
Single-cell transcriptome and metagenome profiling reveals the genetic basis of rumen functions and convergent developmental patterns in ruminants
- Juan Deng1,2,7,
- Ya-Jing Liu1,7,
- Wen-Tian Wei1,
- Qi-Xuan Huang1,
- Li-Ping Zhao3,4,
- Ling-Yun Luo1,
- Qi Zhu2,
- Lin Zhang2,
- Yuan Chen2,
- Yan-Ling Ren5,
- Shan-Gang Jia6,
- Yu-Luan Lin1,
- Ji Yang1,
- Feng-Hua Lv1,
- Hong-Ping Zhang2,
- Feng-E Li4,
- Li Li2 and
- Meng-Hua Li1
- 1College of Animal Science and Technology, China Agricultural University, Beijing 100193, China;
- 2Farm Animal Genetic Resources Exploration and Innovation Key Laboratory of Sichuan Province, Sichuan Agricultural University, Chengdu 611130, China;
- 3CAS Key Laboratory of Animal Ecology and Conservation Biology, Institute of Zoology, Chinese Academy of Sciences (CAS), Beijing 100101, China;
- 4Key Laboratory of Agricultural Animal Genetics, Breeding, and Reproduction of the Ministry of Education and Key Laboratory of Swine Genetics and Breeding of Ministry of Agriculture and Rural Affairs, Huazhong Agricultural University, Wuhan 430070, China;
- 5Shandong Binzhou Academy of Animal Science and Veterinary Medicine, Binzhou 256600, China;
- 6College of Grassland Science and Technology, China Agricultural University, Beijing 100193, China
-
↵7 These authors contributed equally to this work.
Abstract
The rumen undergoes developmental changes during maturation. To characterize this understudied dynamic process, we profiled single-cell transcriptomes of about 308,000 cells from the rumen tissues of sheep and goats at 17 time points. We built comprehensive transcriptome and metagenome atlases from early embryonic to rumination stages, and recapitulated histomorphometric and transcriptional features of the rumen, revealing key transitional signatures associated with the development of ruminal cells, microbiota, and core transcriptional regulatory networks. In addition, we identified and validated potential cross-talk between host cells and microbiomes and revealed their roles in modulating the spatiotemporal expression of key genes in ruminal cells. Cross-species analyses revealed convergent developmental patterns of cellular heterogeneity, gene expression, and cell–cell and microbiome–cell interactions. Finally, we uncovered how the interactions can act upon the symbiotic rumen system to modify the processes of fermentation, fiber digestion, and immune defense. These results significantly enhance understanding of the genetic basis of the unique roles of rumen.
Ruminants have a unique and complex digestive system with a four-chambered stomach that allows them to generate energy from fibrous plant material more efficiently than other herbivores. The rumen, which is the largest compartment, is inhabited by a highly dense and diverse consortium of microorganisms. For millions of years, ruminant animals and ruminal microorganisms have coevolved and worked together to promote the fermentation and degradation of plants (Russell and Rychlik 2001).
The rumen undergoes developmental and structural changes particularly during embryonic and early postnatal stages. The genetic basis underpinning the distinct rumen characteristics has been investigated using transcriptomes and metagenomes (Bush et al. 2019; Li et al. 2019). Given the heterogeneity of rumen tissues and their dynamics during developmental progression (Schmitz-Esser 2021), conventional RNA-seq is confounded by changes in cellular composition and thus has difficulties in accurately revealing cell-specific changes in gene expression for rare cell types and the interactions between microbiota and specific cell types (Cheng et al. 2019). Rumen epithelial single-cell atlases of cattle and sheep obtained with scRNA-seq have been reported (Gao et al. 2021; Wu et al. 2022; Xue et al. 2022; Yuan et al. 2022). However, a comprehensive developmental cell atlas and a metagenomic characteristic map of the rumen have not been constructed, nor have the interactions between the ruminal microbiota and epithelial cells of the host been explored.
To this end, we performed integrated profiling of the single-cell transcriptomes of ruminal cells and metagenomes of the rumen microbial community in sheep and goats over a time course spanning major embryonic and postnatal developmental stages. We profiled about 183,800 sheep and about 124,400 goat ruminal cells at 17 distinct developmental time points, ranging from embryonic day 45 to postpartum day 90. We investigated the difference in transcriptional profiles among various developmental stages and species (sheep, goat, cattle, human, monkey, and mouse) at a single-cell resolution. We also analyzed the rumen microbial metagenomes and explored the interactions between ruminal microbial genomes and ruminal cells. We tested the hypothesis of convergent transcriptomic patterns of rumen between ruminants at the single-cell level as well as close interactions between rumen epithelial cells and microbiomes during development.
Results
Histological changes in rumen walls
The rumen wall at seven postconception (e45, e60, e75, e90, e105, e120, and e135) and 10 postpartum time points (d0, d7, d14, d21, d28, d35, d42, d49, d56, and d90) was dissected from sheep and goats (Supplemental Fig. S1A; Supplemental Table S1). In addition to the three (e.g., early, middle, and late) major embryonic stages, the sheep and goats analyzed here were in three postpartum stages: prerumination (0–14 d), transition (21–49 d), and rumination (after 56 d). Histological examination revealed macro- and microscopic morphological and compositional changes in the rumen walls along the developmental time line (Supplemental Fig. S1B; Supplemental Note S1). We observed significant changes in the histomorphometric measurements, such as the length and width of Ruminal papillae and the thickness of epithelium, lamina propria and submucosal tissue, and tunica muscularis (Tm), during development, particularly among the six stages (Supplemental Fig. S1C; Supplemental Note S2). The measurements showed a distinct developmental pattern and indicated a vital role in immune defense against pathogens.
Single-cell transcriptomic atlas of rumen tissues
For embryonic and postpartum samples, we profiled 6227–17,487 and 5418–11,930 single cells per sample for sheep and goats, respectively. After filtering the low-quality cells, we profiled a total of 183,802 and 124,441 single cells from the different developmental time points of sheep and goats, respectively, for the downstream analyses (Supplemental Fig. S2A,B; Supplemental Table S2). In sheep and goats, we achieved sequencing depths of 28,364–91,904 and 48,900–100,032 reads per cell, which enabled the analyses of 1180 and 1137 genes per cell on average (median), respectively (see Methods section).
We tested three different scRNA-seq data integration methods such as merging with scaling, canonical correlation analysis (CCA) integration, and harmony. The method of merging with scaling used here is the best, which can detect all the different cell types while retaining the variability in both species (Supplemental Fig. S2C,D; Supplemental Note S3). We identified 32 and 37 main cell clusters for sheep and goats, respectively, based on their gene expression profiles (Supplemental Fig. S2C,D; Supplemental Tables S3, S4). In general, most cells from the same developmental stages clustered closely into one to three clusters. Also, we found that most clusters consisted of cells from different developmental stages, and the main distributions of cells differed among the stages (Supplemental Fig. S2C,D). Similar co-occurrences were observed among cell clusters defined by the 17 developmental time points (Supplemental Fig. S2C,D). Notably, cells from the embryonic (e45–e135) and prerumination (d0–d14) stages were clearly separated from those from the transition and rumination stages (d21–d90) (Supplemental Fig. S2C,D). These findings showed a high level of genomic heterogeneity of rumen cells and a dynamic progression of rumen morphogenesis over the developmental period.
Annotation of cell types and subtypes
We subsequently assigned the cell clusters based on three different approaches (see Methods section), and annotated 30 and 31 major cell types (and subtypes) across the 17 time points in sheep and goats, respectively (Fig. 1A,B; Supplemental Tables S5, S6). Epithelial cells consisted of basal cells (BCs), spinous cells (SCs), and granular cells (GCs). In sheep, marker genes such as KRT17, KRT15, KRT5, and TOP2A were highly expressed in proliferating BCs (PBCs), whereas KRT17 and KRT15 also showed high expression levels in the four subtypes of BCs (Fig. 1A; Supplemental Table S5). SCs showed substantially increased expression of multiple genes, namely, KRT15, KRT17, KRT5, and SFN, and the seven subtypes were defined by the combinations of different marker genes. Marker genes such as SFN and S100A8 were highly expressed in the three clusters of GCs, which were divided into two major subtypes (Fig. 1A; Supplemental Table S5). KRT15 was highly expressed in embryonic SCs and BCs but not in GCs after birth, which was confirmed by immunofluorescence staining (Fig. 2A). Additionally, immunofluorescence staining showed that the expression of KRT15 was significantly increased in embryonic stages and then significantly decreased in the postpartum stage in both sheep and goats (Supplemental Fig. S2E,F). The SCs showed high expression of marker genes for both BCs and terminally differentiated GCs, such as the epithelial proliferation marker gene KRT15 and epithelial differentiation marker gene KRTDAP.
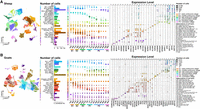
UMAP plots and the number of cell types. Cell number (n > 3) at the 17 time points (middle) and expression levels of representative marker genes shown as violin plots (right) for each cell type in sheep (A) and goat (B) rumen.
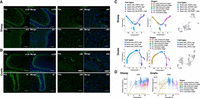
Immunofluorescence staining and pseudotime trajectory analysis. (A) Immunofluorescence of KRT15 in rumen tissues of sheep at the time points e135 and d21 and of goats at the time points e120 and d7. Scale bars, 60 μm (zoomed-in image). (B) Immunofluorescence of LYVE1 in rumen tissues of sheep and goats at the time points e90 and d90. Scale bars, 60 μm (zoomed-in image). (C) Pseudotime trajectory analysis corresponding to the differentiation of basal cells in sheep and goats. Cells are colored by pseudotime, cell types, and sample stages. (D) Pseudotime trajectory analysis corresponding to the marker gene CA1 in sheep and CA3 in goats. Cells are colored by sample stage.
