
The pig pangenome provides insights into the roles of coding structural variations in genetic diversity and adaptation
- Zhengcao Li1,
- Xiaohong Liu1,
- Chen Wang1,
- Zhenyang Li1,
- Bo Jiang1,
- Ruifeng Zhang1,
- Lu Tong1,
- Youping Qu1,
- Sheng He1,
- Haifan Chen1,
- Yafei Mao2,
- Qingnan Li1,
- Torsten Pook3,
- Yu Wu1,
- Yanjun Zan4,
- Hui Zhang1,
- Lu Li1,
- Keying Wen1 and
- Yaosheng Chen1
- 1State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-Sen University, 510006 Guangzhou, China;
- 2Bio-X Institutes, Shanghai Jiao Tong University, 200240 Shanghai, China;
- 3Animal Breeding and Genomics, Wageningen University & Research, Wageningen 6700 AH, The Netherlands;
- 4Key Laboratory of Tobacco Improvement and Biotechnology, Tobacco Research Institute, Chinese Academy of Agricultural Sciences, Qingdao 266000, China
Abstract
Structural variations have emerged as an important driving force for genome evolution and phenotypic variation in various organisms, yet their contributions to genetic diversity and adaptation in domesticated animals remain largely unknown. Here we constructed a pangenome based on 250 sequenced individuals from 32 pig breeds in Eurasia and systematically characterized coding sequence presence/absence variations (PAVs) within pigs. We identified 308.3-Mb nonreference sequences and 3438 novel genes absent from the current reference genome. Gene PAV analysis showed that 16.8% of the genes in the pangene catalog undergo PAV. A number of newly identified dispensable genes showed close associations with adaptation. For instance, several novel swine leukocyte antigen (SLA) genes discovered in nonreference sequences potentially participate in immune responses to productive and respiratory syndrome virus (PRRSV) infection. We delineated previously unidentified features of the pig mobilome that contained 490,480 transposable element insertion polymorphisms (TIPs) resulting from recent mobilization of 970 TE families, and investigated their population dynamics along with influences on population differentiation and gene expression. In addition, several candidate adaptive TE insertions were detected to be co-opted into genes responsible for responses to hypoxia, skeletal development, regulation of heart contraction, and neuronal cell development, likely contributing to local adaptation of Tibetan wild boars. These findings enhance our understanding on hidden layers of the genetic diversity in pigs and provide novel insights into the role of SVs in the evolutionary adaptation of mammals.
Structural variants (SVs) are derived from deletion, insertion, duplication, inversion, and translocation of genome segments >50 bp (Ho et al. 2020). A vast majority of the mutations normally segregate at low frequencies and are depleted from functional regions of the genome, owing to intense purifying selection (Weischenfeldt et al. 2013; Chakraborty et al. 2019; Collins et al. 2020). Despite the general conclusion, structural variations occurring on coding sequences, here called “coding structural variations,” such as gene presence/absence variations (PAVs) and PAVs of transposable elements (TEs) that contain open reading frames (ORFs) (Joly-Lopez and Bureau 2018), have been shown to be important determinants of genome evolution and phenotypic variability in a range of species (Gao et al. 2019; Niu et al. 2019). We have reported that pangenomic coding sequence PAVs explain a substantial proportion of the “missing heritability,” and using such SV information in genomic prediction dramatically improves prediction accuracies of multiple complex traits in the model organism Saccharomyces cerevisiae (Li and Simianer 2020). The pangenome, originally representing the complete genic content of a population, was first proposed in bacteria (Tettelin et al. 2005) and subsequently unlocked in higher organisms including plants (Tang et al. 2022), animals (Crysnanto et al. 2021; Wang et al. 2021a), and humans (Li et al. 2010; Duan et al. 2019), with myriad dispensable genes responsible for agronomic traits and human diseases uncovered (Yu et al. 2022). In contrast to microorganisms, mammals typically have larger genome size and less genome flexibility, where genes in the usual sense constitute a minor fraction of total sequences and are dominated by long introns (Francis and Wörheide 2017). Thus, merely targeting gene PAVs in a mammalian pangenome analysis may obtain limited coding SV information (Sherman and Salzberg 2020). Incorporating both genic and nongenic coding SVs, such as SVs resulting from recently or currently active TE coding sequences (Morgante et al. 2007; Joly-Lopez and Bureau 2018), into a broad sense pangenome presents the opportunity of capturing a wide range of coding SVs not only for mammals but also for other eukaryotes with large genomes. TEs, also known as “jumping genes” (Ravindran 2012), account for approximately half of mammalian genomes, most of which are genome fossils that have lost the capability of transposing (Chuong et al. 2017). Only a minority of TEs still retain activity and are capable of transcription and mobilization, ongoing to generate inter-individual genome variations in a form of TE insertion polymorphisms (TIPs) (Huang et al. 2012; Fueyo et al. 2022). TE coding sequences give rise to alterations in host gene expression (Babarinde et al. 2021) by encoding transposases and transcription factors, etc. (Joly-Lopez and Bureau 2018). A plethora of instances have shown that TE insertions with regulatory functions are preserved by selection, acting as potent agents of adaptation (Schlenke and Begun 2004; Rech et al. 2022).
The pig (Sus scrofa), one of the most widespread mammals globally, is a major source of animal protein and a valuable biomedical model organism for humans (Scherf et al. 1995; Groenen et al. 2012). China and Europe stand out as two prominent centers for pig breed diversity, encompassing more than two-thirds of global pig breeds, the formation of which can be attributed to a combination of natural and artificial selection, following the independent domestication of Asian and European wild boars ∼10,000 yr ago (Giuffra et al. 2000; Larson et al. 2005; Ai et al. 2015). During the late eighteenth and early nineteenth centuries, Chinese indigenous pig breeds were introduced to Europe to enhance the productivity of local pigs, contributing to an expanded gene pool for European breeds (Bosse et al. 2015). Hitherto a few of the resulting European hybrid commercial pig breeds have held a dominant position in the global hog market (Megens et al. 2008; White 2011). In contrast to their domesticated counterparts, wild boars play a vital role as genetic resources for understanding evolution (Frantz et al. 2016); for example, Tibetan wild boars are endemic to the Qinghai-Tibet Plateau and have adapted to numerous harsh environments including hypoxic conditions and cold temperatures, which makes them an ideal model for investigating the genetic foundations of local adaptation (Li et al. 2013). For the sake of characterizing the genetic diversity of the genomic resources, initial pangenome efforts in pigs centered on retrieving sequences and genes missing from the current reference sequence S. scrofa 11.1 (Li et al. 2017; Tian et al. 2020; Warr et al. 2020), whereas these studies are based just on a small number of genome assemblies, incapable of comprehensively ascertaining the catalog of coding sequence PAVs for the animal. Therefore, (1) the biological function of most dispensable genes, (2) to what extent gene PAVs affect genome variability, and (3) the properties of TIPs, such as their population dynamics and contributions to population differentiation and gene expression, remain unclear. Furthermore, much of what is known about the molecular mechanisms of adaptation of pigs comes from research focusing on single-nucleotide variations (SNVs) (Li et al. 2013; Wilkinson et al. 2013; Frantz et al. 2015). Previous work has sporadically documented the biological consequences of TE insertions in the species (Giuffra et al. 2002); for instance, a co-opted DNA transposon encodes ZBED6 transposase, which is a transcriptional repressor with important influences on muscle growth regulation (Volff 2010). However, their potential role in genetic flexibility and environmental adaptation is still poorly understood.
