
Lossless indexing with counting de Bruijn graphs
- Mikhail Karasikov1,2,3,
- Harun Mustafa1,2,3,
- Gunnar Rätsch1,2,3,4,5 and
- André Kahles1,2,3
- 1Department of Computer Science, ETH Zurich, 8092 Zurich, Switzerland;
- 2Biomedical Informatics Research, University Hospital Zurich, 8091 Zurich, Switzerland;
- 3Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland;
- 4Department of Biology at ETH Zurich, 8093 Zurich, Switzerland;
- 5ETH AI Center, ETH Zurich, 8092 Zurich, Switzerland
Abstract
Sequencing data are rapidly accumulating in public repositories. Making this resource accessible for interactive analysis at scale requires efficient approaches for its storage and indexing. There have recently been remarkable advances in building compressed representations of annotated (or colored) de Bruijn graphs for efficiently indexing k-mer sets. However, approaches for representing quantitative attributes such as gene expression or genome positions in a general manner have remained underexplored. In this work, we propose counting de Bruijn graphs, a notion generalizing annotated de Bruijn graphs by supplementing each node–label relation with one or many attributes (e.g., a k-mer count or its positions). Counting de Bruijn graphs index k-mer abundances from 2652 human RNA-seq samples in over eightfold smaller representations compared with state-of-the-art bioinformatics tools and is faster to construct and query. Furthermore, counting de Bruijn graphs with positional annotations losslessly represent entire reads in indexes on average 27% smaller than the input compressed with gzip for human Illumina RNA-seq and 57% smaller for Pacific Biosciences (PacBio) HiFi sequencing of viral samples. A complete searchable index of all viral PacBio SMRT reads from NCBI's Sequence Read Archive (SRA) (152,884 samples, 875 Gbp) comprises only 178 GB. Finally, on the full RefSeq collection, we generate a lossless and fully queryable index that is 4.6-fold smaller than the MegaBLAST index. The techniques proposed in this work naturally complement existing methods and tools using de Bruijn graphs, and significantly broaden their applicability: from indexing k-mer counts and genome positions to implementing novel sequence alignment algorithms on top of highly compressed graph-based sequence indexes.
Introduction
The sequencing of DNA and RNA has become a commodity in the portfolio of biomedical data acquisition techniques, leading to an increase in both the demand and availability of sequencing data (Stephens et al. 2015). Often, independent from the original research questions that individual data sets were created to answer, they find a second life as a valuable source for other analyses (Su et al. 2020; Nayfach et al. 2021). Thus, methods for the efficient storage and indexing of sequence data are urgently needed. In the past years, various approaches have been proposed to address this problem. On the one side, there are methods that extract relevant information, such as expression counts from vast cohorts of RNA sequencing data, and summarize it in aggregated form (Collado-Torres et al. 2017). On the other side, stand approaches that provide a full-text index of the sequencing data and allow to retrieve metadata for arbitrary sequence queries (Bradley et al. 2019; Karasikov et al. 2020a), which is of great practical relevance for projects generating large sequence cohorts (Almeida et al. 2021; Danko et al. 2021). As a balance between compressibility and access, methods using k-mer decompositions of the input sequences have proven very successful (Ondov et al. 2016; Bradley et al. 2019; Karasikov et al. 2020a; Marchet et al. 2021). In this work, we focus on approaches representing such k-mer sets as annotated sequence graphs, which we will briefly review in the following, discussing benefits and limitations of existing approaches.
Annotated genome graphs
To fully represent all information of a sequencing sample for interactive study, two components are necessary: (1) an index representing the sequence information, allowing for the query of presence; and (2) a structure containing additional metadata, such as the biological label of a sequence or the location of a sequence within a genome context (commonly referred to as genome coordinate) (Morgulis et al. 2008; Almodaresi et al. 2018; Li 2018). Both components can be represented jointly or in separate data structures.
A commonly used data structure for representing sequence information is a de Bruijn graph, where k-mers serve as nodes, and every two nodes u and v are connected with a directed edge (u, v) if and only if u[2,k] = v[1,k−1], where u[2,k] and v[1,k−1] denote the suffix and the prefix of length k − 1 of k-mers u and v, respectively. As all edges of a de Bruijn graph can be inferred from the set of its nodes, a de Bruijn graph can be fully represented by the set of its k-mers. In practice, all k-mers must also be indexed and assigned unique numeric identifiers to allow for association with any metadata (e.g., counts or coordinates). Such indexes can be represented explicitly as either a hash table–like structure (e.g., counting quotient filter) (Pandey et al. 2018) or a self-index (e.g., the space-efficient BOSS representation) (Bowe et al. 2012). The label information, on the other hand, can be encoded separately. The numeric k-mer identifiers provided by the k-mer index generate an address-space that can be used by a separate data structure holding the metadata, for example, linking the k-mers to their presence in different input sources (Muggli et al. 2017; Karasikov et al. 2020a; Marchet et al. 2020).
Other approaches, such as Bloom filters (Chikhi and Rizk 2013; Bingmann et al. 2019; Bradley et al. 2019), do not require numeric k-mer identifiers. However, the lack of any address-space for structuring additional metadata limits their uses to only answering approximate k-mer membership queries. Thus, in this work, we consider the approach encoding the metadata in a separate structure called graph annotation and focus on its efficient representation.
In addition, we further extend the notion of genome coordinates and introduce k-mer coordinates, representing the occurrence positions of a certain k-mer in the input stream. This stream may be a single genome, a list of sequencing reads, or an entire collection of arbitrary sequences. All (not only distinct) k-mers from the input are naturally ordered, and knowing this order allows reconstructing the original sequences from their corresponding paths in the graph, which we call sequence traces. Indeed, the first k-mer provides the first k characters of the first sequence, and every k-mer with the following coordinate can be used to reconstruct the next character of the sequence, whereas the end of the sequence can be encoded with a skipped coordinate. Hence, by representing k-mer coordinates, we encode traces of the input sequences in the graph and thereby make the index fully lossless.
Graph annotations
Approaches for representing relations between k-mers and input files have been extensively explored in the past decade (Iqbal et al. 2012; Almodaresi et al. 2017, 2020; Muggli et al. 2017; Karasikov et al. 2020b; Danciu et al. 2021). Motivated by the experiment discovery problem, which is to find a sequencing library within a large collection based on a query pattern, these methods encode binary metadata attributes (e.g., the membership of a k-mer to a certain sequence or file) in a sparse binary matrix. Depending on the number of k-mers and files, this matrix can have up to 1012 rows (corresponding to distinct k-mers) and 107 columns (corresponding to different files or, in general, labels) (Karasikov et al. 2020a). However, it can be highly compressed thanks to its sparsity (Almodaresi et al. 2017, 2020; Muggli et al. 2017; Karasikov et al. 2020b; Danciu et al. 2021).