In goats, we identified three BC subtypes, four SC subtypes, and two GC subtypes (Fig. 1B; Supplemental Table S6). Marker genes for the epithelial cells of sheep, which were associated with epithelial proliferation (e.g., KRT15 and KRT5) and differentiation (e.g., SPINK5, KRTDAP, and DAPL1), were also found in the same cell types of goats. Nevertheless, the high expression levels of some marker genes (e.g., KRT8 and RBP2) in the subtypes basal cells_IGFBP6_high and spinous cells_IGFBP6_high were substantially reduced in sheep (Fig. 1A,B; Supplemental Tables S7, S8).
We found that fibroblasts (Fib) were fewer in number (sheep: n = 34,024; goats: n = 35,274) than epithelial cells (sheep: n = 90,583; goats: n = 48,454). Among Fib, we defined four cell subtypes in sheep and eight subtypes in goats based on canonical marker genes (e.g., LUM and DCN) and subtype-specific gene markers (Supplemental Tables S5, S6). Notably, cells of subtypes such as fibroblast_MFAP5_high of sheep and fibroblast_TNFAIP6_high and fibroblast_MGP_high of goats release inflammatory adipokines (e.g., interleukin 6), suggesting a critical role of these cellular subsets in inflammatory responses (Tanaka et al. 2014). In addition, 12 additional cell types were common between sheep and goats (Supplemental Tables S5, S6). Lymphatic endothelial cells (LECs) were found only in goats, but immunofluorescence experiments showed expression of the marker gene LYVE1 in the ruminal vascular walls of both species (Fig. 2B). This suggested the existence of LECs in the sheep rumen as well, although the discrepancy could be attributed to limitations in the current tissue dissociation and single-cell sequencing technologies (Kotliar et al. 2019). In sheep, the cell numbers obtained at each stage were more than those in goats, but the proportions of cells at each stage were similar between the species. The compositional ratio of each cell type was relatively consistent, with the least numbers of neurons and enteroendocrine cells in both the species. Although the cell types were highly similar between species, the subtypes of epithelial cells and Fib showed large differences in cell number and marker gene. Meanwhile, we detected the cell subtypes specific at particular stages by manual annotation. We observed similar cell subtypes and proportions as the integrated data set (Supplemental Fig. S3; Supplemental Note S4).
In general, the definition of cell types was comparable between sheep and goats, indicating a small species-specific effect on the cell types. Here, we identified a panel of novel marker genes to define these various cell types at different stages of rumen development, including those primarily expressed in epithelial cells and Fib (Supplemental Tables S7, S8).
Reconstruction of the cellular ecosystem of rumen tissues during maturation
To delineate the dynamic changes in cell type (and subtype) composition during rumen development, we compared the cell numbers of individual cell types across the 17 time points and six stages (Fig. 1A,B; Supplemental Tables S5, S6). We observed marked changes in cell numbers of the cell types during rumen development (Fig. 1A,B). Across various cell types, the cell number ranged from 314 (enteroendocrine) to 19,214 (basal cells_CA1_high) in sheep and 95 (enteroendocrine) to 12,220 (endothelial) in goats (Fig. 1A,B; Supplemental Tables S5, S6). Of these cell types, more than 20 were specific to particular stages. For example, PBCs were present only in the embryonic and prerumination stages, and BC_CA1_high cells, a BC subtype, were present only in the transition and rumination stages in sheep (Fig. 1A,B).
Notably, we observed the transition of cell types across the developmental stages (Fig. 1A,B; Supplemental Tables S5, S6). For example, the two stromal cell subtypes, stromal cells_PTN_high and stromal cells_POSTN_high, were present mainly in the early embryonic stage, suggesting their vital roles in the self-renewal and differentiation potentials of stromal cells at this stage (Huang et al. 2019). Fib are a main cell type of rumen connective tissue, and the changes in gene expression in these cells determine the developmental status of connective tissues (Wlaschek et al. 2021). In sheep, fibroblast_DLK1_high and fibroblast_PLAC9_high were the main Fib subtypes in the embryonic stages. However, the abundance of fibroblast_APOD_high increased after birth, making them the main Fib subtype in the prerumination stage (d0–d14), whereas fibroblast_MFAP5_high was the main Fib subtype in the transition and rumination stages (d21–d90) (Fig. 1A). For the epithelial cells, which were composed of BCs, SCs, and GCs, there were marked changes in cell numbers among various subtypes within each cell type during development (Fig. 1A). Endothelial cells (E) and immune cells (T and B cells) appeared at the earliest sampling time of e45, and the number of cells increased after birth. Mural cells (M) were most closely associated with endothelial cells (E). Neurons were present in the early embryonic stage, after which they were rarely or no longer detected. Neuroendocrine cells (NECs), similar to neurons, proliferated significantly after the late embryonic stage (E3) in sheep and the middle embryonic stage (E2) in goats (Fig. 1A,B). Similar developmental patterns were also observed in different goat cell types (Fig. 1B). Altogether, we generated a spatial and temporal representation of cellular diversity.
Trajectory analyses further supported consistency between the inferred pseudotime and the developmental stages of BCs, SCs, and Fib (Fig. 2C; Supplemental Figs. S4, S5). Additionally, we examined the relative gene expression dynamics for marker genes throughout the pseudotime period. The changes in expression of these genes across the pseudotime period were mostly congruent with those observed above (Fig. 2D; Supplemental Figs. S4, S5). For example, the expression of the marker genes carbonic anhydrase 1 (CA1) and carbonic anhydrase 3 (CA3) of BCs, which catalyze the reversible hydration of carbon dioxide and participate in a variety of biological processes, such as cellular respiration, acid–base balance, and saliva formation, peaked during the transition to rumination stages (Fig. 2D; Supplemental Figs. S4, S5). RNA velocity analyses have been implemented accordingly. We found that the analyses supported the differentiation of basal, spinous, and Fib subtypes, which is consistent with the results by the Monocle software (Fig. 2C; Supplemental Figs. S4, S5).
Differentially expressed genes among cell types/subtypes
For cell type–specific gene expression profiling, we identified differentially expressed genes (DEGs) between different cell types of sheep or goats using the Wilcoxon rank-sum test (Supplemental Tables S7, S8). Comparisons have been implemented between the target cell type and the cluster of all the other cell types (see the menu “FindAllMarker” in Seurat). Heatmaps showed the relative expression of the top 10 DEGs in each cell type (Fig. 3A). The two major cell types, namely, epithelial cells and Fib, displayed similar gene expression patterns among various cell subtypes in both species (Fig. 3A). In epithelial cells, Gene Ontology (GO) enrichment analysis of the DEGs revealed several common terms between goats and sheep, whose main functions were related to epithelial cell proliferation (e.g., JUN and SOX2), keratinocyte differentiation (e.g., SPINK5 and KRT17), and fatty acid (e.g., MCEE and PPARG) and ketone (e.g., PSMA5 and COQ7) metabolic processes (Fig. 3A; Supplemental Tables S9, S10). Ketone metabolic processes are considered characteristic indicators of rumen maturation (Pan et al. 2021a); thus, the presence of the epithelial cell subtypes BC_CA1_high and SC_KRT17_high in sheep and GC_RBP2 in goats was indicative of rumination. In Fib, functions of the top DEGs (e.g., LUM and COL1A1) and the significantly (Padj < 0.01) enriched GO terms (e.g., “extracellular matrix organization” and “regulation of canonical Wnt signaling”) indicated their important roles in providing structural support and communication with other cells of rumen tissue (Sorrell and Caplan 2009). Additionally, we found significant (Padj < 0.05) DEGs with important functions in other cell types, such as the genes CARD9 and PAK1 involved in innate immune responses, GPR183 and PTPRC involved in B cell activation, CORO1A and TNFRSF1B involved in T cell activation, and ACTG2 and MYH11 involved in muscle contraction in smooth muscle cells (SMCs) (Supplemental Tables S9, S10). Also, we examined the correlations of the relative expression levels of all the DEGs between sheep and goats. We observed high Spearman's rank correlation coefficients for the different cell types (e.g., epithelial cells, ρ = 0.74–0.85; Fib, ρ = 0.76–0.87; SMCs, ρ = 0.85), indicating convergent development in the gene expression signatures of ruminal cell types between ruminants (Supplemental Fig. S6A).
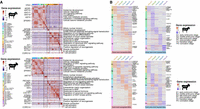
scRNA-seq-based gene expression in rumen cell atlases of sheep and goats. (A) Heatmap showing the top 10 expressed genes (row) in each cell (column) in sheep and goat rumen. The right panel shows the significant (Padj < 0.05) Gene Ontology (GO) terms for the marker genes in each cell type. (B) Heatmaps showing the up-regulated and down-regulated DEGs for each pairwise comparison between different stages (stages E2 vs. E1, E3 vs. E2, P1 vs. E3, P2 vs. P1, and P3 vs. P2) in sheep and goat rumen. The abbreviations of the cell names are shown in Figure 1.