Here we construct a pig pangenome based on 250 sequenced individuals from 32 phenotypically divergent pig breeds in Eurasia. Our objective is the systematic interrogation of coding sequence PAVs by integrating gene PAVs and TIPs into a pangenome framework, and exploring their impacts on genome variability, gene expression, and adaptation at a large population scale. Our findings provide novel perspectives into the previously undisclosed genetic diversity and biological underpinnings of adaptive evolution and create valuable resources that will facilitate future genetic improvements in this species.
Results
Genome assembly and gene annotation
We sampled 160 Chinese local pigs including 114 individuals from 23 phenotypically divergent breeds and 46 Tibetan wild boars to represent the gene pool of Chinese pigs. Additionally, 90 individuals from six European commercial breeds and two European commercial crossbreeds were included, representing the gene pool of major international commercial breeds (Fig. 1A; Supplemental Tables S1–S3). All individuals were sequenced with an average sequence depth of ∼35×, obtaining a total of 20.6 Tb of raw sequence reads. Each of the 250 genomes was de novo assembled, resulting in final assemblies with an average contig N50 value of 34.2 kb and an average assembled genome size of 2.42 Gb. The gene- and sequence-based pangenome was constructed with a “map to pan” strategy (Supplemental Figs. S1, S2; Supplemental Table S4; Wang et al. 2018). After mapping all assembled contigs to the reference genome S. scrofa 11.1 as well as a clustering step filtering out redundant and putative contaminating sequences (Supplemental Table S5), 308.3 Mb nonredundant, nonreference sequences >500 bp were retained (Warr et al. 2020). Combining these novel sequences with the reference genome provided a sequence-based pangenome of 2.8 Gb, comprising 3,107,619 SNPs, 636,812 indels, and 3438 newly identified genes in the nonreference sequences (Supplemental Tables S6, S7). In addition, there were 1051 nonreference genes exclusively found in European pigs, in contrast to the lower 766 in Chinese pigs (Fig. 1B).
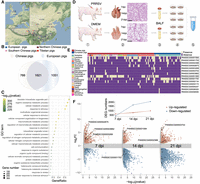
Novel genes identified in the nonreference sequences including 14 genes in the MHC. (A) Geographic distributions of pig breeds used for the construction of pig pangenome. Northern Chinese pigs (dark red) are as follows: Hetaodaer (HTDE), Min (MIN), Laiwu black (LW), and Erhualian (EHL). Southern Chinese pigs (pink) are as follows: Wuzhishan (WZS), luchuan (LC), bamaxiang (BMX), Baoshan (BS), Debao (DB), Diannan small-ear (DNS), Gaoligongshan (GLGS), Guangdong small-ear spotted (GDS), Guanzhuang spotted (GZ), Guizhong spotted (GZS), Huai (HUAI), Lantang (LT), large black-white (LBW), Longlin (LONG), Minbei spotted (MBS), Putian (PT), Saba (SB), Tunchang (TCH), and Yuedong black (YDB). European pigs are as follows: Duroc (DR), Yorkshire (YS), landrace (LR), Pietrain (PTR), Hampshire (HSH), Berkshire (BSH), Landrace × Yorkshire (LRYS), and Duroc × (Landrace × Yorkshire) (DLY). (B) Venn diagram displaying the distribution of 3438 genes from the pangenome not found in the reference genome, including genes shared among or unique to either the Chinese or European breeds. (C) Enriched GO terms of nonreference genes. (D) The pipeline of the PRRSV challenge trial: (1) Piglets from the PRRSV-infected group and noninfected group were injected intramuscularly with PRRSV and 2% DMEM, respectively; (2) lungs of piglets were collected at 7, 14, and 21 days post infection (dpi) and tissue slices examined microscopically by hematoxylin-eosin staining; (3) extraction of bronchoalveolar lavage fluid (BALF); (4) collection of porcine alveolar macrophages (PAMs); and (5) RNA extraction and sequencing. (E) Fourteen nonreference SLA genes suffered from PAV among 103 individuals from 22 pig breeds. The seven nonreference SLA genes in the gray box were differentially expressed after PRRSV infection. (F) Volcano plots show DEGs under PRRSV infection at three time points (7, 14, and 21 dpi). The black circles represent the 60 DEGs identified in the nonreference sequences. Orange and blue points denote the up-regulated and down-regulated DEGs, respectively. The line chart describes that the number of DEGs increases as dpi increase.
Novel nonreference SLA genes are involved in immune responses to PRRSV infection
Nonreference genes identified were significantly enriched in the cellular response to chemical stimulus, response to stress, and macromolecule metabolic process (Fig. 1C; Supplemental Table S8), suggesting that they are likely contributors to the phenotype diversity and environmental adaptations. Functional annotation based on domains and orthologous groups of proteins from the Pfam and eggNOG database (Huerta-Cepas et al. 2016; Mistry et al. 2021) revealed that 14 nonreference genes are the major histocompatibility complex (MHC), also termed swine leukocyte antigen (SLA), class I or class II genes, with varying presence frequencies among 103 individuals from 22 pig breeds, and the genomic position predicted for the novel genes is consistent with the functional annotation (Fig. 1E). SLA class I presents peptides to CD8+ cytotoxic T cells, whereas SLA class II presents exogenous peptides to CD4+ helper T cells for immune recognition, both of which are pivotal in responses to pathogen infection and vaccines. We previously showed that the transcript abundance of SLA II genes in the pig reference genome was markedly increased after infection with productive and respiratory syndrome virus (PRRSV) (Xiao et al. 2010). To validate that the nonreference SLA genes are also functional in immune responses, pairwise comparisons between six PRRSV-infected and six noninfected Yorkshire pigs were performed at three time points (Fig. 1D). In total, 4319 differentially expressed genes (DEGs) were determined in at least one of the pairwise comparisons (Supplemental Tables S9–S11). The number of DEGs increased as days post infection (dpi) increased, indicative of the reliability of the challenge trial (Fig. 1F). We observed that 60 nonreference genes were differentially expressed, with 34 down-regulated and 31 up-regulated. Among them, 18 out of the 60 were supported by immune-related proteins or domains from the Pfam and eggNOG database, providing additional evidence for their immune functions. Of note, seven nonreference SLA genes were differentially expressed after PRRSV infection, suggesting their functional involvement in immune responses (Supplemental Table S12; Mähler et al. 2017). SLAs are xenoantigens when pigs are used as the transplant source for clinical xenotransplantation (Ladowski et al. 2018, 2019). We discovered that two of the seven nonreference SLA genes were present in Wuzhishan miniature pigs, which are a preferred preclinical model for xenotransplant research. Because of the high amino acid sequence identities and 3D structural similarities between human leukocyte antigens (HLAs) and SLAs, HLA-specific antibodies can cross-react with SLAs, leading to xenograft rejection. We aligned the 14 nonreference SLA genes to all HLA sequences in the IPD-IMGT/HLA sequence database (Barker et al. 2023) and observed that the homologous similarities between them range from 75% to 92%. When aligning these SLA genes to all SLA sequences in the IPD-MHC sequence database (Maccari et al. 2017), we discovered that nine of them are missing from the database, representing newly discovered SLA genes for future clinical validation (Supplemental Table S13).
