
Listeria monocytogenes genes supporting growth under standard laboratory cultivation conditions and during macrophage infection
Abstract
The Gram-positive bacterium Listeria monocytogenes occurs widespread in the environment and infects humans when ingested along with contaminated food. Such infections are particularly dangerous for risk group patients, for whom they represent a life-threatening disease. To invent novel strategies to control contamination and disease, it is important to identify those cellular processes that maintain pathogen growth inside and outside the host. Here, we have applied transposon insertion sequencing (Tn-Seq) to L. monocytogenes for the identification of such processes on a genome-wide scale. Our approach identified 394 open reading frames that are required for growth under standard laboratory conditions and 42 further genes, which become necessary during intracellular growth in macrophages. Most of these genes encode components of the translation machinery and act in chromosome-related processes, cell division, and biosynthesis of the cellular envelope. Several cofactor biosynthesis pathways and 29 genes with unknown functions are also required for growth, suggesting novel options for the development of antilisterial drugs. Among the genes specifically required during intracellular growth are known virulence factors, genes compensating intracellular auxotrophies, and several cell division genes. Our experiments also highlight the importance of PASTA kinase signaling for general viability and of glycine metabolism and chromosome segregation for efficient intracellular growth of L. monocytogenes.
Listeria monocytogenes is a foodborne pathogen causing life-threatening infections in humans and animals. The pathogen is transmitted from environmental and animal sources to food processing plants, where it regularly causes food contamination. It can survive the passage through the gastrointestinal tract and may finally cross the gut epithelium to reach the bloodstream (Freitag et al. 2009). Because of the ubiquitous presence of L. monocytogenes in the environment, food contaminations and thus periods of asymptomatic carriage in the gut are quite common. Approximately 5%–10% of the population are considered to carry L. monocytogenes in the gastrointestinal tract, and it is estimated that healthy adults experience two periods of asymptomatic L. monocytogenes carriage per year (Grif et al. 2003; Hafner et al. 2021). L. monocytogenes that crosses the epithelial barrier is rapidly cleared from the bloodstream by macrophages and nonprofessional phagocytes in the liver and the spleen, which represent an important line of defense against the infection (Gregory et al. 1992; Ebe et al. 1999; Vázquez-Boland et al. 2001). L. monocytogenes may proliferate inside macrophages and hepatocytes and spread inside the liver parenchyma, even though this phase of infection is usually limited by innate immune defense and T cell–mediated cytotoxic mechanisms (Cousens and Wing 2000; Gregory and Liu 2000; Vázquez-Boland et al. 2001). In immunocompromised persons, however, in which an adequate immune response cannot be established, L. monocytogenes proliferation in the liver continues, facilitating systemic dissemination and translocation to other organs, primarily the brain and the uterus during pregnancy. Bacteremia, cerebral, and fetal infections are characteristically associated with high case fatality rates (Lamont et al. 2011; Charlier et al. 2017; Scobie et al. 2019; Wilking et al. 2021), and therefore, listeriosis is considered a major public health concern despite its low incidence (Werber et al. 2013).
L. monocytogenes is equipped with specific virulence factors to invade nonphagocytic cells in a phagocytosis-like process and to escape the phagosome after internalization, to multiply within their cytosol, and to spread from cell to cell (Vázquez-Boland et al. 2001; Camejo et al. 2011). Six of the most important virulence factor genes (prfA, plcA, hly, mpl, actA, plcB) cluster in the Listeria pathogenicity island LIPI-1 (Glaser et al. 2001). Although these genes coordinate virulence factor expression (PrfA) and act as hemolysin (Hly) and phospholipases (PlcA, PlcB) for penetration of host cell membranes or as nucleator of host actin polymerization for intracellular motility and cell-to-cell spread (ActA) (Camejo et al. 2011), further proteins are required for intracellular proliferation. These proteins contribute to the biosynthesis of selected nucleotides (Schauer et al. 2010; Faith et al. 2012), aromatic amino acids (Stritzker et al. 2004), and menaquinones (Chen et al. 2017; Smith et al. 2021), as well as to the uptake of certain amino acids (Borezee et al. 2000; Sun and O'Riordan 2010) to compensate for auxotrophies occurring in the cytosol or to ensure efficient separation of daughter cells after completion of cell division as a prerequisite for efficient intra- and intercellular spread (Lenz and Portnoy 2002; Halbedel et al. 2012).