Conserved dynamic patterns of gene expression between goats and sheep
Dynamic changes at developmental time points and between adjacent stages
To analyze the gene expression changes associated with functional transitions during rumen development, we identified up- and down-regulated DEGs by comparing gene expression at a time point to the average of all the other time points (Supplemental Fig. S6B–D; Supplemental Tables S11–S16). In sheep and goats, most of the DEGs (57%–80%) were detected at two and more time points, and the rest (20%–43%) were detected at a particular time point (Supplemental Fig. S6B). The total numbers of up-regulated DEGs were much higher at e45–e75 (stages E1 and E2) than at the other time points and then decreased until birth (Supplemental Fig. S6C), highlighting the two stages as a critical time period during rumen development. The number of down-regulated DEGs decreased during the embryonic stages but showed an increase after birth (Supplemental Fig. S6C). The top 50 up- and down-regulated DEGs showed similar patterns of increased or decreased relative expression during e45–e105 (stages E1 and E2) and d42–d90 (stages P2 and P3), but their expression patterns were different during the late embryonic (E3), prerumination (P1), and transition (P2) stages (e120–d35) (Supplemental Fig. S6D). This suggested that gene expression varied substantially during these stages. Additionally, we identified up- and down-regulated DEGs by comparing gene expression between adjacent stages. The DEGs associated with epithelial development, such as KRT15, KRT4, and KRT19, were up-regulated sequentially at the embryonic (E1–E3), prerumination (P1), and rumination (P3) stages, respectively, and those genes related to smooth muscle contraction, such as MYH11 and ACTA2, were up-regulated at the embryonic stage (Fig. 3B; Supplemental Tables S17, S18).
Next, we performed GO enrichment analysis of the up- and down-regulated DEGs at each time point (Supplemental Fig. S6E; Supplemental Tables S19–S22). In the early and middle embryonic stages (E1–E2), the up-regulated DEGs were significantly (Padj < 0.01) enriched in GO terms associated with cell proliferation (e.g., FGF7 and FGFR1) and differentiation (e.g., PDGFRA and BMP4), embryonic development (e.g., BMP4 and BMPR2), and immune regulation (e.g., RSF1 and POLR2F) (Supplemental Fig. S6E; Supplemental Tables S19–S22). In the late embryonic stage (E3), the up-regulated DEGs were significantly (Padj < 0.01) enriched in GO terms involved in “epidermis development” (e.g., KRT15 and KRT5), “cornification” (e.g., KRT15 and SPINK5), and “smooth muscle contraction” (e.g., MYH11 and ACTA2), which could account for the development of epithelium and Tm. In the prerumination stage (P1), the up-regulated DEGs were significantly (Padj < 0.01) enriched in GO terms related to “aerobic metabolism” (e.g., ATP6 and CYTB), “carbohydrate and lipid metabolism” (e.g., GPT and DECR1), and “immune response” (e.g., “response to interleukin-1,” ETS1, and CCL2). In the transition and rumination stages (P2–P3), the up-regulated DEGs were significantly (Padj < 0.01) enriched in GO terms related to “T cell activation” (e.g., IL6 and CD81), “regulation of epithelial cell apoptotic process” (e.g., IL6 and KDR), “regulation of cellular ketone metabolic process” (e.g., AVPR1A and CAV1), and “fatty acid metabolic process” (e.g., MCEE and PPARG) (Supplemental Fig. S6E), suggesting the progressive development of the rumen to the mature state.
Additionally, we performed GO enrichment analysis of the up- and down-regulated DEGs in four cell types (epithelium, endothelial cells, Fib, and immune cells). We focused on the adjacent stages in sheep and goats (Supplemental Fig. S6F; Supplemental Tables S23, S24). We found a few common GO terms in the enrichment analyses of up-regulated DEGs between adjacent stages in the same cell types, such as “epithelial development” (e.g., KRT15 and KRT5) and “extracellular matrix organization” (e.g., LUM and COL1A1) at the early embryonic stage (E2) in epithelium and stromal cells, respectively, and “neutrophil mediated immunity” (e.g., ATP6 and CYTB) at the ruminant stage (P3) in immune cells.
Convergent gene expression patterns between cell types at particular stages
In sheep and goats, we found common up- or down-regulated DEGs at individual developmental stages (Supplemental Fig. S7; Supplemental Tables S25–S27). For example, in the early and middle embryonic stages (E1–E2), we observed quite a few up- (e.g., TCF12, MDK, and BMP4) and down-regulated DEGs (e.g., SPINK5, KLF5, and KRT5) across multiple cell types (Supplemental Fig. S7). Six common up-regulated DEGs, namely, ACTA2, ACTG2, TPM2, CNN1, MYH11, and SMTN, all of which are related to actin production muscle contraction, were detected in SMCs in the late embryonic stage (E3). Subsequently, the number of up-regulated DEGs decreased, and their expression levels differed among the different cell types (Supplemental Fig. S7).
Gene expression specific to each cell type at individual developmental stages
Furthermore, we defined the DEGs specific for each cell type at individual developmental stages. As indicated by the varying sizes of the circles, rumen development showed specific impacts on various cell types at different developmental stages (Supplemental Fig. S8A; Supplemental Tables S28, S29). In both species, cell types such as epithelial cells (PBCs, BCs, SCs, and GCs), Fib, and nerve cells (Neu and NECs) showed significant variations in the number of DEGs among the six stages (Supplemental Figs. S8B, S9C). In general, the numbers of DEGs in sheep were lower than those in goats. Nevertheless, we identified the DEGs specific for particular cell types across all developmental stages in both species (Supplemental Fig. S8C; Supplemental Tables S30, S31), such as the up-regulated genes HSD17B2, KRT5, and KRT15 in BCs. Additionally, we observed common up-regulated DEGs, such as ANLN, ATAD2, and ATF7IP, and shared down-regulated genes, such as ALDH1A1, CUTA, and DSP, in mitotic cells (MCs) in both species, and these genes might play key roles in mitosis in ruminants (Oegema and Hyman 2006). All together, we found a few common genes that showed differential expression in different cell types at different developmental stages in ruminants, which might be beneficial to our understanding of the gene expression patterns of different cell types during rumen development.
Transcriptional regulatory network analysis
To better understand the underlying molecular mechanisms driving the development of different cell types, we predicted core transcription factors (TFs) regulating these target DEGs during rumen development (Fig. 4; Supplemental Fig. S9; Supplemental Tables S32, S33). Of the 111 (54 up-regulated and 57 down-regulated) TFs in sheep and 99 (52 up-regulated and 47 down-regulated) TFs in goats, we found 23 up-regulated and 23 down-regulated TFs that were shared between the species (Supplemental Fig. S9A,B). Up- and down-regulated TFs as well as their target genes were identified in all the stages in goats. In sheep, up-regulated TFs were not found in the rumination stage, whereas down-regulated TFs were not found in the embryonic E1 and E2 stages (Supplemental Fig. S9C). In particular, a few up- and down-regulated TFs were specific to particular stages of both species, such as six (HDAC2, MEIS1, MEIS2, ETS2, FOSB, and ATF3) at the early embryonic stage (E1), 11 (ATF3, FOS, IRF1, JUN, KLF10, MEIS1, HES1, MEIS2, TRIM28, KLF2, and NFIX) at the middle embryonic stage (E2), one (JUN) at the prerumination stage (P1), eight (MEIS2, RBPJ, TCF21, TCF7L2, TCF4, MEIS1, TCF12, and SMARCC1) at the transition stage (P2), and six (CUX1, FOS, JUN, TCF12, TCF21, and YY1) at the rumination stage (P3) (Fig. 4A). To better understand the underlying molecular mechanisms driving the development of different cell types, AUC scores were calculated for these above TFs in various cell types (Fig. 4B; Supplemental Tables S34, S35). These TFs were found to act in the same or different cell types at different stages in both species. For example, TFs such as ATF3, ETS2, and RBPJ were all expressed in B cells, whereas TFs such as HDAC2 and KLF2 were expressed in epithelial cells, Fib, or neurons, suggesting that they could play multiple roles in regulating epithelial integrity, inflammation, cell cycle progression, or developmental events (Tanigaki et al. 2002; Li et al. 2018). These data revealed cross-species TFs with stage-specific expression and differential expression among different cell types.
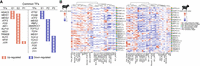
Changes in core regulatory transcription factors (TFs) during rumen development. (A) Common up-regulated and down-regulated TFs across the six major developmental stages. (B) Heatmap of the area under the curve (AUC) scores of TF motifs estimated per cell of the cell types across the six major developmental stages. The abbreviations of the cell names are shown in Figure 1.
Further, we performed the TF target gene enrichment analysis at the six developmental stages. The motifs ranking the top 50 potential target genes of the TFs were included in the analysis, and significant GO terms were predicted at each stage. In both sheep and goats, target genes for most of the TFs (e.g., MEIS1 [+], MEIS2 [+], and YY1 [+]) showed similar GO terms at different stages, and multiple TFs (e.g., FOS [+], FOSB [+], and ATF3 [+]) were observed to be involved in the same functions (Supplemental Fig. S9D). Target genes for some TFs (e.g., JUN [+] and CEBPD [+]) were significantly enriched in different biological processes at different stages, with more genes related to embryonic development and cell proliferation in the embryonic stages and more genes for the up-regulated TFs involved in immune-related functions in the postnatal stages (Supplemental Fig. S9D; Supplemental Tables S36, S37).
Cellular communication patterns associated with rumen functions
To quantify the ligand–receptor (LR) interactions during rumen development, we computed the numbers of LR pairs among all pairs of cell types as well as in autocrine signaling. We identified 560–729 and 700–741 kinds of potential pairs of LR interactions at the six stages of sheep and goats, respectively (Supplemental Tables S38, S39). In general, goats showed more LR interactions than sheep. We observed that the cellular communication patterns of the two species were similar at and before the transition stage but became markedly different at the rumination stage, which could be owing to the difference between the forages typically fed to sheep and goats (Supplemental Fig. S10). In addition, cell–cell communications were enhanced in the prerumination stage (P1) compared with the late embryonic stage (E3), whereas many kinds of communications were abolished when the animals began to ferment and digest forages in the transition and rumination stages (P2–P3) (Supplemental Fig. S10).