Supplementing a de Bruijn graph with this type of binary graph annotation provides an excellent tool for answering k-mer membership queries. However, any quantitative information of the original data is lost. In particular, queries relating to the exact occurrence position in a sequence or relating to how often the queried sequence is present in a sample cannot be answered with binary annotations.
To address this problem, methods for representing nonbinary graph annotations have recently started to emerge, but very few have been proposed so far. On the one end stands gPBWT, which supplements genome graphs and enables lossless encoding of haplotypes (Novak et al. 2017). On the other end, REINDEER (Marchet et al. 2020) represents approximate k-mer counts in genome graphs by averaging them within each unitig of the sample de Bruijn graphs and compressing them via run-length encoding. Unfortunately, these methods do not cover the entire spectrum of needs. In particular, REINDEER does not represent genome coordinates and does not provide a lossless representation of input sequences, such as reads. In contrast, gPBWT does provide a lossless representation of input sequences; however, the time complexity of querying quantitative information on a pattern would be linear in the number of its occurrences, making it less well suited for indexing large collections of reads.
Sequence-to-graph alignment
Many tools have used de Bruijn graphs as indexes for alignment to collections of sequences (Liu et al. 2016; Rautiainen and Marschall 2020; Karasikov et al. 2020a; Almodaresi et al. 2021; Luhmann et al. 2021; Schulz et al. 2021), applying the seed-and-extend paradigm with varying seed filtration and extension strategies. Some strategies extract sequences from the index onto which a sequence-to-sequence alignment is performed (Liu et al. 2016; Almodaresi et al. 2021), whereas others traverse the graph and compute (Karasikov et al. 2020a) or approximate (Luhmann et al. 2021; Schulz et al. 2021) an alignment score. However, very few of these methods index global coordinates in reference genomes to avoid alignments to spurious paths in the graph, which would be especially helpful when aligning to complex regions in the graph with many short overlapping unitigs. To our knowledge, deBGA (Liu et al. 2016) and PuffAligner (the aligner from Pufferfish) (Almodaresi et al. 2018; Almodaresi et al. 2021) are the only de Bruijn graph–based tools that index global coordinates. The more recent PuffAligner uses a colinear chaining approach inspired by minimap2 (Li 2018) to effectively select a good candidate location for alignment and to limit alignment to query regions between seed hits. However, both deBGA and PuffAligner are designed for indexing long reference genomes and optimize for query performance, thus making only limited use of compression techniques and reducing their scalability. Lastly, both use k-mer hash tables to index the unitig set, restricting the minimum seed length to k.
In this work, we consider the problem of representing numeric attributes assigned to each k-mer–label relation in graph annotations. We call such annotations extended graph annotations, emphasizing that each label is supplemented with an attribute, which may include a single or multiple numeric values. In particular, we focus on indexing (1) k-mer counts, representing the number of times a k-mer occurs in a certain sequencing sample, and (2) k-mer coordinates, where the attributes represent all the occurrence positions of a k-mer in a sequence, a genome, or a collection thereof. The latter makes a fully lossless representation of the input sequences. Together with the underlying de Bruijn graph, such extended graph annotations make up an abstract data structure that we call a counting de Bruijn graph. We show the advantage of such an index by devising a sequence-to-graph alignment algorithm called Trace-Consistent Graph-based Aligner (TCG-Aligner) that avoids spurious paths and correctly estimates the alignment score even when aligning sequences with repeats to loops in the graph.
Methods
In this section, we present methods and techniques ultimately used to efficiently represent extended graph annotations in compressed data structures that can be queried without full decompression. Assume each node–label relation (i, j) is supplemented with an attribute ai,j, representing a single or multiple numeric values. Naturally, such annotations can be represented as a sparse matrix, and thus, the first question to be answered is how such matrices can be represented to minimize the memory footprint, while still allowing for efficient queries without full decompression.
Succinct representation of sparse matrices
Here we propose a general approach for the efficient compressed representation of sparse matrices and, in particular, extended graph annotations, which supplement each binary relation k-mer–label (i, j) with an attribute ai,j (Fig. 1A, left).
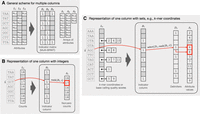
The proposed representation of sparse matrices in compressed form. (Panel A) General scheme for sparse matrices with abstract attributes, where the nonassigned attributes are eliminated by an indicator binary matrix stored in a compressed representation (e.g., multi-BRWT), supporting the rank operation on its columns to enable the access to the corresponding attribute for any given cell of the matrix. These attributes are stored separately, typically in the form of compressed arrays. (Panel B) The scheme applied to a single column with integer values (e.g., k-mer counts) and the query algorithm (e.g., the count of k-mer i is retrieved as A1[rank(B1, i)]). Empty cells in gray represent zeros. (Panel C) The scheme applied to a single column where each cell is a set of numbers, or a tuple (e.g., representing k-mer coordinates). The “zero” attributes (empty sets) are eliminated with an indicator bitmap, and the nonempty sets are encoded in an array that holds all numbers together with a delimiting bitmap.
This attribute may be a single numeric value (e.g., the number of times k-mer i occurs in experiment j) or a set of numbers (e.g., all positions where k-mer i occurs in genome j). Without loss of generality, we assume a very high sparsity of the annotation matrix and decompose the initial annotation into two components schematically shown in the right part of Figure 1A: (1) a binary indicator matrix representing the indexes of the entries present in the matrix, and (2) the relation attributes ai,j stored in a separate data structure, typically in form of a compressed array. The indicator matrix is then represented with a scheme supporting the rank operation on its columns or rows (depending on the layout of the attribute values), defining an ordering on the (i, j) pairs and enabling access to the attribute values stored in separate arrays in the consistent order.
Note that the layout of the attribute arrays can be different depending on the rank operation supported by the indicator matrix. Namely, this can be the rank on its columns (shown in Fig. 1), rows, or their concatenation. Thus, this scheme allows for compressing the indicator matrix using a large class of approaches for the compressed representation of binary relations, which have already been applied for representing binary graph annotations: ColumnCompressed (Karasikov et al. 2020b), multi-BRWT (Karasikov et al. 2020b), BinRelWT (Barbay et al. 2013), and RowFlat (the scheme used in VARI) (Muggli et al. 2017), all of which support the rank operation on the nonzero entries in a certain layout.