Characterization of gene PAVs
All 24,718 genes were categorized according to their presence frequencies. Among these, 20,583 genes (83.2%) were classified as core genes, which are considered essential and are shared by all individuals. Additionally, there were 4135 dispensable genes, including 363 softcore genes, 2534 shell genes, and 1238 cloud genes present in >98%, between 98% and 1%, and <1% of all individuals, respectively. (Fig. 2A,B). Amid the dispensable genes, 98.8% of them contained InterPro domains, which was slightly higher than that of core genes (95.8%). We investigated evolutionary pressures on dispensable genes by calculating the number of nonsynonymous and synonymous SNPs in each individual (Fig. 2C) and observed that dispensable genes are less subject to stabilizing selection than core genes. Softcore genes showed a higher average ratio of nonsynonymous SNPs to synonymous SNPs, compared with core, shell, and cloud genes, indicating that they are subject to stronger selective pressures and are less functionally conserved.
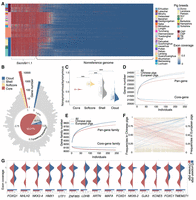
Characterization of gene PAVs. (A) The heatmap illustrates the exon coverage of dispensable genes identified in the reference genome S. scrofa 11.1 and nonreference sequences. The colors listed on the left side of the heatmaps represent different pig breeds. (B) The circle histogram depicts the distribution of gene counts with different presence frequencies in the pig population. The pie chart shows the proportion of core, softcore, shell, and cloud genes in the pig genic pangenome. (C) The ratio of nonsynonymous SNPs to synonymous SNPs in core, softcore, shell, and cloud genes. Significance values were calculated: (***) P < 1 × 10−6, Wilcoxon test. (D) Saturation curve modeling the genic pangenome and core-genome size in Chinese pigs, European pigs, and 250 individuals. The solid curve denotes fitting to the maximum gene number of sampled individuals, and the dashed curve depicts the extrapolation of the fitting. (E) Modeling of gene families in the pangenome and core genome. (F) The left y-axis and the right y-axis are occurrence frequencies of dispensable genes in Chinese pigs and European pigs, respectively. Pink and blue lines denote the genes that are favorable for Chinese pigs and European pigs, respectively. Light brown lines denote the genes with a change of presence frequency that is less than 0.4 between the two populations. (G) Comparison of probability distribution for exon coverage among populations of Chinese and European pigs for 15 genes having higher presence frequency in European pigs. The blue and red bars denote the presence frequencies of the dispensable genes in Chinese pigs and European pigs, respectively.
The pangene space in pigs has not been saturated within 250 individuals (Fig. 2D), which was rarely observed in plant pangenome analyses (Gao et al. 2019; Sun et al. 2020). The number of both pan- and core-genes for European commercial pigs was substantially larger than that for Chinese domestic pigs, even though distinctly fewer European breeds were sampled. Ontology-based classification identified 8869 gene families. If an individual possessed at least one gene from a particular gene family, we considered that gene family to be present in that individual. In total, we obtained 8049 core gene families and 820 dispensable gene families. The number of pangene families for European commercial pigs was higher than that for Chinese domestic pigs, and the gene families identified in European commercial pigs encompassed nearly all genes present within the 250 individuals (Fig. 2E). Based on the historical evidence that the increased genetic diversity of European commercial pigs was mainly owing to the introgression of genetic material from Chinese breeds ∼200 yr ago (Bosse et al. 2014b), and some studies quantified that ∼30% of the genomes for European commercial pigs originated from Chinese breeds (Groenen et al. 2012; Bosse et al. 2014a), the aforementioned observations suggest that the hybridization-driven increment in genetic diversity could be a contributing factor to the augmentation of the gene repertoire within the population.
Despite the recent admixture, enduring natural and artificial selection have resulted in profound phenotype differentiation between Chinese and European modern pigs. Some dispensable genes with phenotypic effects may have been gained or lost during this process. To identify favorable genes that may be selected in each population, we performed a comparison between Chinese pigs and European pigs according to the presence frequency of dispensable genes. We defined that the gene with a significant change of presence frequency that is greater than 0.4 between the two populations was regarded as positively selected. We identified 109 and 38 positively selected genes for Chinese and European pigs, respectively (Fig. 2F), with the former enriched for regulation of macromolecule metabolic process and the latter for regulation of transcription factor activity and peripheral nervous system development, etc. Among them, only 15 favorable genes in European pigs have known functions annotated in the reference genome S. scrofa 11.1 (Fig. 2G). Two genes, NHLH2 and MAFA, were present in all European pigs but absent from ∼30% of Chinese pigs. NHLH2 was found to be associated with a failure to attain puberty in pigs and delayed pubertal development and age at first estrus in mice (Ranawade et al. 2013; Nonneman et al. 2014). The overexpression of MAFA was shown to influence the differentiation and proliferation of pancreatic stem cells (You et al. 2011). HMX1 was a favorable gene in European pigs, which was previously found to be a candidate gene associated with ovulation rate in pigs (Campbell et al. 2003).