To further identify the functions of the LR interactions affected during rumen development, we performed GO analyses of the common gene pairs involved in the up- or down-regulated interactions between sheep and goats. Most of the common interactions occurred between cytokines and cytokine receptors, which were associated with the cell proliferation and robust immunomodulatory functions of rumen tissues (Fig. 5A,B). For example, amphiregulin (AREG) interacts with the epidermal growth factor receptor (EGFR) to trigger numerous signaling cascades that mediate cell survival, proliferation, and motility (Wang et al. 2020). AREF-induced EGFR signaling was increased during the early and middle embryonic stages (E1–E2) (Fig. 5B). Additionally, the interactions between interleukin 1 receptor antagonist (IL1RN; also known as IL1RA) and interleukin 1 alpha (IL1A; a highly active proinflammatory cytokine) and the PROS1-AXL pathway, which regulates intrinsic mesenchymal signaling as well as the extrinsic immune microenvironment (Sadahiro et al. 2018; Kitanaka et al. 2019), were enhanced in multiple stages (e.g., E2, P2 and P3) (Fig. 5B).

Changes in ligand–receptor (LR) interactions between different cell types in the comparisons of adjacent stages (stages E2 vs. E1, E3 vs. E2, P1 vs. E3, P2 vs. P1, and P3 vs. P2) in sheep and goats. (A) Functional enrichment analysis showing the GO terms for the common up- or down-regulated LR interactions (Padj < 0.05). (B) Common interaction pairs and their frequencies (Freq).
Conserved marker genes, TFs, and LR interactions among ruminants
We sourced single-cell data of stomach or rumen tissue from humans, mice, monkeys, cattle, and sheep from the public repositories (Supplemental Table S40). After quality control (QC), we obtained the final data sets of 39,874, 30,000, 5355, 45,303, and 53,260 cells for the above five species, respectively. UMAP analysis revealed 11–17 major cell types (Supplemental Fig. S11A–E). We identified 17 and 12 newly added cell types/subtypes in the cattle and sheep rumen tissues, respectively. We found a set of the same marker genes across the three ruminants (sheep, goats, and cattle), such as KRT5 for BCs and DAPL1 for BCs (Supplemental Fig. S11F; Supplemental Table S41). In the nonruminant species (human, monkey, and mouse), the cell types were mainly composed of epithelial cells (e.g., gastric chief cells in humans and Pit cells in monkeys), endothelial cells, stromal cells, SMCs, neurons, and immune cells (e.g., B cells in humans and monkeys and macrophages in mouse). We found similar expression patterns of the marker genes in the same cell types, such as PGC and CLDN18 in epithelial cells, COL3A1 and COL1A2 in stromal cells, and JCHAIN and CAP3 in immune cells (Supplemental Fig. S11F; Supplemental Table S41).
To identify the conserved cellular communications among the ruminants, we compared the LR interactions among the three ruminants and those among the three nonruminant species. We found four EGFR-related LR interactions only expressed in the ruminants, particularly in the rumen epithelial cells (Fig. 6A; Supplemental Table S42). Of the LR interactions associated with immune responses, the expression of CD44-HBEGF was much higher in cattle and goats than in the three nonruminants. In addition, we predicted the core TFs and calculated the AUC values among the cell types (Fig. 6B; Supplemental Table S43). Eight TFs (ATF4, CEBPD, FOS, IRF1, JUN, NFIX, PITX1, and YY1) were shared among ruminants, and three TFs (ATF3, FOSB, and JUNB) were shared across the six species (Fig. 4B; Supplemental Table S33). These TFs were highly active in distinct cell categories. For example, ATF3, which functions in embryogenesis, a regulator of metabolism and immunity (Ku and Cheng 2020), was highly active in embryo cells. YY1, which functions in embryogenesis, differentiation, and cellular proliferation (Gordon et al. 2006), was highly active in ruminants.

Conserved TFs and cell communications in rumen or stomach tissues across species of sheep (including download data), goats, cattle, monkeys, mice, and humans. (A) Dot plots showing the cell–cell interaction pairs in the indicated cell types. The size of the dots indicates the −log10 P-value, and the colored dots show the mean expression levels. (B) Heatmap of the area under the curve (AUC) scaled scores of TF motifs estimated per cell of the cell types.
Patterns of microbial community change
We built metagenome assemblies and predicted 18,103,515 open reading frames (ORFs) with an average length of 538 bp (Supplemental Table S44). To reveal the rumen microbial composition and relative abundances, we identified 23 bacterial phyla (Supplemental Fig. S12A) and 278 bacterial genera (Supplemental Table S45). We examined the dynamic temporal patterns of ruminal microbial communities along the six main developmental stages in goats and sheep (Supplemental Fig. S12B). The microbial communities at the early embryonic stage consisted mainly of aerobic bacteria (e.g., Phyllobacterium spp., Sphingomonas spp., and Agrobacterium spp.). At the transition and rumination stages, the communities were dominated by anaerobic bacteria (e.g., Prevotella spp., Succiniclasticum spp., and Clostridium spp.), which ferment carbohydrates to produce volatile fatty acids (VFAs). The microbial compositions and abundances showed similarity between sheep and goat samples, particularly in the transition and rumination stages (Supplemental Fig. S12B). Following the changes in microbial communities, the functional annotation of ORFs also showed dynamic patterns across these successional stages (Supplemental Fig. S12C). Gene annotation identified a total of 410 CAZy families, 414 KEGG pathways, and 25 clusters of orthologous groups (COGs). We detected, in contrast to those at the postpartum stages (P1, P2, and P3), fewer specific functional categories of CAZymes, KOs, and COGs at the embryonic stages (E1, E2, and E3), suggesting that the internal environment and microbiota in the embryonic rumen were relatively stable.
Hierarchical clustering of correlation scores between the expression of DEGs in single-cell transcriptome sequencing and the abundances of bacterial genera revealed five bacterial clusters, which were dominant at the corresponding embryonic and prerumination stages (Supplemental Fig. S12D). One bacterial cluster (e45–e75) consisted of Sphingomonas, Cutibacterium, Phyllobacterium, Agrobacterium, Rhizobium, and Pseudomonas. It was strongly positively correlated with the expression of 67 development-related (e.g., IGF2, BMP4, and FSTL1) and immune-related (e.g., the COL gene family and CCND1) genes, whereas it was highly negatively correlated with the expression of 36 DEGs (e.g., COX1, COX2, COX3, and ACAA2) related to energy metabolism, cell migration, and immune and fatty acid metabolic processes. Conversely, another bacterial cluster (d21–d90) contained species mainly from the genera Prevotella, Butyrivibrio, Clostridium, Succiniclasticum, and Alloprevotella and showed reversed patterns of correlation with the relative expression of the same two sets of DEGs (Supplemental Fig. S12D).
To dissect the correlations between ruminal cell types and the ruminal microbiota or their predicted KEGG pathways, we calculated their Spearman's correlation coefficient based on the first eigenvector of principal component analysis (PCA) during the developmental stages. We observed two clusters of cells with different correlation patterns with bacterial genera and pathways (Fig. 7A,B), suggesting the responses of particular ruminal cells to specific microbiomes. One cluster, which consisted of cell types such as subtypes of BCs (e.g., Basal cells_CA1_high and Basal cells_SELENBP1_high), SCs (e.g., Spinous cells_KRT15_high and Spinous cells_KRT17_high), GCs (Granular cells_S100A8_high), Fib (Fibroblast_APOD_high and Fibroblast_MFAP5_high), and T, B, and endothelial cells, was positively correlated with seven bacterial genera, including Alloprevotella, Bacteroides, Butyrivibrio, Clostridium, Mannheimia, Prevotella, and Succiniclasticum (Fig. 7A), and 12 pathways involved in biosynthesis and metabolism in the rumen microbiome (Fig. 7B). The other cluster consisted of stromal cells, SMCs, enteroendocrine cells, and neuron cells and subtypes of BCs, SCs, and Fib and was negatively correlated with 12 bacterial genera (Agrobacterium, Brachybacterium, Cutibacterium, Enterococcus, Escherichia, Klebsiella, Phyllobacterium, Pseudomonas, Rhizobium, Serratia, Sphingomonas, and Streptococcus) in both sheep and goats. Meanwhile, the second cluster in general showed negative correlations with the above 12 pathways but showed positive correlations with the other six pathways, such as “arginine and proline metabolism,” “quorum sensing,” “butanoate metabolism,” “fatty acid degradation,” “oxidative phosphorylation,” and “glyoxylate and dicarboxylate metabolism.” In particular, 25 up-regulated DEGs (P2 vs. P1) in epithelial cells and 83 up-regulated DEGs (P2 vs. P1) in Fib were identified to be significantly (P < 0.05) correlated with Prevotella copri (Supplemental Tables S50, S51).
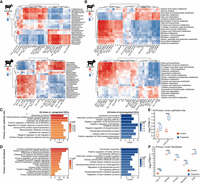
Ruminal microbiota correlations with single-cell transcriptome sequencing in sheep and goats, and transcriptomic analysis of co-cultured rumen cells with Prevotella copri. (A) Correlations of host rumen cell types with the dominant ruminal microbiota. Heatmap shows the Spearman's correlation coefficient between the Eigengene/PC1 value of cell types and the abundances of dominant ruminal microbiota (P < 0.05). (B) Correlations of host rumen cell types with microbial KEGG pathways. Heatmap shows the Spearman's correlation coefficient between the Eigengene/PC1 value of cell types and the abundances of selected microbial KEGG pathways (P < 0.05). The abbreviations of the cell names are shown in Figure 1. (C,D) GO terms (P < 0.05) for up- and down-regulated DEGs (P < 0.05 and |logFC| > 0.75) between the treatment group (co-cultured with P. copri) and control group in primary rumen epithelial cells (C) and primary rumen Fib (D). (E,F) Boxplots showing expression levels of overlap between up-regulated DEGs (treatment vs. control) in primary rumen epithelial cells (E) and primary rumen Fib (F) with up-regulated DEGs (stages P2 vs. P1; Padj < 0.05 and |logFC| > 0.25), which were significantly (P < 0.05) correlated with P. copri. (****) P < 0.0001, (**) P < 0.01, (*) P < 0.05.