Succinctness of the proposed scheme
The proposed decomposition does not change the entropy of the data, which suggests that it also does not change the theoretical minimum of the number of bits required to store the matrix by representing the two components separately.
To prove this formally, consider the problem of representing a sparse matrix of size n × m with s “nonzero” entries from universe A and other nm − s entries set to a fixed “zero” value that does not belong to A. As there are 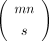 ways to pick s out of mn positions for nonzero values, where each can store one of
ways to pick s out of mn positions for nonzero values, where each can store one of 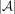 possible values, the total number of such matrices is
possible values, the total number of such matrices is 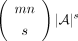 . Hence, the minimum number of bits required to encode any such matrix is
. Hence, the minimum number of bits required to encode any such matrix is 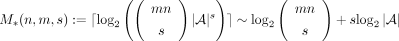 . On the other hand, the indicator matrix in the proposed scheme (Fig. 1A) can be reshaped into a vector encoded in the succinct Raman–Raman–Rao (RRR) representation (Raman et al. 2002) taking asymptotically
. On the other hand, the indicator matrix in the proposed scheme (Fig. 1A) can be reshaped into a vector encoded in the succinct Raman–Raman–Rao (RRR) representation (Raman et al. 2002) taking asymptotically 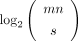 bits, which together with an optimal coding of the attributes asymptotically makes up the same space complexity
bits, which together with an optimal coding of the attributes asymptotically makes up the same space complexity 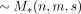 . We now can make the following claim (see the proof in the Supplemental Material).
. We now can make the following claim (see the proof in the Supplemental Material).
Theorem 1. If both the indicator matrix and the arrays of attributes are represented succinctly, the proposed scheme also is a succinct representation of the matrix. That is, there is no other data structure that could represent any such matrix with asymptotically fewer bits.
Base representations
Now, we will show that the commonly used compressed sparse column (CSC) format (e.g., used in NumPy) (Harris et al. 2020) is a special case of our proposed scheme. CSC stores the matrix entries in three arrays: (1) an array containing all nonzero values in the order they appear in the rows of the matrix, (2) an array with their column indexes, and (3) a compressed array delimiting the positions in the first two arrays corresponding to different rows of the matrix. One can see that the first array corresponds to the attribute arrays in our scheme, whereas the other two essentially encode the indicator matrix, making our scheme at least as efficient as the basic CSC format. Our scheme, however, allows for additional freedom in choosing specific encodings for the attribute arrays and the indicator matrix.
Encoding the columns of the indicator matrix with succinct RRR bit vector representations (Raman et al. 2002) asymptotically achieves the theoretical minimum in space. Extra compression can be achieved in practice by exploiting column correlations using the multi-BRWT scheme (Karasikov et al. 2020b).
In practice, we use succinct bitmaps to represent the columns of the indicator matrix during construction. Subsequently, we convert the matrix to the multi-BRWT representation to reduce its final size and enhance the query speed. Typically, we compress the attribute arrays with simple bit-packing or universal coding such as directly addressable codes (dac_vector) (Brisaboa et al. 2013), supporting the direct access.
Representing attributes with multiple numeric values
The scheme described above effectively erases zero entries from the matrix and stores any nonzero entries in a separate data structure (Fig. 1A). We note that this approach generalizes beyond single integers as matrix entries shown in Figure 1B. In particular, it allows entries to be number sets and, hence, can be used for representing k-mer coordinates, where each k-mer may occur in multiple positions of a genome or, more generally, a collection of input sequences.
Without loss of generality, Figure 1C schematically shows how the entries of such matrices can be encoded and queried, using a single column as example. All tuples (entries) are concatenated into a single dense array storing all values together, and an additional delimiting bitmap is used to separate the different tuples. The dense array together with the delimiting bitmap represents a single array of attributes in the general scheme as shown in Figure 1A.
Diff-compression of extended graph annotations
Similar to the case of binary annotations, extended graph annotations often possess a certain structure that can be exploited for their compression. Indeed, attributes of nodes in the graph can often be approximated with high accuracy from the attributes of their neighbors. For example, k-mer counts, being an aggregate function of contiguous paths induced from reads, usually change incrementally and, hence, can be approximated by averaging the counts of their adjacent k-mers. Another example is k-mer coordinates, which simply shift by one at each node along the paths of the de Bruijn graph derived from the input sequences, which we call traces. Hence, one can construct an expected set of coordinates at a node if the set of coordinates at its adjacent node is known or can be reconstructed recursively.
Leveraging similarity of annotations of neighboring nodes
For the case of binary annotations, transformations assuming likely similarity between annotations of adjacent nodes in the graph and replacing them with relative differences have been explored in Mantis-MST (Almodaresi et al. 2020) and RowDiff (Danciu et al. 2021). The RowDiff algorithm conceptually consists of two parts. First, for each node with at least one outgoing edge, it arbitrarily picks one of them and marks its target node as a successor. The subset of edges leading to the assigned successor nodes forms a spanning tree of the graph. Second, it replaces the original annotations at nodes with their differences to the annotations at their assigned successor nodes. This delta-like transform is applied to all nodes in the graph except a small subset of them (called anchors). These anchor nodes keep the original annotation unchanged and serve to terminate every path composed of successors and break the recursion when reconstructing the original annotations (inverse transform).
Here, we devise a generalization of the RowDiff scheme to the case of extended graph annotations. We design an invertible transform, which losslessly compresses them by effectively removing the information that can be reconstructed from a neighborhood in the graph. Our generalization goes in two directions. First, in the following section, we generalize the diff-operation to act on arbitrary sets and define specific functions for the two specific cases considered in this work: k-mer counts and k-mer coordinates (genome positions). Second, motivated by the idea illustrated in Supplemental Figure 1, we propose a data-driven procedure for optimizing successor nodes instead of assigning them randomly as in RowDiff (see Supplemental Section 1). In addition, we propose a more efficient algorithm for the anchor assignment (see Supplemental Section 2). Note that these improvements are also applicable to binary annotations and enable better compression. In addition, we consider schemes admitting the aggregation of multiple successors at forks before computing the diff (see Supplemental Section 3). This essentially replaces the diff-paths with trees and, by design, helps improve the compression at forks, where some of the traces branch out and carry their annotations away, increasing the diff.