The identification of genes required for intracellular proliferation of L. monocytogenes and for growth of the bacterium in general is an important task to facilitate development of novel antilisterial therapies or to design more efficient strategies to prevent growth of the bacterium in food matrices. Up to now, such L. monocytogenes genes were identified either in stepwise gene-by-gene approaches or by laborious screening of plasmid insertion mutant libraries (Joseph et al. 2006; Schauer et al. 2010), which, however, did not reach genome-wide saturation. Transposon insertion sequencing (Tn-Seq) is a technique that allows identification of genes, which do not accept transposon insertions without loss of viability. Such genes can be collectively identified by massive parallel sequencing of transposon insertion libraries, which do not need to be separated into individual clones (van Opijnen et al. 2009). Tn-Seq has been used to identify growth-promoting genes in a wide variety of bacteria. We here present the application of this technique to L. monocytogenes and have used it to identify genes required for growth of this important pathogen under standard laboratory cultivation conditions and during macrophage infection.
Results
Tn-Seq-based identification of genes required for growth under standard laboratory cultivation conditions
A highly saturated mariner transposon library was generated in the L. monocytogenes reference strain EGD-e with approximately 79,000 individual clones. Thus, >30% of the 216,636 TA sites (0.07 TA sites per base pair), which serve as possible transposon insertion sites, were factually hit by a transposon. The library was cultivated in BHI broth at 37°C as described in the Methods section (15 generations in total), and chromosomal DNA was isolated. Massive parallel sequencing of the transposon DNA junctions (Tn-Seq) (van Opijnen et al. 2009) was used for identification of all Tn insertion sites. This uncovered 66,708 unique insertion sites, confirming that 30.8% of all possible insertion sites were hit. Insertion sites were uniformly scattered around the chromosome as a consequence of the uniform distribution of TA sites (Fig. 1A). For the identification of genes required for growth under standard cultivation conditions, transposon insertions in the N-terminal 5% of the open reading frames (ORFs) and C-terminal 20% of the ORFs were neglected, and insertions were only counted if they were replicated at least three times. In addition, genes with an insertion density as defined by Langridge et al. (2009) below 0.01 (average insertion density in the data set, 0.32) were added, summing up to 394 genes (Supplemental Table S1; Supplemental Fig. S1). An assignment of functional categories showed that most of these genes encode components of the translation machinery; act in replication, segregation, and maintenance of the chromosome; or are involved in biosynthesis of the cell envelope (Fig. 1B). The function of 29 of these genes is not known (Supplemental Table S1); however, homologs of several of these 29 genes are also described as required for viability/growth in other Gram-positive bacteria (Supplemental Fig. S2).
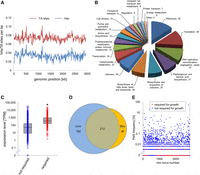
Identification of L. monocytogenes genes required for growth in BHI broth by Tn-Seq. (A) Distribution of TA sites and Tn insertion sites along the L. monocytogenes EGD-e genome. TA sites/hits per base pair were calculated using a coverage window of 10,000 bp. (B) Functional classes of the 394 L. monocytogenes EGD-e genes that are required for growth in BHI broth at 37°C according to Tn-Seq. (C) Expression levels of the 394 genes required for growth in comparison to all other L. monocytogenes EGD-e genes according to a previously published RNA-seq experiment also performed under standard growth conditions (Hauf et al. 2019). The asterisk indicates statistical significance (t-test, P < 0.0001). (D) Venn diagram illustrating the intersection between the 394 L. monocytogenes genes required for growth and the essential genes of B. subtilis 168 (n = 258). (E) Frequency of naturally occurring premature stop codons (PMSs) in each of the L. monocytogenes EGD-e genes as observed in a data set of 27,118 L. monocytogenes genomes from clinical and environmental isolates that were downloaded from the NCBI server.