Validation of the interactions between microbes and cells
To validate the interactions between specific microbes and cells, we co-cultured primary rumen epithelial cells and Fib with P. copri and routine media in vitro, separately (Supplemental Fig. S13A). Then, we performed RNA-seq for the total RNA from cultured cells and conducted differentially expression analysis between treatments and control in each cell types. We observed that t-SNE was driven primarily by the cell types, followed by the treatment (Supplemental Fig. S13D,E). We identified 295 (140 up- and 155 down-regulated) and 507 (218 up- and 289 down-regulated) DEGs (P < 0.05 and |logFC| > 0.75) in primary rumen epithelial cells and Fib, respectively. GO enrichment analysis suggested that P. copri stimulates the development of rumen epithelial cells (Fig. 7C) and enhances the immunity response and energy metabolism of rumen Fib (Fig. 7D). Genes in association with P. copri above (Supplemental Tables S50, S51) and the up-regulated DEGs (treatment vs. control) were significantly (P < 0.05) enriched in several common GO terms, which were mainly involved in catabolic process (cellular nitrogen compound, aromatic compound, and ribonucleotide) in epithelial cells (Supplemental Fig. S13F) and multiple cell responses (e.g., molecule of bacterial origin, xenobiotic stimulus, and nutrient and chemical stress), cell chemotaxis, and extracellular matrix organization in Fib (Supplemental Fig. S13G).
In particular, two up-regulated genes (CA1 and UGT1A1) in epithelial cells and five up-regulated genes (PTGS2, CCL11, HSPA8, TNFAIP6, and TXN) in Fib showed significantly (P < 0.05) positive associations with P. copri in the microbiota-gene association analyses above (Fig. 7E,F). Compared with the overlap expected by chance, there was a significant (P < 0.001) excess of overlapping genes that were shared between experimental validation and association analyses (Supplemental Fig. S12H,I; Lv et al. 2014). We validated the associations between P. copri and of the two cell types (i.e., epithelial cells and Fib). Overall, we showed the robustness and validity of the classifications and dynamic patterns through correlation across cellular types, developmental stages and microbiomes, and convergence across species. Our results showed that the rumen microbes had different effects on the various types of rumen cells (Fig. 8).

A summary model of the genetic basis underlying the rumen functions along the development stages.
Discussion
We produced a comprehensive map of the cell type composition of the rumen during the developmental time course, including as many as 30 cell types (and subtypes). In addition to the major cell types, such as epithelial cells, we revealed other cell types, such as stromal cells, T cells, and B cells, which were not reported previously (Gao et al. 2021; Xue et al. 2022; Yuan et al. 2022) but play vital roles in the immunological regulation of the rumen. This is especially relevant for the rumen, which is exposed to anaerobic bacteria and dietary pathogens. Additionally, the cell types such as NECs, neurons, interstitial cells of Cajal, SMCs, and endothelial cells detected here provide structural and physiological (e.g., neurotransmission) support for rumen mobility and contraction (Wang et al. 2018). In particular, we identified several novel subtypes in BCs (four), SCs (seven), GCs (two), and Fib (four) based on their respective highly expressed genes (e.g., KRT15, KRT17, IGFBP6, IGFBP2, MFAP5, TNFAIP6, and MGP), which played important roles in mediating rumen keratin epithelial growth, epithelial cell proliferation, VFA intake, and inflammatory responses. Additionally, we defined a few new marker genes for the cell types. We observed a high concordance of the emergence time for different cell types as inferred from the expression patterns of the marker genes, cell trajectory, and histological structure. These marker genes could therefore be used to resolve ambiguous cell type annotation in other single-cell data sets and in tissue analysis (Muhl et al. 2020). The functions of the marker genes and the highly expressed genes have broad implications for physiology, metabolism, and medicine (e.g., gastric mucosal fibrosis), providing insights into the functions, regulation, and interactions of the known and new cell types (Travaglini et al. 2020). The marker genes and cell resources annotated here can be used in future similar studies on the stomachs of other animals. In particular, the resources pave an important foundation in deciphering the genetic mechanisms of ruminants, including domestic and wild ruminants, in adapting to different climatic environments. In addition to the highly conserved cell types and marker genes, our results showed high correlations of gene expression patterns between sheep and goats, suggesting evolutionary conservation of the gene expression patterns of homologous cell types.
We characterized the changes in the number and functions of DEGs along the six main developmental stages and observed conserved patterns of dynamic gene expression in the developing rumen between sheep and goats, indicating that these cell types had comparable developmental maturity. We also identified critical features of the temporal expression patterns. First, the early and middle embryonic stages showed the largest number of up-regulated DEGs, acquiring the largest gene expression signatures and highlighting the two stages as a critical time period during rumen embryogenesis. Second, relative gene expression varied substantially during the late embryonic and prerumination stages during this period, which was consistent with the changes in the cell type and microbial components and suggested the potential occurrence of changes in gene regulation during these stages. Third, the genes with significantly up-regulated expression had specific functions at particular developmental stages, shedding light on the main structural (e.g., FGF7, FGFR1, PDGFRA, BMPR2, BMP4, KRT15, and KRT5; E1–E3 stages) and metabolic (e.g., ATP6, CYTB, GPT, and DECR1; P1–P3 stages) functional changes during the physiological stages of rumen maturation. Notably, we revealed quite a few highly expressed genes (e.g., IL6, CD81, RSF1, POLR2F, ETS1, and CCL2) related to innate immune responses across all developmental stages; these genes affect the rumen microbial community, rumen health, and eventually animal health. Neurons expressed abundant transcripts of ATP6 and CYTB in the early postnatal stage (e.g., P1 and d21 in P2) (Supplemental Figs. S14, S15; Supplemental Table S46), indicating more ATP production and mitochondrial function required in the early development after birth. Previous bulk RNA-seq studies have profiled rumen tissue samples from sheep, goat, and cattle to understand the gene expression dynamics in the postpartum developmental stages (Bush et al. 2019; Malmuthuge et al. 2019; Pan et al. 2021b). These studies observed increased expression of ATP6 and CYTB in rumen tissue from young and adult cattle, which was supported by the results of this study, showing increased expression of ATP6 and CYTB after birth using the scRNA-seq.
Additionally, the previous bulk RNA-seq studies found that COL1A1 and COL1A2, which are associated with biosynthesis of connective tissue and collagen, showed more than twofold up-regulation during 1–8 wk after birth in the rumen of sheep and goats (Bush et al. 2019). However, no significantly increased expressions of these two genes were found in any associated cell types (e.g., Fib) after birth (Supplemental Figs. S16, S17). Instead, the numbers of some associated cell types (e.g., Fib) increased gradually after birth in rumen tissues (Fig. 1B), which suggested that the increase of cell number affects the expressions of COL1A1 and COL1A2 in developing rumens of sheep and goats (Bush et al. 2019). KRT4, which associated with epithelial development, showed higher expression in the rumens of breast-milk-fed lambs in contrast to starter-feed-fed lambs (Wang et al. 2016). In our study, KRT4 showed up-regulated patterns around the birth in spinous (Spinous cells_CA3_high and Spinous cells_DAPL1_high), granular (Granular cells_KRT4_high and Granular cells_RBP2_high), and basal (Basal cells_CA3_high) cells in goats (Supplemental Fig. S18). Our results improved understanding of the gene expression dynamics in various cell types within the rumen around the birth.
Normal rumen development is achieved by the orchestrated regulation of the TF network that is synergistically controlled by different signaling pathways at different developmental stages. We revealed critical TFs that are potentially important for the differentiation and maturation of different cell types. During the early embryonic stage (E1), genes (e.g., ATF3, ETS2, and RBPJ) regulate immune competence to promote the intensity of immune responses from immune B cells. During the rumination stage when the rumen has matured, we found that the TCF ligands TCF12 and TCF21 may activate the TCF signaling pathway in epithelial cells and Fib to regulate fiber digestion and VFA absorption. Moreover, we observed that eight conserved TF regulons (ATF4, CEBPD, FOS, IRF1, JUN, NFIX, PITX1, and YY1) were highly active in distinct cell types across the ruminants (sheep, goats, and cattle). Thus, the TF regulatory programs validated the hypothesis that the similarities in gene expression among cell types can also arise from convergent or concerted evolution (Arendt et al. 2016). TFs such as RBPJ, HDAC2, and KLF2 were active in multiple cell types, suggesting that they could play multiple roles in regulating epithelial integrity, inflammation, cell cycle progression, or developmental events (Tanigaki et al. 2002; Li et al. 2018).
We found that intercellular communications were altered from the embryonic to postpartum stages, which could be explained by the metabolic changes that occur after birth. Additionally, goats had an increased number of communications and more cell types involved in the communications than sheep. Differences between the forages typically fed to goats and sheep could account for the marked difference in postpartum patterns of communications among cell types between the two species. Additionally, cellular interactions associated with the immune response (e.g., PROS1-AXL pathway), cytokine secretion (e.g., IL1 signaling pathway), and cell proliferation (e.g., AREF-induced EGFR signaling) were identified during the early and middle embryonic stages (E1 and E2), indicating that cells in these stages were critical for developing essential immune functions and structural tissues.