Generalized diff-transform for graph annotations
Suppose the nodes in the graph are annotated with attributes from a set A. Consider a node v holding an attribute 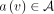 and its successor node vsucc holding an attribute
and its successor node vsucc holding an attribute 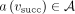 . To define a diff-transform of the graph annotation, we need to specify an invertible diff-operation
. To define a diff-transform of the graph annotation, we need to specify an invertible diff-operation 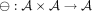 acting on pairs of attributes and replacing the original annotation a(v) with its delta relative to the annotation at the successor:
acting on pairs of attributes and replacing the original annotation a(v) with its delta relative to the annotation at the successor: 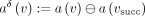 . The invertibility of this transform entails the existence of an inverse transform ⊕, such that
. The invertibility of this transform entails the existence of an inverse transform ⊕, such that 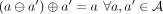 , which makes it always possible to reconstruct the original annotation a(v) from the delta
, which makes it always possible to reconstruct the original annotation a(v) from the delta 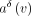 and the original annotation a(vsucc), which is, in turn, either reconstructed recursively (or stored explicitly if vsucc is an anchor).
and the original annotation a(vsucc), which is, in turn, either reconstructed recursively (or stored explicitly if vsucc is an anchor).
For sparsifying k-mer count annotations, where the labeling at each node is encoded by a row of the integer count matrix, we use the simple
vector difference as the diff-operation. For the case of coordinate annotations, each attribute a is a set of natural numbers (occurrence positions of a k-mer in a genome or a file); that is,  . At the same time, we naturally expect the coordinates to shift by one when transitioning from a k-mer to its adjacent successor. Thus, the diff-operation ⊖ in this case is the symmetric set difference between the coordinates
at the successor node vsucc and the incremented coordinates at node v:
. At the same time, we naturally expect the coordinates to shift by one when transitioning from a k-mer to its adjacent successor. Thus, the diff-operation ⊖ in this case is the symmetric set difference between the coordinates
at the successor node vsucc and the incremented coordinates at node v:
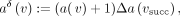 (1)
where operator Δ denotes the symmetric set difference:
(1)
where operator Δ denotes the symmetric set difference: 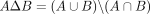 . Note that we chose Equation 1 over a probably more intuitive formula,
. Note that we chose Equation 1 over a probably more intuitive formula, 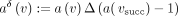 , to avoid negative numbers and keep the result
, to avoid negative numbers and keep the result 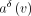 in the same set
in the same set  of subsets of positive coordinates.
of subsets of positive coordinates.
Compressed extended graph annotations
In this section, we combine the techniques presented in the sections above and propose two memory-efficient representation schemes for encoding quantitative data for two important practical cases of nonbinary graph annotations: k-mer counts (e.g., in read sets, representing gene expression levels) and k-mer coordinates. Coordinates may represent positions of k-mers in genomes, any collections of sequences, or in a file in general.
Representation of k-mer counts
Formally, the task is to represent a matrix with integer entries, where each entry corresponds to a k-mer–label pair and encodes the k-mer's count in the respective label.
We use the techniques presented above and, first, transform the initial count matrix with the generalized diff-transform (see
the preceding Methods subsection “Diff-compression of extended graph annotations”). The proposed method is schematically illustrated
in Figure 2. Assuming that adjacent nodes in the graph are likely to have identical or similar counts, we use the following formula to
compute the diff between two rows of the integer annotation matrix: 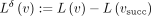 . After this operation, the diff-values
. After this operation, the diff-values 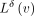 are often either zeros or integer values close to zero, thus requiring fewer bits to represent compared with the original
counts L(v). If the diff
are often either zeros or integer values close to zero, thus requiring fewer bits to represent compared with the original
counts L(v). If the diff 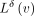 does not require fewer bits to store it than the original counts L(v) (as for nodes GGC and AGC in the example graph in Fig. 2), such nodes are explicitly made anchors and no transform is applied to them (for more details, see the anchor assignment
procedure described in Supplemental Section 2).
does not require fewer bits to store it than the original counts L(v) (as for nodes GGC and AGC in the example graph in Fig. 2), such nodes are explicitly made anchors and no transform is applied to them (for more details, see the anchor assignment
procedure described in Supplemental Section 2).

A schematic diagram illustrating the encoding of k-mer counts in m columns with the proposed approach. Circles represent nodes of a de Bruijn graph. Edges are shown as arrows. Red nodes represent anchor nodes, and red edges represent paths to row-diff successors. The transformed counts are shown in red (e.g., cf. L1: − 1 for k-mer GCT after the transform and L1:17, L2:11 before; for k-mer TAT, the transformed counts are not shown because they depend on TAT's successors, not shown in the graph). Then, the diff-transformed matrix is decomposed into an indicator binary matrix stored in the compressed multi-BRWT representation, supporting the rank operation on the columns and arrays storing nonzero diffs.
Note, in work (Italiano et al. 2021) developed independently in parallel to ours, a similar delta-like coding was considered for compression of k-mer counts in the framework of weighted rooted trees. This solution is close to our diff-transform for the case of a single annotation column with k-mer counts.
After performing the diff-transform, we decompose the transformed count matrix into a binary matrix of the same shape, indicating the nonzero entries and a set of additional arrays containing those entries (Fig. 2, right) according to the scheme shown in Figure 1, A and B. The binary matrix is stored in the compressed multi-BRWT representation, and the count arrays are stored separately.
Lastly, we note that the diff-transformed values in the count arrays may be negative. We, thus, map them to nonnegative integers to enable further compression with variable-length codes, using the following invertible mapping: 2(|x| − 1) + 1[x < 0], where 1[A] is a Boolean predicate function, which evaluates as one if the statement A is true and as zero otherwise. After this mapping, we further compress the count arrays using directly addressable codes (dac_vector) (Brisaboa et al. 2013).
Representation of k-mer coordinates
Finally, in this section we describe how to efficiently encode k-mer coordinates and thereby create a lossless index of the input sequences. We observe that aggregating all sequences from a single source (or label) and encoding the enumerations of each k-mer is sufficient to reconstruct the original sequences. We, thus, will consider this simplified case for brevity of the description. However, our implementation does support the explicit indexing of multiple sources with multiple annotation columns.