Pig mobilome
Active TEs as potent insertional mutagens propagate in the genome, being one of the most important causes of structural variations. We profiled the pig mobilome, the atlas of recently or currently active TEs in pigs, by investigating TIPs that are presumed to reflect insertions occurring after divergence from a common ancestor (Huang et al. 2012). Overall, 490,480 TIPs were identified, of which 99,037 were missing in the reference genome (Fig. 3A; Supplemental Tables S14, S15). The TIPs were composed of 458,683 retrotransposons (106,547 LTRs, 294,058 LINEs, and 58,078 SINEs) and 31,797 DNA transposons and were contributed by 970 TE families that may retain the ability to transpose, with 284 newly annotated TE families being absent from current Repbase/Dfam database (Fig. 3B,C; Wheeler et al. 2013). A majority of TIPs showed very low frequency. Only 5.9% of TIPs were present in >95% of all individuals, most of which were LINE and LTR, indicating that they inserted before the divergence of subpopulations from their progenitor. In contrast, few SINE insertions had >95% frequency, which suggested that they occurred much later compared with other TE types (Fig. 3D,E). The number of potentially active TE families plateaued with the inclusion of merely around 100 pig genomes, and the TE family types identified for Chinese pigs are almost equivalent to those for European pigs, according to a bootstrap resampling of genomes (Fig. 3F). However, at the individual level, we found that the former contained significantly more TE families with TIPs than the latter did (904.01 ± 34.6 vs. 845.6 ± 28.9, t-test, P = 1.06 × 10−8) (Fig. 3G), suggesting that there exists a disparity in the activity levels of certain TE families between Chinese pigs and European pigs. Genomes with a minimum coverage of 30× were used in the comparison to minimize biases that might result from variable depth of coverage. However, the inherent limitations of short reads, particularly in the identification of long repetitive sequences such as LINEs, should not be disregarded. Principal component analyses (PCAs) using all TIPs shared the same pattern with PCAs only using LINE-1 insertions, suggestive of the dominant role of active LINE-1 in the pig mobilome (Supplemental Fig. S6). tRNAGlu-derived SINEs are also referred to as porcine repetitive elements (PREs), the expansion of which was presumed to occur exclusively in the porcine lineage during the first half of the Tertiary period (Groenen et al. 2012). We discovered that PRE insertions accounted for 63.3% of the total SINE insertion polymorphisms. PRE insertion polymorphism–based PCA revealed that Chinese pigs and European pigs were well separated (Fig. 3H), suggesting PRE with TIPs significantly contribute to population differentiation of pigs. PCA based on non-PRE-SINE insertion polymorphisms or other types of TIPs can also distinguish between the two populations to some extent, but with partial overlaps (Supplemental Fig. S3).
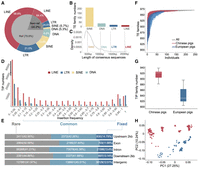
The composition of pig mobilome. (A) Percentage of four major TIP types (DNA transposons, SINE, LINE, and LTR) in the reference (Ref) and nonreference (Non-ref) genome. (B) The number of newly identified TE families in four major TE types. (C) The distribution of the length of newly identified TE families. The vertical dotted lines denote the average length of consensus sequences of four major TE types in Repbase. (D) Frequency distribution of four types of TIPs—LINE, SINE, LTR, and DNA—in the 250 genomes. (E) Proportions of rare, common, and fixed TIPs in intergenic and intragenic regions. TIPs observed in <5% of animals are designated as “rare,” between 5% and 95% as “common,” and >95% as “fixed.” (F) The cumulative number of TE families identified with increasing numbers of individuals by iterative resampling of individuals. Red, blue, and black represent Chinese pigs, European pigs, and all 250 individuals, respectively. (G) Comparison of the number of TE families with TIPs in Chinese and European genomes. Only genomes with sequencing coverage >30× were included (904.01 ± 34.6 in Chinese and 845.6 ± 28.9 in European genomes, respectively). (H) Principal component analysis based on PRE with TIPs. Chinese pigs and European pigs were well separated. Colors represent the populations as indicated in F.
Uneven distribution of TIPs across the pig genome
Various types of TIPs were disseminated widely but not at random throughout the pig genome (Fig. 4A,B). To quantify the relationship of TIP density (TIP count within nonoverlapping 2-Mb windows along the genome) with two established factors shaping their distribution in eukaryotic genomes, recombination rate and gene density (the proportion of gene sequences within nonoverlapping 2-Mb windows along the genome), we calculated the pairwise correlations between TIP densities among various TE types, as well as their correlations with both parameters (Fig. 4B). There was a positive correlation between LTR and LINE insertion densities (Spearman's ρ = 0.34), and both LTR and LINE insertion densities showed negative correlations with gene densities and recombination rates, suggesting that strong purifying selection acts against them. This probably resulted from two scenarios: (1) selection acting against deleterious effects caused by TIPs within or nearby genes and (2) selection acting against chromosomic rearrangements caused by ectopic recombination (Rizzon et al. 2002; Dolgin and Charlesworth 2008). In contrast, SINE insertion densities were negatively correlated with both LINE (ρ = −0.58) and LTR insertion densities (ρ = −0.22) across the entire genome. SINE with TIPs tended to enrich in genes, especially their flanking regulatory regions (2 kb upstream of or downstream from genes) (Fig. 4C; Supplemental Table S16), with the Spearman's ρ between the SINE insertion densities and gene densities reaching 0.46. The correlation on each chromosome varied, in particular, with notable higher correlation coefficients observed on Chromosome 2 (ρ = 0.66), Chromosome 8 (ρ = 0.68), and Chromosome 11 (ρ = 0.64) (Supplemental Fig. S4). Likewise, SINE insertion densities were also highly positively correlated with recombination rates (ρ = 0.58), with higher correlation coefficients on Chromosome 9 (ρ = 0.72), Chromosome 11 (ρ = 0.77), and Chromosome 18 (ρ = 0.64) (Supplemental Fig. S5). On Chromosome 11, there is a strong positive correlation between PRE insertion density and both recombination rate and gene density, with correlation coefficients of 0.8 and 0.62, respectively (Fig. 4D; Supplemental Figs. S6, S7). The highly positive correlations mentioned above implied that SINE, especially PRE, went through a recent transposition burst and might have contributed to recent adaptations. Additionally, significant positive correlations between recombination rate and gene density were observed in five out of the 18 chromosomes (Supplemental Fig. S8). To further quantify to what extent recombination rate and gene density contribute to TIP distribution, we fitted a linear model to estimate the variance components explained by the two variables. Gene density captured 95% of the variations in the distribution of SINE with TIPs, with recombination rate only explaining 4% (Fig. 4E). In contrast, for DNA transposons and LTR, the recombination rate captured 44.8% and 40.3% of the total variations, whereas gene densities captured 55.1% and 4.3%, respectively. Evidently, the distributions of the three TIP types above are determined by the combined effects of proximity to genes and recombination. However, no variance component explained by recombination rate was captured when LINE insertion densities were treated as a dependent variable in the model.
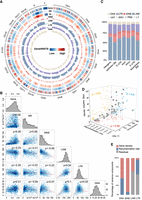
Forces driving genomic distribution of TIPs. (A) The circle plot reports the distribution of TIPs in the pig pangenome. The outermost circle denotes the number and size of chromosomes (gray), followed by gene density, recombination rate, and TE density on the reference genome. Red, green, purple, and orange represent the distributions of insertion densities of SINE, LINE, LTR, and DNA transposon, respectively. (B) Spearman's rank correlations of TIP densities between major TE classes, and their correlations with gene density and recombination rate. (C) Percentage of major TIP types (DNA transposons, LTR, SINE, and LINE) locating in genomic regions. (D) The 3D scatter plot of correlations among gene densities, recombination rates, and PRE insertion density on Chromosome 11. (E) Percentages of variance components explained by gene density and recombination rate in SINE, LINE, LTR, and DNA transposon insertion densities.