To date, the majority of bulk RNA-seq on the stomachs of herbivores have focused on ruminants but seldom on nonrumination herbivores. This could be ascribed to the fact that the rumen and stomachs are not the main sites for peptide and amino acid absorption. However, some genes (e.g., PPARG and MCT1) showed similar expression patterns in the cecum and rumen, two important fermentation organs, in herbivores (Supplemental Tables S46, S47).
Here, we identified a variety of conserved cellular connections exclusively present in ruminants (Supplemental Table S42). Although the evolutionary mechanism of the ruminant rumen is still disputed, the emergence of the rumen was suggested to be a consequence of ecophysiological adaptation (Hofmann 1989). However, there remains a large gap between rumen structural evolution and cellular communication modifications. Thus, our study could provide some clues regarding the mechanisms underlying the origins and evolution of the rumen at the cellular and molecular levels.
It was discovered that beneficial or nonpathogenic microbes were found in the fetus before birth in multiple tissues such as placental tissue (Aagaard et al. 2014), umbilical cord blood (Jiménez et al. 2005), amniotic fluid (DiGiulio 2012), fetal membranes (Steel et al. 2005), and meconium (Moles et al. 2013). We observed a low microbial diversity in the rumen exactly before birth (i.e., e120 for goat and e135 for sheep) (Supplemental Fig. S12A), which was consistent with that in cecal microbiota of lambs during parturition (Bi et al. 2021). However, a dynamic microbial community, especially for even higher microbial diversity in the early embryonic developmental stage (Supplemental Fig. S12B), was found in prenatal rumens of sheep and goats. Furthermore, Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes were the core phyla dominating the rumen microbiota, with Phyllobacterium, Sphingomonas, and Cutibacterium as dominant genera from e45 to e75. As nonfermenting bacteria, Phyllobacterium, Sphingomonas, and Cutibacterium could survive in low concentrations of nutrients (Liu et al. 2014) from placenta (Leoni et al. 2019). The human fetus was believed to swallow amniotic fluid early from the late second trimester of pregnancy (DiGiulio 2012), for prenatal microbial colonization inside rumen. Also, methanogens and fibrolytic bacteria were identified in the GIT and feces of calves <20 min after birth (Guzman et al. 2015; Supplemental Table S48).
Rumen microbiota dysfunction for lambs would increase the risk of diarrhea from sucking and weaning to grass feeding. We revealed dynamic changes of the ruminant rumen microbiome of lambs of sheep and goats in the postpartum developmental stages (from d0 to d90). The d0 is the key point during the transition from embryonic rumen development to postpartum rumen maturation. At the day of birth (d0), Escherichia was the most prevalent genera in goats, which is consistent with the observation that phylum Proteobacteria far exceeded the other phyla, with the majority from the genus Escherichia at d0 (Jiao et al. 2015). Also, Escherichia coli showed the highest relative abundance in all the prenatal cecal content samples (Bi et al. 2021). Bacteroidetes and Firmicutes were also found to be predominant phyla in postpartum early developmental stages in other ruminants (Wang et al. 2016). Bacteroides have the effects of protein and fat fermentation (Wu et al. 2011) and could be helpful for milk digestion during the early rumen development, as after weaning, Bacteroides was found with a significant reduction in both ruminants and nonruminants.
Bacteroides increased significantly from d0 to d15 after birth, which was followed by a decrease to lower levels for the later age groups in buffalo (Koringa et al. 2019). The bacterial community abruptly changed between d2 and d3 after birth, and Bacteroides became the dominant genera until d12 in cattle (Rey et al. 2014) and around weaning in rabbit gut (Fang et al. 2020). Here, cellulosic bacteria were the most popular genera in the late developmental stage (d21 to d90) of both sheep and goats. All these observations reflected the increase of a plant-rich diet during the transition from the preruminant to the ruminant digestion phases. The dominant genus Prevotella, which could produce propionates (Koringa et al. 2019), was found at birth and increased gradually up to the maximal relative abundance at d135 after birth in buffalo (Koringa et al. 2019) and yaks (Huang et al. 2021). With increasing solid food intake, Prevotella were found to become dominant in cattle and goats as well (Rey et al. 2014; Jiao et al. 2015). The relative abundance of the Prevotella genus was correlated with the rumen maturation of buffalo (Koringa et al. 2019) and was also positively correlated with the rumen weight of goats (Jiao et al. 2015). Diet potentially manipulated rumen microbiota, and meanwhile, the fermentation products of solid diet and associated microbiota could regulate gene expression to improve rumen tissue development (Wang et al. 2016). Also, butyrate by the microbiota played a role in physical and metabolic development of ruminal epithelium (Chai et al. 2021; Supplemental Table S48). Taken together, the rumen microbiota composition was associated with rumen development and diet.
In comparison, nonrumination herbivores (e.g., horse, donkey, and rabbit) showed significantly different stomach microbiota compositions during the development, as enzyme-based chemical and fermentative digestions were found in the stomach of nonruminants and ruminants, respectively. Actually, the cecum and gut seem to be more important in the studies of nonruminants, for example, rabbits (Paës et al. 2022). Previous studies have shown that Firmicutes and Bacteroidetes were the dominant microbes in the stomach or rumen of adult herbivores (Supplemental Table S49; Jin et al. 2018; Guo et al. 2022).
We correlated microbiota and cell types, as well as their gene expression levels, and detected interconnections among microbial metabolism and cell functions (Supplemental Fig. S12D). On the one hand, we obtained a better understanding of the rumen microbial groups encoding the vital gene functions essential for carbohydrate utilization and metabolism or mucosal immunity. We also characterized the specific interactions between the microbes and various types of ruminal cells identified in this study (Fig. 7A,B). In addition to the metabolite-mediated interactions between microbes and ruminal cells, our integrative analyses were helpful for inferring the potential indirect effects of microbial proteins on host cells via a signaling network. In particular, we observed interactions between immune cells (T and B cells) and microbiomes that were involved in biosynthesis and metabolism. Thus, our results provide us with a fundamental framework to better understand the functions of microbial genomes by their interacting genes and to design strategies for selecting and manipulating rumen microbiomes to improve the metabolic and immune traits of ruminant animals (Xue et al. 2022).
In conclusion, we combined comprehensive scRNA transcriptomics and metagenomics to explore the histomorphometric, genetic, and microbial development of the rumen at high-molecular and temporal resolution. Our results collectively provide key insights into the genetic mechanisms (e.g., critical genes, key TFs, and regulatory pathways) underlying the particular functions of the rumen. Additionally, the findings will enable the development of future interventions for improving the performance and health of ruminant animals and will be useful for investigations of the innate immune functions and dietary fiber digestion in the stomachs of other mammals, including humans. These resources thus lay the foundation for future research (e.g., research on mammalian gastrointestinal organogenesis, differential digestive functions of the stomach, and gastric diseases) and enhance our understanding of rumen biology.
Methods
Animals and tissue sampling
All animal experiments were performed in accordance with guidelines and regulations approved by the institutional animal care and use committee of China Agricultural University (CAU20160628-2) and the local animal research ethics committee. Native Chinese populations of sheep (Wadi sheep) and goats (Chengdu gray goat) were included in the study. Wadi sheep were housed in the experimental farm of the Shandong Binzhou Academy of Animal Science and Veterinary Medicine, and Chengdu gray goats were housed in the Chengdu gray goat breeding farm. All animals were fed hay and silage corn and had ad libitum access to water and mineral salt.
We collected 17 rumen tissues from 17 sheep embryos or cubs and 17 rumen tissues from 17 goat embryos or cubs (Supplemental Table S1). Rumen tissues were mainly obtained from sheep and goats at six main developmental stages (the early embryonic [E1: 45–60 d after conception], middle embryonic [E2: 75–105 d after conception], late embryonic [E3: 120–135 d after conception], prerumination digestion [P1: 0–14 d postpartum], transition [i.e., the period covering the move from the prerumination to the rumination stage; P2: 21–49 d postpartum], and rumination digestion [P3: 56–90 d postpartum]). These stages were dissected from embryos or animals at 17 time points: e45 (45 d after conception), e60, e75, e90, e105, e120, e135, d0 (the day of birth), d7, d14, d21, d28, d35, d42, d49, d56, and d90. One rumen tissue sample was harvested at each time point in each of the two species (Supplemental Table S1).
Immediately after carotid artery sacrifice, we dissected the rumen from embryos or animals. The whole-rumen tissues were collected in the embryo and prerumination stages, and the wall tissues at four locations were collected in transition and rumination, representing various parts of rumen such as Saccus dorsalis, Saccus ventralis, and Saccus cecus caudoventrali (Supplemental Fig. S1A). Ruminal tissues from the embryos and the animals in the prerumination stage were obtained by Cesarean delivery (C-section). Tissue samples were harvested and then washed with PBS. Ruminal tissues from the animals at the transition and ruminant stages were stripped of the muscularis and serosa, rinsed with precooled PBS containing four antibiotics (4% penicillin-streptomycin solution and 4% gentamicin-amphotericin B solution), and then cleaned with PBS only (Klotz et al. 2001). The mature rumen walls developed into four layers, including an internal epithelium (E), a middle layer of lamina propria and submucosal tissue (Lp + Sb), a Tm, and an external layer or serosa (S). The rumen epithelium acts as a protective barrier between the rumen and the host, and has been the focus of previous researches.