After all the k-mers are enumerated (Fig. 3A), the underlying de Bruijn graph is annotated, and the coordinates are stored in an array of lists (Fig. 1C). It is easy to see that most of the adjacent pairs of k-mers are crossed by the same sequences, and hence, the successor k-mer usually has the same coordinates as its predecessor k-mer incremented by +1. Thus, it is most natural to define the diff-operation for sparsifying coordinate annotations as described
in the Methods subsection “Generalized diff-transform for graph annotations,” Equation 1. If this delta 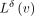 is an empty set, very few bits are needed to encode it. Otherwise, it would contain the information necessary to losslessly
reconstruct the coordinates at the predecessor from the known (or recursively reconstructed) coordinates at the successor.
This transformation step is schematically shown in Figure 3B and shows how well the predictability of the coordinates at the successor nodes can be exploited to compress the coordinate
annotations.
is an empty set, very few bits are needed to encode it. Otherwise, it would contain the information necessary to losslessly
reconstruct the coordinates at the predecessor from the known (or recursively reconstructed) coordinates at the successor.
This transformation step is schematically shown in Figure 3B and shows how well the predictability of the coordinates at the successor nodes can be exploited to compress the coordinate
annotations.

Extraction of k-mer coordinates from sequence ACTAGCTAGCTAG for k = 3 (panel A) and subsequent compression with a diff-transform (panel B), where the coordinates at a node's successor are expected to be the same but incremented by +1, as these nodes are likely
to be consecutive in the input sequence(s). The symmetric set difference 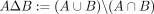 is used as a diff-operation. Thus, for example,
is used as a diff-operation. Thus, for example, 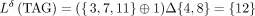 . The inverse transform is performed losslessly by
. The inverse transform is performed losslessly by 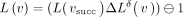 , which follows from the following property:
, which follows from the following property: 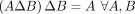 .
.
After the coordinate annotation is transformed, the new attributes 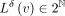 are still subsets of natural numbers, and hence, the full diff-transformed annotation matrix can be encoded with the same
general scheme as shown in Figure 1C, right.
are still subsets of natural numbers, and hence, the full diff-transformed annotation matrix can be encoded with the same
general scheme as shown in Figure 1C, right.
Trace-consistent graph-based aligner (TCG-Aligner)
The usage of counting de Bruijn graphs encoding k-mer counts or k-mer coordinates can greatly broaden the range of problems to which de Bruijn graphs are currently applied. In this section, we extend the sequence-to-graph alignment algorithm introduced in MetaGraph (Karasikov et al. 2020a). With this, we can not only ensure that all aligned paths in the graph are trace-consistent but also construct seed chains (detailed below) to more efficiently select good candidate positions for alignment and to reduce the number of base pairs from the query that need to be aligned.
We start by generating the initial seed set for a query sequence q. For low-error reads, these seeds consist of all maximal unique matches of the query that are contained in graph unitigs (called uni-MEMs) (Liu et al. 2016) with a minimum length of 19, as previously described (Karasikov et al. 2020a). For error-prone reads, we use all matches of length 19 as the seeds. For cases in which the k-mer size is greater than 19, all matches are made to the suffixes of k-mers (Karasikov et al. 2020a).
We then use the coordinates to discard all seeds that are not contained in a graph trace. Each remaining seed is associated
with one or more coordinate ranges. Then, we apply a dynamic programming seed chaining algorithm similar to the approaches
from minimap2 (Li 2018) and PuffAligner (see Supplemental Algorithm 2) (Almodaresi et al. 2021) to produce an initial partial alignment composed of a sequence of seeds (a chain). Let 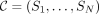 be the highest-scoring chain. For a given seed Si matching ℓi characters with initial node vi, we denote the corresponding position in the query by yi. We complete the alignments between each pair Si and Si+1 by extending Si using a modification of the extension algorithm from the MetaGraph aligner (Karasikov et al. 2020a) on the region of the query from yi to yi+1 + ℓi+1. To complete the alignment of q, we extend SN forward and S1 backward until the end and beginning of q, respectively.
be the highest-scoring chain. For a given seed Si matching ℓi characters with initial node vi, we denote the corresponding position in the query by yi. We complete the alignments between each pair Si and Si+1 by extending Si using a modification of the extension algorithm from the MetaGraph aligner (Karasikov et al. 2020a) on the region of the query from yi to yi+1 + ℓi+1. To complete the alignment of q, we extend SN forward and S1 backward until the end and beginning of q, respectively.
Our modification of the extension algorithm ensures that paths traverse along the corresponding graph trace of the starting
coordinates L(vi) (in practice, only a subset originating from the top labels detected among the seeds is used for faster alignment). More
precisely, we construct a trace-consistent alignment tree 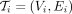 rooted at vi during graph traversal similar to the one defined in MetaGraph (Karasikov et al. 2020a), where Vi ⊂ V × ℕ contains all the nodes along the traces originating at vi:
rooted at vi during graph traversal similar to the one defined in MetaGraph (Karasikov et al. 2020a), where Vi ⊂ V × ℕ contains all the nodes along the traces originating at vi:
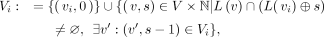 and Ei: = {((v, s), (v′, s + 1)) ∈ Vi × Vi|(v, v′) ∈ E} ⊂ Vi × Vi contains all the edges within these paths. To avoid querying coordinates on each traversal step, we collect all reachable
nodes during the forward pass of seed extension with their adjacent edges. Then, we discard noncoordinate consistent paths
to obtain
and Ei: = {((v, s), (v′, s + 1)) ∈ Vi × Vi|(v, v′) ∈ E} ⊂ Vi × Vi contains all the edges within these paths. To avoid querying coordinates on each traversal step, we collect all reachable
nodes during the forward pass of seed extension with their adjacent edges. Then, we discard noncoordinate consistent paths
to obtain 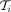 at forks in the graph (i.e., nodes with outdegree > 1). Finally, during the backtracking pass, we only extract the highest-scoring
coordinate-consistent alignment (i.e., a path along
at forks in the graph (i.e., nodes with outdegree > 1). Finally, during the backtracking pass, we only extract the highest-scoring
coordinate-consistent alignment (i.e., a path along 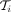 ) to correct for cases in which the alignment terminates before the next fork node is reached. With this, we can reduce the
alignment search space by more effectively filtering seeds, refining the traversal search space, and only performing alignment
on defined substrings of the query.
) to correct for cases in which the alignment terminates before the next fork node is reached. With this, we can reduce the
alignment search space by more effectively filtering seeds, refining the traversal search space, and only performing alignment
on defined substrings of the query.