Gene expression differentiation by TIPs
When landing in the vicinity of genes, TIPs can modify the expression of adjacent genes by altering gene structure or by affecting regulatory sequences (Fueyo et al. 2022). In total, 154,173 (31.4%) TIPs were located in gene regions, including 5862 in exons and 917 in promoter regions (Supplemental Table S17). To investigate the impacts of TIPs on the expression of particular genes, we explored the expression levels of genes with TIPs using transcriptome data of five tissues (including spleen, muscle, subcutaneous adipose, kidney, and liver) from 16 individuals. We compared the expression fold changes between genes with TE insertions and genes without TE insertions. We determined that breed is not a significant factor affecting transcript abundance in addition to the presence/absence of a TE (Supplemental Tables S18–S21). Specifically, 765 TE insertions localized in introns, followed by upstream and downstream regulatory regions (103) and exons (24), markedly altered the expression of corresponding genes (Supplemental Tables S18–S21). The genes affected by TIPs were differentially expressed in only one or fewer of these tissues, with subcutaneous fat, dorsal muscle, and liver being the most affected, suggesting that they regulate mRNA transcription in a tissue-dependent manner (Fig. 5A). Furthermore, both up-regulated and down-regulated differential expression was observed, indicating that TIPs influence host genes in both directions. Such influences may result from novel enhancers, transposases, alternative splice sites, or polyadenylation signals imbedded in TE insertions, some of which may be of adaptive significance or may contribute to phenotype divergence (Fueyo et al. 2022). It is noteworthy that PRE insertions occupied a significantly higher proportion in the set of TIPs affecting gene expression (20.6%) compared with their prevalence in the pig mobilome (7.6%), suggesting they may play important role in the modulation of gene expression (Fig. 5B). For instance, a PRE insertion was absent in the reference genome but present in the intron of gene HSPH1 (heat shock protein family H [Hsp110] member 1) ∼12.6 kb downstream from the transcriptional start site on Chromosome 11 in some individuals (Fig. 5C; Oh et al. 1997). HSPH1 has been recognized as one of the primary heat shock proteins in mammalian cells, which is ubiquitously expressed in multiple tissues (Oh et al. 1997). It protects cellular and molecular targets from heat damage and is involved in the body temperature regulation of cattle in the presence of climatic stress (Howard et al. 2014). This gene showed significant down-regulation when the PRE insertion was present, suggesting that the PRE insertion may affect the response networks of the living cells under environmental stresses. We observed that 51 and 20 population-specific TE insertions that affect gene expression are exclusively present in Chinese pigs and European pigs, respectively, which may have significant effects on the genetic activity of corresponding genes and phenotype differentiation (Fig. 5D; Supplemental Fig. S9), for example, a PRE insertion located in 3′ UTR of gene MMP2 (matrix metallopeptidase 2) (Fig. 5E). We found that the PRE insertion was lost in Chinese pigs, but present in 47% of European ones, and the expression of MMP2 was significantly higher in individuals harboring the insertion than individuals devoid of the insertion. MMP2 is expressed predominantly in subcutaneous adipose tissue and encodes an extracellular matrix-degrading enzyme associated with meat quality. In pigs, MMP2 was identified to be associated with lean meat production (Onteru et al. 2009).
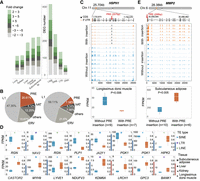
Gene expression differentiation by active TEs. (A, left) The number of differential expression genes affected by TIPs in five tissues (spleen, muscle, subcutaneous adipose, kidney, and liver). (Right) Comparison of the number of differential expression genes affected by different TE types. (B, left) Percentage of major TIP superfamilies or families affecting gene expression. (Right) Percentage of major TE families of pig mobilome. (C) The sketched gene structure of HSPH1, and the comparison of HSPH1 expression levels (FPKM) between individuals with a PRE insertion and individuals without a PRE insertion. (D) The sketched gene structure of MMP2, and the comparison of the MMP2 expression level (FPKM) between individuals with a PRE insertion and individuals without a PRE insertion. (E) Sixteen genes harbor population-specific TE insertions with a presence frequency > 0.4; comparisons of the gene expression level (FPKM) of these genes between individuals with population-specific TE insertions (orange) and without population-specific TE insertions (blue). The red and blue bars on the right side represent the presence frequencies of TE insertions present only in Chinese pigs and European pigs, respectively.
Detection of candidate TE insertions contributing to local adaptation
To examine TE insertions likely to contribute to local adaptations of Tibetan wild boars, we used SNPs as an indicator to identify signals of selective sweeps with three well-established haplotype-based metrics: iHS, nSL, and XP-EHH (Fig. 6A). The first two methods identified recent and incomplete within-population selective sweeps in Tibetan wild boars, whereas XP-EHH detected selection signatures that have almost or have fully risen to fixation, by comparing extended haplotype homozygosity between Tibetan wild boars and a reference population composed of 23 lowland Chinese domestic breeds. A total of 1501 genomic windows showing extremely high Z-scores (top 5% percentile of distribution as threshold) in the three methods were considered to be the genomic regions targeted by positive selection, which encompassed 2719 candidate selected genes. We considered that TE insertions being nearly fixed or at very high frequencies in Tibetan wild boars, while at much lower frequencies in Chinese domestic breeds and European breeds (Fig. 6B), and located within or nearby positively selected genes were putatively adaptive. To further distinguish selection from genetic drift, we required potential candidate TE insertions located to be <10 kb from SNPs showing the top 5% high Z-scores in at least one method. In total, 23 TIPs fulfilled all these criteria and were candidates contributing to adaptation. Among these TE-inserted genes, 20 have known functions involved in responses to hypoxia, skeletal development, regulation of heart contraction, neuronal cell development, etc. (Fig. 6C). Furthermore, we found that three of the candidate TE insertions affected the expression of corresponding genes (SVIL, PLXNA4, and NELL2), which are more likely to be the actual targets of positive selection. PLXNA4 is known to play a role as a guide for axons in the development of the nervous system and in the positive regulation of axonogenesis (Cho et al. 2022), whereas NELL2 is multifunctional in neural cell growth and is strongly linked to the urinary tract disease (Liu et al. 2021).
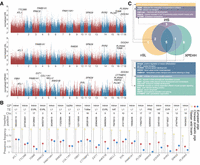
Detection of candidate TEs contributing to high-altitude adaptation. (A) Genome-wide scans for selective sweeps using iHS, nSL, and X P-EHH. iHS, nSL, XP-EHH values are normalized as Z-scores, which are shown in a Manhattan-inspired plot, with significant SNP Z-scores >2 plotted against genomic positions. (B) Presence frequency differences of 20 candidate adaptive TEs between Tibetan wild boars, Chinese domestic pigs, and European commercial pigs. (C) The Venn plot shows 20 genes with candidate adaptive TEs identified in the three methods.