During the embryonic and the prerumination stages, it is difficult to dissect the muscularis from the middle layer of the lamina propria and submucosal tissue. At the transition stage for the rumen, the muscularis thickens rapidly, whereas the epithelium is highly keratinized. Because of the limitation of the pore size of the Chromium Controller instrument (10x Genomics), single-cell transcriptome sequencing is not suitable for the muscle fibers. To obtain as many epithelial cell subtypes as possible, we removed the muscularis and took only ruminal epithelial tissue at the transition and ruminant stages. The sliced rumen tissues of each sample were then divided into three parts: One was stored in tissue storage solution (Miltenyi Biotec) at 0°C for single-cell sequencing; the second was placed in a frozen storage tube, kept in liquid nitrogen for >30 min, and then transferred to a −80°C freezer for frozen section histological analysis; and the third was fixed in 4% paraformaldehyde solution (Beijing Solarbio Science & Technology) for standard histological examination.
Rumen digesta collection and metagenomic sequencing
Whenever possible, rumen content (∼10–30 mL) was taken immediately after the rumen was opened and was drained using four layers of sterile medical gauze. Thirty-one rumen digesta samples were collected from the same embryos or postnatal animals as the rumen tissues were dissected (Supplemental Table S44). All collected fresh samples were frozen in liquid nitrogen for >30 min and then stored in a −80°C freezer until needed for DNA isolation. After DNA extraction, assessment of DNA integrity, concentration, and quality (Supplemental Methods), metagenomic libraries with an insert size of 350 bp were then prepared from high-quality genomic DNA using an Illumina TruSeq DNA PCR-free library preparation kit (Illumina) and sequenced on an Illumina NovaSeq6000 platform (Illumina, Novogene Corporation). From the 31 samples, we generated 709.75 Gb of Illumina data and approximately 2.37 billion reads with a length of 150 bp (Supplemental Table S44).
Histological analysis
Frozen or normal rumen tissues for hematoxylin and eosin (H&E) staining were processed following the normal procedures such as fixation, dehydration, incubation, embedding, sectioning, and staining. Five histomorphometric variables (e.g., papillae base width and length, epithelium thickness, lamina propria and submucosa thickness, and Tm thickness) of the rumen of the sheep and goats were measured on the sections at the 17 time points. For each variable, 200 measurements were examined in five to 10 fields of view per section using ImageJ software v1.8.0 (Schindelin et al. 2015).
Single-cell RNA-seq library construction and sequencing
A single-cell RNA-seq library was constructed following a modified protocol from previous investigations (Supplemental Table S1; Supplemental Methods; Ma et al. 2020). The generated libraries were sequenced on an Illumina NovaSeq 6000 (Illumina), and paired-ended 150-bp (PE150) reads were produced for downstream analysis (Supplemental Methods).
scRNA-seq data preprocessing
Chromium scRNA-seq data were processed for demultiplexing, alignment, and read counting following the set of 10x Genomics official analysis pipelines using CellRanger v4.0.0 software (https://www.10xgenomics.com/) with default settings (see also Supplemental Information). We obtained a mean number of 10,327 cells per sample (range: 6033–21,074), a mean fraction of reads mapped confidently to the genome of 87.88% (±SD, 3.58%), a mean number of 569,230,327 (±84,664,892) reads for each scRNA library, a mean number of 60,473 (±20,840) reads per cell, and a mean sequencing saturation of 64.73% (±10.82%) (Supplemental Table S2). The pipeline output files containing gene expression matrices and UMI information from CellRanger software were then used in downstream analysis and visualization.
Post CellRanger assessment QC
Post CellRanger assessment QC to remove the low-quality cells was conducted based on the matrices, such as UMI counts, the number of genes, and the percentage of mtDNA genes, using the Seurat v4.0 package in R v4.2.0 (R Core Team 2022). After QC, a total of 183,802 cells remained with the capture of 6227–17,487 cells for sheep (a mean number of 10,812 cells per library) and 5418–11,930 cells for goats (a mean number of 7414 cells per library) (Supplemental Table S2). To reduce the technical batch effect, the gene expression data were then natural log-transformed and normalized for scaling the sequencing depth to 10,000 molecules per cell using the “NormalizeData” function of the Seurat package v4.0 with a scale factor of 10,000 (Hao et al. 2021).
Single-cell clustering and annotation of cell types
Preprocessing of each sample data set
First, the top highly variable genes (n = 3000) from the data set were selected using the default “vst” selection method in the “FindVariableFeatures” function of the Seurat package v4.0 (Hao et al. 2021). We applied a linear transformation (scaling), a standard preprocessing step before PCA, using the “ScaleData” function. This sets an equal weight in downstream analyses, preventing highly expressed genes from dominating the data. PCA was performed on the highly variable genes using the “RunPCA” function. Functions such as “VizDimReduction,” “DimHeatmap,” and “ElbowPlot” were used to visualize the cells and features in defining the PCA, decide the PCs included for further downstream analyses, and determine the “dimensionality” of the data set sequentially.
Next, we refined the edge weights between any two cells based on the shared overlap in their local neighborhoods (i.e., the Jaccard Index) by running the “FindNeighbors” function, which took the previously defined dimensionality of the data set as input. Then, a k-nearest neighbor (KNN) graph with the Euclidean distance was constructed in the PCA space. Afterward, we used the “FindClusters” function to cluster the cells by grouping cells together iteratively. Finally, the R package DoubletFinder v2.0.3 (McGinnis et al. 2019) was used to detect and filter potential doublets in the scRNA-seq data (see Supplemental Information).
Cell clustering analysis of merged data
We used the “merge” function to merge the preprocessed data sets of sheep and goats separately. Cells in each data set were time-stamped. The merged data sets were subjected to the steps such as normalization, selection of top highly variable genes, feature scaling and centering, PCA plotting, dimension reduction, calculation of the Jaccard similarity, cell clustering, and visualization by using Seurat package v4.0 (see Supplemental Methods). Meanwhile, CCA integration in Seurat package v4.0, and Harmony package v0.1.1 (Korsunsky et al. 2019) were used to test the scRNA-seq data integration.
Identification of various cell types and novel cell type marker genes
A good protocol for cell type identification should be both sensitive and selective. Here, we used three complementary approaches to define cell types (or subtypes) from rumen tissues. First, we searched the top 10 highly expressed genes for each cell cluster (subcluster) to find the cell type (or subtype) in the online PanglaoDB database (https://panglaodb.se/search.html), which contains 1368 scRNA-seq data sets. Then, the cell clusters (or subclusters) were annotated manually based on the expression of previously reported canonical marker genes of known cell types (or subtypes). Additionally, cell types were delineated according to the specific functions of the top 100 highly expressed genes and the significantly enriched signaling pathways of the highly expressed genes for each cell cluster. Furthermore, to identify genes specifically expressed in various cell types (or subtypes), we implemented the “FindAllMarkers” function with the settings “test.use = “wilcox” || logfc.threshold = 0.25 || min.pct = 0.1” and an Padj < 0.05. Novel marker genes for each cell type or subtype were identified by specific expression analysis.
Analysis of DEGs
To compare transcriptomic gene expression profiling among the 17 time points or six main developmental stages, we performed an analysis of DEGs using Seurat. GO and pathway enrichment analyses of DEGs were performed using the ClusterProfiler R package v3.18.1 (Yu et al. 2012) and Metascape (Zhou et al. 2019). The results were visualized with the “ggplot2” v3.3.5 (Wickham et al. 2021) and “barplot” functions of the Graphics R package v4.1.0 (Murrell 2005). Heatmap analysis and hierarchical clustering of gene expression profiles were performed using the ComplexHeatmap R package v2.8.0 (Gu et al. 2016).
Single-cell trajectory and RNA velocity analysis
We built single-cell pseudotime trajectories based on the UMI matrix and the variable genes detected above using the Monocle package v2.20.0 (Trapnell et al. 2014). The cell subtypes of interest were subjected to the processing steps detailed in the Supplemental Information. RNA velocity analysis of loom-annotated matrices of the 10x data set was implemented as previously (see also Supplemental Methods; Han et al. 2020).
TF-target gene regulatory network analysis
Core regulatory TFs were determined based on the scRNA-seq data using the program AnimalTFDB v3.0 (http://bioinfo.life.hust.edu.cn/AnimalTFDB/#!/). We compared the results with the SCENIC v1.2.4 R package (Aibar et al. 2017), following the protocols detailed in the Supplemental Information.
Cell–cell communication inference
To investigate how the context-dependent cross talk of different cell types enables physiological processes to proceed, we implemented the command “cellphonedb method statistical_analysis” of CellPhoneDB software v1.1.0 (Efremova et al. 2020). The interaction was absent if either ligand or receptor was not detected. To identify LR pairs expressed specifically in particular cell types, the average expression of LR pairs was compared between all pairs of various cell types. The pairs with P < 0.05 were retained in the cell–cell communication analysis of each cell type in sheep and goats. Changes in the number of LR pairs were visualized using Cytoscape v3.7.1 (Shannon et al. 2003).
Immunofluorescence staining
For immunofluorescence staining, rumen tissues were processed following the normal procedures such as perfuse, embedding, washing, dehydration, deparaffinization, dewaxing, incubation, and staining (see also Supplemental Information). In parallel, negative-control sections were processed identically to the tissue sections except for the use of the KRT15/LYVE1 antibody instead of the secondary antibody. Images were captured using a computer-controlled Zeiss Axio imager microscope (Zeiss) and were further processed and analyzed using ImageJ software v1.8.0 (Schindelin et al. 2015) with the IHC profiler plugin.