Results
Indexing k-mer counts from 2652 RNA-seq read sets
For comparing our approach to the current state of the art, we used a set of 2652 RNA-seq read sets from different human tissues that was originally composed by Solomon and Kingsford (2018) and has since been widely used for benchmarking methods indexing raw sequencing data (Pandey et al. 2018; Solomon and Kingsford 2018; Almodaresi et al. 2020).
We counted all 21-mers in each read set with KMC3 (Kokot et al. 2017) and extracted canonical k-mers (defined as the lexicographical minimum of a k-mer and its reverse complement) occurring at least a certain number of times, using frequency thresholds from Pandey et al. (2018). Sixty-six out of the 2652 read sets contained only reads shorter than k = 21 and, hence, could not be indexed. The remaining 2586 read sets resulted in a de Bruijn graph with a total of 3.9 billion canonical k-mers and an annotation matrix of density 0.27%.
We compared the counting de Bruijn graph to REINDEER (Marchet et al. 2020), which, to the best of our knowledge, is the only published tool for indexing collections of samples with k-mer counts. We also compared against two state-of-the-art methods limited to binary annotations: Mantis-MST (Almodaresi et al. 2020) and RowDiff (Danciu et al. 2021). The latter was used to highlight the effect of modifications made to the diff-transform presented in this work.
The results for all methods are summarized in Table 1. When compressing binary data, our approach achieved a 4.5- and 5.5-fold size improvement over Mantis-MST and REINDEER, respectively. Compared with the original RowDiff scheme, it achieved a 20% improvement in annotation compression, which resulted in an index-size reduction from 7.7 GB to 6.6 GB, thanks to our methodological improvements. The advantage is maintained when indexing k-mer counts. Applying local neighborhood smoothing of counts along the unitigs of single-sample de Bruijn graphs, as introduced in REINDEER, our approach reduces the state-of-the-art index size of 59 GB to only 11 GB. This effect becomes even more pronounced when querying, as REINDEER needs to inflate its index for access. As we maintain our representation as compressed at all times, we achieve an eightfold reduction in memory usage during the query. Even when the smoothing step is omitted, which is not possible in REINDEER, our index takes only 21 GB while performing a lossless compression of the full k-mer spectrum of the input. In all three our indexes (6.6, 11, and 21 GB), the de Bruijn graph representation (the BOSS table) takes up 1.8 GB (27.3%, 16.4%, and 8.6%, respectively), and the annotation matrix takes up the rest. Our indexing workflow is also the fastest among all the compared methods in all indexing scenarios (see Supplemental Table 1) thanks to its careful implementation and parallelization, as well as leveraging the efficiently implemented KMC3 (Kokot et al. 2017) and MetaGraph (Karasikov et al. 2020a) tools.
Comparison of the state-of-the-art methods for indexing raw sequencing data and the proposed approach across three scenarios of indexing 2586 RNA-seq read sets: (1) encoding k-mer presence/absence only (binary), (2) encoding k-mer counts averaged over unitigs for each read set (smooth counts), and (3) encoding the original k-mer counts (raw counts)
Indexing read sets from the SRA
Indexing all viral HiFi reads
In this experiment, we fetched from NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (Leinonen et al. 2011) all viral sequencing samples sequenced with the Pacific Biosciences (PacBio) single-molecule real-time (SMRT) technology, including many recently sequenced SARS-CoV-2 samples. Out of all 152,957 samples (accessed on October 3, 2021), we could download 152,884 (99.95%) successfully. We will refer to this data set as Virus PacBio SMRT. Next, we filtered this set by selecting only high-fidelity read sets to ensure a low sequencing error rate (see Supplemental Section 4; Supplemental Fig. 2). This left 152,418 read sets (99.7%), which we refer to as Virus PacBio HiFi. Note that here and in all other experiments, the headers of the reads (sequence names) and their quality scores were removed before indexing or compressing with the tools tested, including gzip.
All 152,418 read sets combined contained a total of 717 Gbp and were compressed with gzip-9 down to 112 GB, which corresponds to 1.25 bits per base pair (bits/bp), or a compression ratio of 6.4× over the ASCII coding (8 bits/bp). Far better compression of 38× was achieved by Spring (Chandak et al. 2019), a specialized method for read compression. Note, neither gzip nor Spring enable search or alignment against the input data. Then, we individually constructed lossless searchable representations of the read sets with counting de Bruijn graphs over the {A, C, G, T, N} alphabet with coordinate annotations, as well as BLAST databases and sparse PufferFish indexes. To compare the alignment speed for counting de Bruijn graphs with TCG-Aligner (see the Methods subsection “Trace-consistent graph-based aligner (TCG-Aligner)”) and other methods that support queries (BLAST, MegaBLAST, and PufferFish), we assembled a list of nine defining mutations of the SARS-CoV-2 21A (Delta) variant spike protein. This included six sequences of length 59 and one of length 53 (to cover the deletion variants, see Supplemental Section 6 for a list of the sequences). The results are shown in Table 2.
The summary of different representations constructed from Virus PacBio HiFi read sets
The BLAST database required on average 2.68 bits/bp, and constructing an additional MegaBLAST index on top increased its size to an average of 125.85 bits/bp, whereas PufferFish required on average 21.8 bits/bp. We note, however, that these tools are intended for indexing reference genomes (Altschul et al. 1990; Morgulis et al. 2008; Almodaresi et al. 2018), and hence, their representations are not intended to take full advantage of the degree of similarity among the reads in the indexed read set for compression. In contrast, the compression performance of counting de Bruijn graphs was even better than gzip-9. They required on average only 0.54 bits/bp (57% <1.25 bits/bp for gzip-9) while at the same time being fully searchable. Of these 0.54 bits/bp, on average 28% is taken by the de Bruijn graph representation, and the remaining 72% is taken to represent the positional annotations. For a more detailed comparison of the top four methods, see Supplemental Figure 3. Despite the considerably higher compression ratio, our method achieves an alignment speed that is comparable to the state-of-the-art methods (Table 2). We also did a similar evaluation of the methods on the full Virus PacBio SMRT data set (see Supplemental Table 3).
Indexing Illumina RNA-seq reads
To evaluate compression performance on short reads, we used the RNA-seq read sets described in the preceding Results subsection “Indexing k-mer counts from 2652 RNA-seq read sets.” However, instead of indexing k-mer counts, here we constructed counting de Bruijn graphs with coordinate annotations. We indexed all 31-mers in each read set without any other filtering. In total, 2411 read sets (7.55 Tbp) were indexed, with the remaining samples discarded owing to containing reads of variable length or only reads shorter than 31.