Discussion
Systematic and comprehensive assessments of structural variation have been challenging and elusive owing to the intricate and multifaceted features of SVs (Alkan et al. 2011). The advent of the pangenome expands the SV spectrum particularly by deciphering gene PAVs that underpin phenotypic diversity in bacteria, crops, and humans (Yu et al. 2022). However, our understanding on gene PAVs in animals has lagged behind owing to their large genome size making large population-scale de novo genome assembly computationally demanding. To our knowledge, this is the first pangenome study providing a gene PAV catalog for domesticated mammals. Because of the inherent limitation of short-read length in “next-generation” sequencing technologies, it is unrealistic to explore full types of SVs using short reads, especially in long repetitive regions (Goodwin et al. 2016). Although long-read sequencing has emerged as superior to short-read sequencing in profiling structural variations, the currently low throughput and high cost requirements hinder the practical application in a large population context. Comparatively, taking advantage of short-read sequencing genomes with high read coverage (>20×) is realistic and feasible to assemble coding sequences with relatively high quality for mammals (Kofler et al. 2016; Duan et al. 2019; Yu et al. 2022). The question of whether dispensable genes are adaptive remains open for organisms across the tree of life (Golicz et al. 2020). Despite the fact that mammals have less genome flexibility compared with bacteria and plants (Schnable 2012), we found that there is still a substantial proportion (16.8%) of total pig pangenes being variable among individuals, more than that (5%) in humans (Li et al. 2010), potentially reflecting the hidden ability of the animal to respond to environmental changes. We identified 14 nonreference SLA genes belonging to the dispensable gene, seven of which displayed changes in expression in response to PRRSV infection, suggesting that these genes might have been incorporated into existing immune-related biological pathways and regulatory networks, and their functions may be beneficial for survival and may confer selective advantages (Golicz et al. 2020). Similarly, a multitude of dispensable genes in plants have been found to be responsible for complex traits (Zanini et al. 2022), such as those responding to biotic and abiotic stress as well as controlling flowering time (Gao et al. 2019), being speculated to be adaptive (Tao et al. 2021).
TEs have the ability to proliferate their copy numbers and insert into a new region of the genome. The distribution of TEs in various species was suggested to be controlled by the combined effects of insertion preferences and differential natural selection (Medstrand et al. 2002; Kent et al. 2017). We discovered that TIPs show deviations from random distribution in the pig genome, and their distributions were highly variable across different TE superfamilies and orders, implying a diverse adaptive capacity and expansion history during host genome evolution (Kent et al. 2017). LINE and LTR insertions are generally clustered in intergenic and low recombination rate regions, whereas SINE insertions preferentially accumulate in gene-rich and highly recombining regions across the pig genome. A possible explanation about the distribution tendencies is that de novo insertions of longer LINEs and LTRs are autonomous elements encoding their own regulatory elements, which are more likely to disrupt the proper expression of nearby genes, whereas SINEs belong to nonautonomous elements, using the molecular machinery of the autonomous TEs (LINEs) to transpose (Kent et al. 2017). When inserted in coding sequences of genes, TIP-derived variants are usually detrimental. In fact, ample evidence from studies on disease-causing insertions in humans and other organisms serves as prime examples of this assertion (Burns 2020). However, when landing in regulatory regions within or between genes, TIPs may have immediate or latent consequences on host gene expression and beneficial effects on organismal fitness and then rewire gene regulatory networks (Chuong et al. 2017), potentially being recruited by the host for rapid adaptation and plasticity of relevant traits (Jordan et al. 2003; Schrader et al. 2014). The consequences of TE insertions on mRNA expression levels have been studied comprehensively in fruit flies (Villanueva-Canas et al. 2019), mice (Miao et al. 2020), and humans (Cao et al. 2020), yet such adaptive changes at a single insertion level could be so subtle that cannot be captured phenotypically in large animals (Fueyo et al. 2022). We quantified that a total of 892 TE insertions localized within or nearby genes, markedly altering the expression of corresponding genes, among which 71 TE insertions were present exclusively either in Chinese pigs or in European pigs. It can be speculated that some of the population-specific TE insertions are more prone to be adaptive mutations. Additionally, we uncovered several potentially positively selected TE insertions in Tibetan wild boars, which have been co-opted into genes involved in responses to hypoxia, regulation of heart contraction, and other functions. An EVRL inserted in the intron of gene PRKCE, being fixed in Tibetan wild boars. This gene can be specifically activated under hypoxia and can contribute to increasing in Ca2+ and the contraction of pulmonary artery smooth muscle cells. Besides, loss of PRKCE would decrease the contractile and/or structural response of the murine pulmonary circulation to chronic hypoxia (Littler et al. 2005). It is likely that positive selection under hypoxia might drive the insertion to fixation. Tibetan wild boars have developed prominent pulmonary blood vessels, enhancing the efficiency of oxygen exchange in the pulmonary artery and alleviating pulmonary artery pressure. A common hAT inserted in DOCK4, which is involved in blood vessel lumen morphogenesis and in positive regulation of vascular associated smooth muscle cell migration. Experimental validation of TE co-option for local adaptation remains a challenging endeavor owing to the subtle phenotypic effects caused by regulatory changes driven by TEs. As a result, it is imperative to approach these putatively adaptive TEs with increased insertion frequencies cautiously.
Taken together, we construct a pig pangenome based on 250 sequenced individuals of 32 phenotypically divergent pig breeds in Eurasia and establish a pangenome framework to characterize coding sequence PAVs for eukaryotes with large genomes. The dispensable coding sequences provide a reservoir of genetic variability with adaptive potential, rendering the genome a more dynamic source of variations to gain evolutionary advantages for pigs. The systematic identification of coding sequence PAVs allows us to unveil hidden layers of genetic diversity. In addition, an adequate understanding of the genetic mechanism underlying coding sequence PAVs and effective utilization of the genetic resource is of critical importance for the future conservation of biodiversity, breeding practice, and human health (Li and Simianer 2020).
Methods
Genome sequencing
We sequenced 64 pigs comprising three European commercial breeds and 18 Chinese domestic breeds selected from geographically diverse regions across China (Supplemental Table S1). Genomic DNA was extracted from ear tissues using the standard phenol/chloroform method and assessed by agarose gel electrophoresis and A260/280 ratio. DNA libraries were constructed according to the Illumina library preparation protocols with an insert size of 350 bp and were sequenced using an Illumina NovaSeq 6000 platform with 150-bp paired-end sequencing kits. The sequencing coverage of each individual was ∼40–50×. In addition, we downloaded the genome data of 186 individuals comprising Tibetan wild boars, seven Chinese domestic breeds, six European commercial breeds, and two European crossbreeds from NCBI database with an average sequence depth of ∼30×, leading to a total of 250 pigs with 90 animals from European breeds and 160 animals from Chinese breeds. (Supplemental Tables S2, S3).