Cross-species comparison of scRNA-seq data
The raw gene counts or normalized gene expression matrix for each single cell was downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) and China National GeneBank DataBase (CNGBdb; https://www.db.cngb.org/) databases. In total, we collected single-cell gene expression data of stomach tissues from five species, including humans (n = 4), mice (n = 3), monkeys (n = 3), cattle (n = 6), and sheep (n = 7; Supplemental Table S40). To facilitate integration of the cross-species single-cell stomach data sets, we converted genes from nonhuman species into human homologs. We downloaded the species-specific annotation packages of the four species (org.Hs.eg.db, org.Mm.eg.db, org.Mmu.eg.db, org.Bt.eg.db) based on R v4.2.0 (R Core Team 2022) and a table was created for all potential homologs between species using the Orthology.eg.db package. The genes were converted in the following cases: (1) a 100% match existed between a nonhuman gene and a human gene, and (2) the given gene had multiple potential human homologs, but one of those homologs was simply the uppercase symbol of the nonhuman gene in question. For all the other remaining genes, their original names were maintained.
For the retrieved single-cell data sets, functions such as NormalizeData, FindVariableFeatures, ScaleData, FindNeighbors, FindClusters, and FindAllMarkers were implemented as described originally (Han et al. 2020, 2022; Wu et al. 2022; Yuan et al. 2022). We collected marker genes for each cell type and then defined core regulatory TFs and predicted cell–cell communications as described above. A panconserved cellular interaction was identified only when an interacting pair was present in all six species investigated (human, monkey, mouse, sheep, goat, and cattle). We defined an interaction as a ruminant-specific conserved cellular interaction when an interacting pair was present in all three ruminant species (sheep, goat, and cattle) but absent in the three nonruminant species.
Metagenomic assembly and analysis
Metagenomic assembly and gene prediction
Adapters from the raw metagenomic sequencing data were first trimmed using fastp v0.19.7 (Chen et al. 2018) with the parameters “fastp -g -q 5 || -u 50 || -n 15 || -l 150” to obtain clean reads. To decrease potential DNA contamination from the host, we then mapped the trimmed clean reads to the sheep and goat reference genomes of ARS-UI_Ramb_v2.0 (GCA_016772045.1) and ARS1 (GCA_001704415.1), respectively, using Bowtie 2 v2.4.4 (Langmead and Salzberg 2012). Each sample data set was assembled using the IDBA-UD assembler v1.1.3 (Peng et al. 2012) with the parameters “‐‐pre_correction || ‐‐min_contig 300.” Prodigal v2.6.3 (Hyatt et al. 2010) was used to predict ORFs. ORFs obtained from the data assembled for the 31 samples were clustered using CD-HIT v4.8.1 (Fu et al. 2012) with the options “-n 9 || -g 1 || -c 0.95 -G 0 || -M 0 || -d 0 || -aS 0.9” to obtain nonredundant microbial gene catalogs of each sample.
Taxonomic annotation and functional annotation
Taxonomic assignment for all gene entries in the catalog was performed using the BLSTSP command of DIAMOND v2.0.13 (Buchfink et al. 2015) with the options “‐‐evalue 0.00001 || ‐‐max-target-seqs 10.” Comprehensive reference sequences of the bacteria, fungi, archaea, and viruses were extracted from the integrated Nonredundant Protein Sequence (NR) database (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/). Then, the taxonomic level of each gene was determined based on the lowest common ancestor (LCA) algorithm implemented in MEGAN v6.21.7 (Huson et al. 2007). The program assigned genes to taxa such that the taxonomic level of the assigned taxon reflected the conservation levels. Carbohydrate-active enzymes (CAZymes) were annotated to match protein sequences to entries of the CAZy database (http://www.cazy.org/) using the Python script “run_dbcan.py” of the program dbCAN2 v3.0.2 (Huang et al. 2018) with the parameters “‐‐dia_eval 1e-5.” Entries in the gene catalogs were annotated to the specific COGs using the Python script “emapper.py” of eggNOG-mapper v2.1.6 (Cantalapiedra et al. 2021) with the parameters “-m diamond ‐‐evalue 1e-5.” Additionally, we used kofam_scan v1.3.0 (Aramaki et al. 2020) to search against the KEGG database and map the KEGG Ortholog IDs (KO IDs) of the best hits in the gene catalogs with the options “exec_annotation -f mapper -e 1e-5.”
The abundance profiles of genes were normalized by the transcripts per million (TPM) using Salmon v1.5.2 (Patro et al. 2017), and in our case, transcripts correspond to reads (Ribicic et al. 2018). The relative abundances of taxa, COGs, KOs, and CAZymes were calculated from the abundances of annotated genes. Briefly, for the taxonomic profiles, we used assignment of each annotated gene from the gene catalogs and summed the relative abundances of genes from the same phylum or genus to produce the abundance of each phylum or genus. The relative abundance of a COG category, KEGG pathway, and CAZyme family was calculated from the sum of the relative abundances of the contained COGs, KOs, and CAZymes, respectively.
In vitro co-culture of rumen cells with P. copri and RNA-seq
In vitro co-culture of rumen cells
P. copri (BNCC354512) obtained from the BeNa Culture Collection was cultured at 37°C in brain heart infusion broth (BHI) liquid medium under anaerobic conditions. Primary rumen epithelial cells and Fib from 2-mo-old sheep were ordered from Delf. At 37°C and under 5% CO2 atmosphere, primary rumen epithelial cells were cultured in the primary epithelial cell basal medium with 2% fetal bovine serum and 1% penicillin/streptomycin, whereas primary rumen Fib were cultured in the primary Fib basal medium with 10% fetal bovine serum and 1% penicillin/streptomycin. Bacterial suspensions were centrifuged and resuspended in antibiotic-free medium to an OD600 = 1, approximately 1 × 109 colony forming units (CFU)/mL (Yu et al. 2019). At ∼90% confluence, primary rumen epithelial cells and Fib were then cultured in the antibiotic-free medium with alive P. copri (1 × 107 CFU/mL) for 4 h, separately. Meanwhile, the controls were cultured in routine media. The above experiment was repeated four times. Final concentration and duration for co-culture of cells with P. copri were evaluated by the cell counting kit-8 (CCK-8) assay (Supplemental Fig. S13B,C).
RNA sequencing
After the total RNA extraction, examination of RNA quality and integrity, and library preparation (Supplemental Methods), we sequenced the libraries on an Illumina NovaSeq platform, and 150-bp paired-end reads were generated.
Differential gene expression analysis
Raw reads of the total of 16 RNA-seq samples were trimmed using the program fastp v0.23.2 (Chen et al. 2018). Program STAR v2.7.10a (Dobin et al. 2013) was then used to map the above clean reads to the sheep reference genome Oar_rambouillet_v1.0 with the default parameters of “‐‐outFilterMismatchNmax 3.” The mapping rate of the clean reads ranged from 85.24%–90.53% with an average rate of 88.79%. Read counts were calculated for each annotated gene using featureCounts v2.0.2 (Liao et al. 2013). Raw counts of annotated genes were normalized, and DEGs were determined using DESeq2 (Love et al. 2014) with the thresholds of P < 0.05 and |logFC| > 0.75.
Statistical analysis
No statistical method was used to predetermine the sample size. Data are presented as the mean ± SD. Measurement data were analyzed by Prism software (GraphPad 6 Software) and the R package “ggplot2” to show the change trend of gene expression and each layer between the sheep and goat rumens. The Wilcoxon signed-rank test was used to compare differences between groups. The correlation analysis adopted a nonparametric measure of Spearman's rank correlation coefficient. The following were considered to indicate statistical significance: (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, and (****) P < 0.0001. The numbers of experimental repeats are indicated in the figure legends.
Data access
Raw sequencing reads from scRNA-seq and metagenome generated in this study have been submitted to the NCBI BioProject database (https:www.ncbi.nil.nih.gov/bioproject/) under accession numbers PRJNA919098 for scRNA-seq data and PRJNA933244 for metagenome sequencing data. Metadata and details of the samples have been deposited in the NCBI BioSample database (https://www.ncbi.nlm.nih.gov/biosample) under the accession numbers SAMN32622164–SAMN32622197 for scRNA-seq and SAMN33230168–SAMN33230198 for metagenome data. The codes used in this study have been uploaded as Supplemental Code and are also available at GitHub (https://github.com/Dengjuansicau/SingleCellSeq-analyses).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Xin Li, Ya-Xi Xu, Chao Zhao, Jia-Nan Jing, Sen Zhao, Guo-Lin Chen, He Cai, Miao Xiao, and Jian-Jun Wang for help in sampling; Qiang-Hui Zhu and Xing Wan for technical help in the laboratory. This study was financially supported by grants from the National Key Research and Development Program-Key Projects (2021YFD1200900), the National Natural Science Foundation of China (nos. 31825024, 31661143014, 31972527, 32061133010, and 32320103006), and the Second Tibetan Plateau Scientific Expedition and Research Program (STEP; no. 2019QZKK0501). The computation was supported by High-performance Computing Platform of China Agricultural University.
Author contributions: M.-H.L. designed the study. M.-H.L. and L.L. supervised the study. J.D., J.-Y.L., W.-T.W., and Q.-X.H. performed the genome data analyses. J.D., J.-Y.L., W.-T.W., Q.-X.H., L.-P.Z., L.-Y.L., Q.Z., L.Z., Y.C., Y.-L.R., S.-G.J., Y.-L.L., J.Y., F.-H.L., and H.-P.Z. performed the laboratory and/or farm work. M.-H.L., J.D., J.-Y.L., W.-T.W., Q.-X.H., L.-P.Z., Q.Z., L.Z., Y.C., Y.-L.R., H.-P.Z., and L.L. prepared the samples or provided help during the sample collection. J.D. and M.-H.L. wrote the manuscript with main contributions from Y.-J.L., Q.-X.H., and W.-T.W. All authors reviewed and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278239.123.
-
Freely available online through the Genome Research Open Access option.
- Received June 30, 2023.
- Accepted September 17, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