Again, we compared the presented approach with two alternatives commonly used for compressing read data: the general-purpose compressor gzip and the domain-specific Spring. The results are presented in Supplemental Figure 4 and discussed in Supplemental Section 5. Counting de Bruijn graphs generated on average 27% smaller representations of the input reads compared with gzip-9 (1.488 bits/bp vs. 2.030 bits/bp) (see Supplemental Table 4).
Dependence on k-mer length
To investigate the relationship of k-mer length on index size, we compressed several representative read sets for 12 different k-mer lengths (Supplemental Fig. 5). We chose one of the human RNA-seq samples (SRA: SRR805801), representing low-error Illumina sequencing, and a Sinorhizobium genome sequencing sample, representing higher-error PacBio long-read sequencing. Although for short, low-error reads, the graph size slightly increases with k, the number of paths in the graph grows as well, which makes the annotation size drop steadily, owing to longer unitig length. As a result, the overall index size (combining graph and annotation) decreases with k. In contrast, the higher error rate in PacBio reads (SRA: SRR3747284) leads to a very large number of k-mers in the graph and, hence, its size. However, long reads with lower error (SRA: SRR13577847, PacBio HiFi) benefit from an increase of k. As a practical consequence, the choice of a large k is beneficial for compression in most scenarios.
Lossless index of RefSeq with k-mer coordinates
To show the use of counting de Bruijn graphs with coordinate annotations for indexing reference genomes, we used a data set consisting of all 32,881,422 reference sequence accessions from release 97 of the NCBI RefSeq database (O'Leary et al. 2016). Each sequence has been annotated with its associated accession ID along with all k-mer coordinates (k = 31). This approach forms an alternative to the commonly used MegaBLAST search tool, which requires an additional database index (Morgulis et al. 2008) for competitive high-throughput search. The summary of both indexes is presented in Table 3 (an extended version is available in Supplemental Table 5).
Lossless indexing of RefSeq (rel. 97) with k-mer coordinates for the complete data set (32,881,422 accessions, 1.7 Tbp, 483 GB compressed with gzip-9) and the set of all fungi (69,034 accessions, 8.8 Gbp, 2.6 GB compressed with gzip-9)
Using our method, the input of 1.7 Tbp was losslessly represented in a self-index comprising 509 GB, which is 4.6× smaller than the corresponding MegaBLAST index (Table 3). We also present the performance for the subset of RefSeq containing all fungi genomes, which we have used for measuring the sequence alignment performance in the following section.
To measure the alignment speed against the constructed indexes, we aligned reads from a metagenomic sequencing sample (SRA: SRR10002688) in two settings. Because the entire MegaBLAST representation did not fit in our SSD, we split it into five parts, which we queried separately and aggregated the total query time and peak RAM from these five separate queries. To benchmark the alignment speed in an online regime where only a single read is aligned, we randomly selected 100 reads and aligned each of them to each of the constructed representations (BLAST, MegaBLAST, and counting de Bruijn graph). Then we averaged the alignment time and the peak RAM across these queries. In the second setting, we measured the alignment speed in the batch mode when aligning 1000 random reads from the same metagenomic sample. One can see that our approach has a favorable alignment speed while being orders of magnitude faster when aligning a single read. In contrast to MegaBLAST, our current implementation does not support memory mapping of the index and, hence, has to load the entire compressed index into RAM. However, because of its high compression, the representation size is still moderate, and in addition, ways for offloading it to disk and using memory mapping can be considered in future work.
Sequence-to-graph alignment with k-mer coordinates
We evaluated the accuracy of our algorithm for alignment to counting de Bruijn graphs and compared it to the state-of-the-art aligners. We used simulated reads as queries and defined the desirable ground truth for these alignments as the corresponding segments of the reference from which the query reads were simulated. Given the human GRCh38 reference Chromosome 22 (Schneider et al. 2017) and the Escherichia coli NC_000913.3 (Riley et al. 2006) genomes, we use ART (Huang et al. 2012) and pbsim (Ono et al. 2013) to simulate 2000 Illumina-type and 200 PacBio-type reads of lengths 150 and 10,000, respectively. After aligning each read back to their respective reference (or graph for sequence-to-graph aligners), we compute the edit distance of the matching sequence (alignment) to the ground-truth sequence (i.e., the original reference segment from which the read was simulated) to measure alignment accuracy. In our evaluation, all nucleotides that are clipped from a query sequence by the aligner contribute as edits when measuring distance. Note, this measure is agnostic to the choice of the scoring approach used by the aligners. In addition, even a simulated read with a large number of errors can still contribute an edit distance of zero in the evaluation if an aligner matches it to the exact ground-truth sequence.
We computed the measure described above and compared our TCG-Aligner (see the Methods subsection “Trace-consistent graph-based aligner (TCG-Aligner)”) to other state-of-the-art methods (Morgulis et al. 2008; Garrison et al. 2018; Li 2018; Rautiainen and Marschall 2020; Karasikov et al. 2020a; Almodaresi et al. 2021), run with default settings. As shown in Figure 4, the degree to which incorporation of coordinates in the alignment procedure improves accuracy and query execution time (see also Supplemental Table 2) is dependent on the complexity of the target genome.
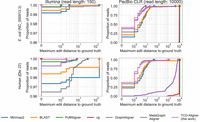
Alignment accuracy on simulated Illumina- and PacBio-type reads (E. coli NC_000913.3 and human Chr 22). The edit distance is measured between the alignment (the returned path in the graph) and the ground-truth sequence. In the top left subplot, the curves of vg- and TCG-Aligner are superimposed.
This is evident for the alignments of simulated E. coli reads, where the use of coordinates provides a limited improvement in accuracy and query time owing to the simplicity of the genome (Fig. 4, top row; Supplemental Table 2). On the other hand, as can be seen in the bottom row of Figure 4, it significantly improves alignment accuracy and query time for human reads: 40.2% of human PacBio reads align to the exact ground-truth sequence when using TCG-Aligner compared with 0.49% with the MetaGraph aligner.
For MetaGraph and vg, the large edit distances of the PacBio read matches relative to the ground truth are owing to the aligners reporting shorter local alignments, rather than alignments of the full reads. By incorporating coordinates to more effectively filter seeds and by restricting the alignment search space to graph traces, TCG-Aligner provides improved accuracy and query execution time when aligning against reference genomes.