Genome assembly and pangenome construction
Raw reads of all 250 individuals were de novo assembled using MaSuRCA (Zimin et al. 2013). The quality of the assemblies was assessed using QUAST with default parameters (Gurevich et al. 2013). The pig pangenome was constructed using a “map to pan” strategy (Wang et al. 2018). The assembled contigs with lengths > 500 bp were aligned to the S. scrofa 11.1 genome using minimap2 (Li 2018; Warr et al. 2020). The unaligned contig was defined as a contig without a continuous alignment longer than a defined threshold of 500 bp with sequence identity >90%. For aligned contigs, if they contained a continuous unaligned region >500 bp, the unaligned region was also extracted as unaligned sequences. The unaligned contigs and sequences were then combined and searched against the GenBank nucleotide database using BLASTN, and the potential contaminations from those sequences whose best hit with an E-value lower than 0.00001 were microorganisms, plants, and non-Artiodactyla animals were removed (Supplemental Table S4). The resulting unaligned sequences were pooled, and the redundant sequences were discarded by CD-hit (Fu et al. 2012). To further ensure the nonredundancy of the novel sequences, the pooled nonredundant sequences were subsequently self-compared and aligned against the reference genome using BLASTN. In all of the filtering steps, the sequence identity cutoff was set to 90%. The sequence-based pangenome was obtained by combining the final nonredundant nonreference sequences and the pig reference genome S. scrofa 11.1. The 14 newly identified SLA genes were verified by aligning them to the SLA sequences in IPD-MHC sequence database using BLAST (Kent 2002; Maccari et al. 2017).
Gene annotation
An annotation pipeline integrating ab initio prediction, homologous-based prediction, and expression evidence-based prediction was performed for gene model prediction. Repeat sequences were first masked using RepeatMasker with a repeat library downloaded from Repbase (Tarailo-Graovac and Chen 2009; Bao et al. 2015). A de novo species-specific repeat library based on nonreference sequences was constructed using RepeatModeler2 for the second-round masking. EST and protein sequences belonging to S. scrofa were downloaded from GenBank, separately. RNA-seq data from 12 tissues obtained through sequencing and downloaded from NCBI (Supplemental Tables S1, S6) were aligned to the pangenome using HISAT2 (Kim et al. 2019). Protein-coding genes were predicted from the masked nonreference genome by BRAKER2 (Brůna et al. 2021). Specifically, RNA-seq alignments protein sequences were used for gene model training with GeneMark-EP+. The good gene models predicted by GeneMark-EP+ combined with expression and homologous evidence were used to train AUGUSTUS. Multiple prediction evidence was finally integrated by BRAKER2, and a set of high-confidence gene models supported by RNA-seq alignment, EST, and/or protein evidence was generated. Protein-coding genes were also predicted using the MAKER2 pipeline (Holt and Yandell 2011). The genes predicted by both BRAKER2 and MAKER2 were used as the final result. The GO and KEGG annotations were predicted using eggNOG-mapper v2 (Huerta-Cepas et al. 2016).
RNA sequencing and differential expression analysis of nonreference SLA genes
To identify nonreference SLA genes that were deferentially expressed under PRRSV infection, RNA sequencing based on porcine alveolar macrophages (PAMs) from PRRSV-infected and noninfected Yorkshire pigs was also performed. Six piglets aged 6 wk were intramuscularly challenged with 2 mL of a viral suspension of HP-PRRSV GDBY1 strain (2 mL: 4.4 × 105 TCID50mL), and six noninfected piglets treated as control were challenged with an identical volume of Marc-145 cell culture supernatant, DMEM, in the same way. At 7, 14, and 21 d dpi, we took two pigs from the control and the GDBY1-infected groups, respectively, which were humanely euthanized via pentobarbital overdose. For the pigs that died after the PRRSV challenge, we took pictures and tissue samples immediately. PAMs were obtained from the bronchoalveolar lavage fluid (BALF) as previously described for RNA sequencing (Wang et al. 2021b). Sequencing libraries of all 12 samples were constructed using a NEBNext library prep kit with an insert size of 250∼350 bp. The library preparations were sequenced on an Illumina NovaSeq 6000 platform for 150-bp paired-end reads. The quality of raw sequencing reads was accessed using FastQC and trimmed to remove low-quality bases and adapters using Trimmomatic (Bolger et al. 2014). The clean reads were mapped to the pangenome using HISAT2 (Kim et al. 2019). The alignments of reads were sorted using SAMtools (Li et al. 2009) and then assembled by StringTie2 using the pangnomic gene annotation models as guidance (Pertea et al. 2015). Transcript abundances were normalized with the FPKM value of each gene. Differential expression analysis was performed between the control and infected pigs for each time point using DESeq2 (Love et al. 2014). Genes were deemed significantly differentially expressed with a twofold change cutoff and a Benjamini–Hochberg-corrected false-discovery rate (FDR) threshold of 0.05.
Gene family analysis
The protein sequences from the S. scrofa 11.1 reference and nonreference genome were combined and inferred gene families using OrthoFinder (Emms and Kelly 2015). Specifically, the protein all-versus-all alignment was conducted using DIAMOND (Buchfink et al. 2015). A reciprocal best hit was then obtained for each protein using the length-normalized score. All scores were used to delimit an inclusion threshold. All hits above this score were assigned to the same orthogroup.
Detection of gene PAV
To detect the gene PAV information, raw reads from each individual were aligned to the pangenome using BWA-MEM (Li 2014). The PAV of each gene in each individual was determined by calculating the depth of mapping coverage against a gene using Mosdepth (Pedersen and Quinlan 2018). For the >30% of the exon regions in which the coverage was ≥2×, the gene was considered present in that individual; otherwise, it was regarded as absent.
SNP calling
DNA sequencing reads were aligned to the S. scrofa 11.1 reference genome using BWA-MEM (Li 2014), and the mapping results were filtered and sorted using SAMtools (Li et al. 2009). Potential PCR duplicates were marked with Picard MarkDuplicates (http://picard.sourceforge.net/). Genotypes of all sites in the genome were called individually on each sample using the GATK HaplotypeCaller algorithm in the GVCF mode. A joint genotyping process was then conducted to call SNPs and short indels using GATK GenotypeGVCFs. To remove false positives, the dbSNP for pig was downloaded from NCBI, and the HD chip (670k) data set was also obtained. These two sets of high-confidence SNVs were used to train the GATK's VariantRecalibrator for variant quality score recalibration (VQSR) to filter low-quality SNPs from raw variant calls with a sensitivity threshold of 99%. Based on the aligned reads obtained in the gene PAV step, the pangenome-based variant calling process was also conducted using BCFtools. To remove potential false-positive SNPs, calls with “QUAL < 30|INFO/FS > 60.0|INFO/MQ < 40.0” were filtered. The gene-based SNP/InDel annotation process was conducted using SnpEff (Cingolani et al. 2012). The pair-wise IBS similarity scores were calculated using PLINK and then converted to a distance matrix through an in-house Python script.
