Nordisk Foundation (Novo Nordisk Foundation Center for Stem Cell Biology, DanStem; grant number NNF17CC0027852).
Author contributions: S.P. conceived the project and analyzed the data with insights from B.T.P. in the interpretation and presentation of the results. J.S. and K.H. performed ChIP-seq experiments in mESCs with assistance from M.T., A.M.H., and J.S.H. J.S. performed 4sU nascent RNA-seq experiments with input from M.T. B.T.P. supervised the data analysis and ChIP-seq experiments during the course of the project. S.P. and B.T.P. wrote the manuscript with input from all coauthors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277089.122.
-
Freely available online through the Genome Research Open Access option.
- Received July 2, 2022.
- Accepted January 20, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