Searching for the delta variant of SARS-CoV-2 in SRA
Finally, to enable fast search in the entire collection of all 152,884 viral PacBio SMRT read sets from SRA (see the Results subsection “Indexing all viral HiFi reads”), we also indexed them in a joint counting de Bruijn graph with coordinate annotation. Being 178 GB in size and only 19% larger than the input reads without quality scores compressed with gzip-9 (150 GB, 875 Gbp), our index provides a fully lossless representation of them and can be used for search and alignment of arbitrary sequences. A total of 152,272 (99.6%) of the indexed read sets originate from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA716984, consisting of PacBio Sequel II sequencing runs from SARS-CoV-2 samples.
We queried DNA sequences flanking the nine defining mutations of the SARS-CoV-2 21A (Delta) variant spike protein (the same mutations as used for the alignment benchmark in the Results subsection “Indexing all viral HiFi reads”) against this joint index to retrieve all the occurrences of its specific mutations within the reads. In total, six sequences of length 59 and one of length 53 (to cover the deletion variants, see Supplemental Section 6 for a list of the sequences) were queried. Each sequence was matched to an average of 107,892 samples and 7.68 million positions in the joint index. Despite the enormous number of returned hits, the query took under 4 min on a single thread.
In this experiment, for a given sample, we classified it as containing SARS-CoV-2 21A if, for each of the defining mutations of the spike protein, there exists at least one read in the sample supporting that variant. If a sample is classified as such, we enumerate all reads containing any of the defining mutations. As shown in Figure 5, 90.5% of samples deposited after July 2021 contain each of the Delta variants of the spike protein, leading to a sharp growth in the number of reads containing these variants. We would like to note that this analysis is derived only from the dates on which these samples were uploaded to the SRA and, hence, cannot be used to determine when these variants actually emerged. Although these metadata can be derived for further analysis, it is outside the scope of this work.

Detection of the delta variants of the SARS-CoV-2 spike protein in SMRT virus sequencing samples deposited on the SRA. (Left) The number of samples detected to contain (orange) and not contain (blue) the SARS-CoV-2 delta variant. (Right) The number of reads containing defining mutations contributed by samples containing the SARS-CoV-2 delta variant.
Discussion
We have presented a novel approach for the efficient and compressed representation of quantitative annotations on de Bruijn graphs. Together with the underlying graph, these annotations make up a data structure which we call a counting de Bruijn graph. It can be used to represent quantitative information and, in particular, encode traces of the input sequences in de Bruijn graphs. This not only provides a much higher flexibility of graph annotations but also allows for the truly lossless representation of any set of input sequences in them. This offers a practical solution to a long-standing problem of many methods using de Bruijn graphs as a base structure and opens the doors to implementing novel sequence alignment algorithms on top of them. The method is agnostic to the alphabet and, hence, can be used for indexing not only nucleotide sequences but also protein sequences or sequences over any other alphabet.
In addition to the presented approach for the compressed representation of sparse nonbinary matrices, we have generalized the RowDiff scheme (Danciu et al. 2021) to nonbinary graph annotations and optimized it by improving the algorithms for anchor and successor assignment. We have considered and have shown the advantages of the coding where multiple successor nodes may be assigned to each node. In future extensions, the aggregating operator g could act not only on the immediate successors of the node but on the whole tree of all successors spanning from it until the terminating anchor nodes. This would lead to a significantly better compression, for instance, in the case of k-mer counts linearly increasing within the paths in the graph. Moreover, an adaptive model can be trained with machine learning methods to predict annotation at a node from its successors and their annotations to further reduce the deltas stored explicitly in compressed data structures. We believe this has promising potential for this coding to benefit from the recent advances in machine learning.
Finally, we devised an algorithm for aligning sequences to counting de Bruijn graphs with coordinate annotations, which avoids spurious paths. This algorithm correctly estimates the alignment score even when aligning sequences with repeats to loops in the graph, which would be impossible with de Bruijn graphs alone. Because sequences shared by many samples are represented by a simple path, de Bruijn graph–based approaches can greatly reduce the overhead of aligning to collections of highly similar sequences, whereas more traditional database search methods would align to each database entry independently. The added availability of k-mer coordinates to the MetaGraph alignment framework (Karasikov et al. 2020a) allows for various other seeding or extension heuristics to be implemented, such as those used in MegaBLAST (Morgulis et al. 2008). Although the alignment method and evaluation presented here are restricted to graphs constructed from assembled reference genomes, such seed extension methods can be adapted for alignment to de Bruijn graphs constructed from raw read sets, which, we believe, is a promising direction for future work.
Playing a similar role for indexing sequences in de Bruijn graphs as gPBWT does in the realm of variation graphs and the indexing of pangenomes, we believe our approach is a significant step forward for the representation of and the search in very large collections of sequences, addressing a still increasing demand for interactive access to growing archives of biological sequences. We envision this as the first step toward enhancing the performance of BLAST-based sequence searches using graph-based approaches.
Software availability
The methods presented in this work were implemented within the MetaGraph framework (https://github.com/ratschlab/metagraph), which provides a means and base data structures for indexing very large collections of sequences in annotated de Bruijn graphs with a modular support of different graph and annotation representations, including the space-efficient BOSS table (Bowe et al. 2012), the optimized multi-BRWT (Karasikov et al. 2020b) scheme, and routines for the RowDiff (Danciu et al. 2021) sparsification, graph cleaning, differential assembly, and sequence-to-graph alignment (Karasikov et al. 2020a). We also used implementations of basic compression algorithms and data structures from the sdsl-lite library (Gog et al. 2014). The resources and scripts used to create figures and start experiments presented in section sec:results are available as Supplemental Code or at GitHub (https://github.com/ratschlab/counting_dbg).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Mario Stanke, Matthis Ebel, Daniel Danciu, and Stefan Stark for their helpful feedback. M.K. and H.M. are funded by the Swiss National Science Foundation grant 407540_167331 “Scalable Genome Graph Data Structures for Metagenomics and Genome Annotation” as part of Swiss National Research Programme (NRP) 75 “Big Data.” M.K., H.M., and A.K. are also partially funded by ETH core funding (to G.R.).
Author contributions: The idea and design of counting de Bruijn graphs and their representation techniques were conceived and developed by M.K.; the TCG-Aligner was conceived and developed by H.M.; and A.K. and G.R. conceptualized and supervised the research and acquired funding. All authors contributed to writing the manuscript and provided feedback on the algorithms, theory, and experiments.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276607.122.
- Received January 17, 2022.
- Accepted May 5, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