Whole-genome recombination map estimations
Fine-scale recombination rates were estimated using pyrho (Spence and Song 2019), which is a computationally efficient and more accurate way of accounting for population size history. To estimate the population demographic history, genotypes were phased using Beagle (Browning and Browning 2016). The program smc++ was used to infer population history using the phased SNPs (Terhorst et al. 2017). The per-generation mutation rate was assumed to be 2.5 × 10−8 (Groenen et al. 2012). To simplify the recombination rate estimation process, 50 samples were randomly selected. Combined with the demographic history results, a lookup table was computed using the pyrho make_table command. The main hyperparameters of pyrho, the window size and the smoothness penalty, were then fine-tuned using the pyrho command hyperparam. The best hyperparameters of the window size and the smoothness penalty, which were estimated to be 200 and 100, respectively, were then used to infer recombination maps using pyrho optimize.
Identification of TIPs
We downloaded 953 consensus sequences of TE families from Repbase (Bao et al. 2015). In addition, 622 TE families were de novo identified from the nonreference sequence with RepeatModeler2 (Flynn et al. 2020). These two parts of TE sequences were combined and clustered using CD-hit (Fu et al. 2012), resulting in a custom TE library with 1286 TE families. TE sequences in the sequence-based pangenome were detected and then masked with RepeatMasker 4.1.2 (Tarailo-Graovac and Chen 2009). Then a modified pangenome consisting of the TE library and the pangenome with the masked TE sequences was obtained. Sequencing reads of 250 individuals were aligned to the modified pangenome using BWA-MEM (Li 2014). The mapping reads were sorted, and duplicates were marked with SAMtools. TE insertions with recent activity were detected using PoPoolation TE2 (Kofler et al. 2016). A physical pileup file was generated with the parameter -map-qual 15. Signatures of TE insertions were identified with the joint mode, with the parameter -mode joint -mincount 3 -signature-window fix100 -min-valley fix100. The population frequency of TE insertions was estimated with a default parameter as the ratio of physical coverage supporting a TE insertion to the total physical coverage. We analyzed the local coverage around the insertion sites to genotype the PAV of TE insertions. We defined insertions with frequency >5% to be present in the individual. Alternatively, insertions were judged to be absent in the individual. The TE insertion allele file was converted to BED format with PLINK (Purcell et al. 2007). TIP-based PCA was performed using FlashPCA2 (Abraham et al. 2017).
Statistical model
We partitioned a variation of each type of TIP distributions (DNA transposons, LTR, LINE, SINE) into recombination rate and
gene density components using a linear model: y = 1μ + r + g + e, where y is the vector of TIP densities for each type, μ is the overall mean. 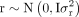 , and
, and 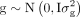 are vectors containing the random effects of recombination rate and gene density, respectively.
are vectors containing the random effects of recombination rate and gene density, respectively. 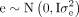 is a vector of residual effects. The variance components were estimated using restricted maximum likelihood with R package
“lme4” (Bates et al. 2015; R Core Team 2021).
is a vector of residual effects. The variance components were estimated using restricted maximum likelihood with R package
“lme4” (Bates et al. 2015; R Core Team 2021).
Detection of active TEs associated with gene expression
Transcriptome data newly generated from spleen, longissimus dorsi muscle, subcutaneous adipose, kidney, and liver of 16 individuals across five Chinese pig breeds and three European breeds were used for gene model training and identification of differential gene expression caused by TIPs. RNA sequencing and quantification of the FPKM value of expression genes followed the same procedure described above. To determine the transcriptomic impact of TIPs on corresponding genes, genes with or without TE insertions were divided into two groups. For each gene, we calculated the ratio between the average gene expression level for the individuals harboring the TE insertion and the average gene expression level for individuals without the insertion. Genes were deemed significantly differentially expressed with a twofold change cutoff.
Detection of candidate TEs contributing to high-altitude adaptation
To examine adaptive TE candidates contributing to high-altitude adaptations of Tibetan wild boars, we used genome-wide common SNPs (minor allele frequency > 5%) as an indicator to identify genomic regions for evidence of selective sweeps. Three haplotype-based approaches—iHS, nSL, and XP-EHH—were used to detect genomic regions targeted by SNPs in Tibetan wild boars, all of which were implemented by selscan (v.1.3) (Szpiech and Hernandez 2014). iHS and nSL tests identify hard sweeps, although they have some power to detect soft sweeps as well. XP-EHH is a statistical test detecting nearly fixed selection signatures by contrasting extended haplotype homozygosity between Tibetan wild boars and a reference population composed of 23 lowland Chinese domestic breeds. Beagle (v.5. 1) was performed with argument burnin = 5 and iterations = 20 for haplotype phasing (Browning et al. 2021). Candidate regions under positive selection were identified using 100-kb nonoverlapping sliding window with significantly high Z-scores (top 5% percentile of distribution as threshold) in the three methods. Normalization of each Z-score across all chromosomes was performed using the norm program distributed along with Selscan (Szpiech and Hernandez 2014). The genes that overlapped with these regions were regarded as genes of positive selection. TE insertions fulfilling three criteria were considered to be putatively adaptive or co-opted: (1) at high frequency (>0.6) or being nearly fixed in Tibetan wild boars, while at relatively low frequency in Chinese domestic pigs; (2) located within or nearby (<2 kb) positively selected genes; and (3) located <10 kb from SNPs showing top 5% high Z-scores in at least one of the three methods.
Data access
The WGS data of 64 individuals from 21 pig breeds and RNA-seq data from 92 tissues generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA530874. The sequences, genes, gene annotations, and proteins identified in the nonreference genome in FASTA format and the TIP data set in VCF format are provided as Supplemental Data and can be downloaded at Zenodo (https://doi.org/10.5281/zenodo.6791874). All codes used in this study are provided as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Jing Guo from Wellcome Sanger Institute for feedback on the manuscript. This research was supported by the National Natural Science Foundation of China (no. 32030102), National Pig Industry Technology System (CARS-35), and the Special Project for Research and Development in Key areas of Guangdong Province (no. 2018B020203003).
Author contributions: Y.C. and Z.L. conceived and designed the project; Z.L. performed the data analysis and wrote the manuscript; R.Z., B.J, Z.Y.L., and L.T. assisted in visualization and data processing; S.H., H.Z., L.L., and Y.W. performed the PRRSV challenge trial; C.W., B.J., Z.Y.L., R.Z., S.H., H.Z., Y.Q., Q.L, H.C., and K.W. collected the samples; Y.Z., Y.M., T.P., and X.L. contributed comments; and Y.C. revised the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277638.122.
-
Freely available online through the Genome Research Open Access option.
- Received December 31, 2022.
- Accepted September 12, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