Using previously published RNA-seq data (Hauf et al. 2019), we found that the 394 genes are on average fivefold stronger expressed, and not a single unexpressed gene was among them (Fig. 1C), which is in good agreement with their necessity for growth. For 330 out of the 394 genes, experimental information on their essentiality is not available. Fifteen genes could not be deleted in previous studies, or their depletion was lethal; however, deletion had been reported for 38 genes (Supplemental Table S1). The set of 394 genes largely overlaps with the 258 Bacillus subtilis essential genes (Fig. 1D; Zhu and Stülke 2018). Thus, our list mostly comprises real essential genes; however, conditional essential genes, growth-promoting genes, and genes contributing to stationary phase survival are likely included to a certain extent. Hence, we refer to the 394 genes as “genes required for growth” throughout the paper. The 46 essential B. subtilis genes that are specific to B. subtilis either do not have a homolog in L. monocytogenes (n = 13), are not essential in L. monocytogenes (n = 22), or are considered nonessential by the Tn-Seq-Explorer algorithm because Tn insertions are present in certain domains only (n = 3) or just above the cutoff (n = 8). We categorize these latter eight genes as potentially required for growth (Supplemental Table S1; Supplemental Fig. S1).
One hundred eighty-two genes were required for growth in L. monocytogenes according to Tn-Seq without being annotated as essential in B. subtilis. Twenty-two of these genes had no homolog in B. subtilis. The remaining 160 genes comprise (1) genes required for growth at increased temperature used for generation of the Tn mutant library (n = 7), (2) genes whose inactivation results in growth defects (n = 15), and (3) genes that are indeed essential in L. monocytogenes but not in B. subtilis (n = 4). A minority of nine genes was found to be required for growth even though previous work reported their deletion without adverse effects on growth (e.g., codY, cshA, cspD, fvrA, lexA, recA, relA, ribU, secDF; see Discussion) (Supplemental Table S1; Supplemental Figs. S1, S3; Bennett et al. 2007; van der Veen and Abee 2011; McLaughlin et al. 2012; Markkula et al. 2012b; Burg-Golani et al. 2013; Jiang et al. 2018; Rivera-Lugo et al. 2022).
Genes with naturally occurring premature stop codons
Genome sequences of L. monocytogenes isolates from environmental, food, and clinical samples currently are becoming available in a high number in the course of pathogen surveillance programs. Allelic information of these genomes is stored on genomic subtyping servers such as core genome MLST (cgMLST; https://cgmlst.org) (Ruppitsch et al. 2015). We extracted the allelic variations known in the L. monocytogenes population for all 2857 EGD-e genes from the cgMLST.org server and identified alleles containing premature stop codons (PMSs) located within 5% and 80% of their sequence relative to their start. Next, we counted the number of genomes that carried such PMSs for each of the 2857 EGD-e ORFs using a collection of 27,118 L. monocytogenes genomes from clinical and environmental isolates available at the NCBI Pathogen Detection (https://www.ncbi.nlm.nih.gov/pathogens/) pipeline at the time of analysis (NCBI 2020). This showed that no naturally occurring premature stop codons are known for 1424 ORFs, an observation that can be interpreted as functional gene essentiality under real-life conditions. Three hundred forty-eight (87%) of the 394 genes required for growth did not contain PMSs, and 42 (10%) of them had a PMS only once (Fig. 1E). Thus, the majority of all laboratory-confirmed genes that are required for L. monocytogenes growth also do not lose their functionality outside the laboratory. In contrast, higher PMS frequencies were observed among the nonrequired genes (Supplemental Table S2; Fig. 1E). Several genes, particularly those coding for extracellular proteins (among them, inlA), showed PMS frequencies >1%, likely reflecting natural antigenic variation. Thus, our Tn-Seq-based assignment of genes required for growth is in congruence with the observed frequency of gene functionality loss through spontaneous allelic variations leading to PMSs under real-life conditions.
Necessity of genes acting in central carbon metabolism for L. monocytogenes growth
L. monocytogenes generates ATP from glycolytic metabolism of hexoses or glycerol. In good agreement with the redundancy of phosphotransferase systems (PTSs) or glycerol uptake facilitators, genes for the uptake systems for any of these carbon sources were found to be inactivated by transposon insertions. In contrast, the genes for the complete glycolytic reaction chain were required for growth (Figs. 2, 3) with the exception of pykA, coding for pyruvate kinase (Fig. 3), suggesting that pyruvate can be generated from phosphoenolpyruvate via alternative routes. Pyruvate is further catabolized to lactate, acetate, acetyl-CoA, oxaloacetate, or acetoin (Pine et al. 1989; Romick et al. 1996; Schär et al. 2010). The ackA1 gene encoding acetate kinase for acetate formation and several pyruvate dehydrogenase component genes for acetyl-CoA formation were required for growth. With pyruvate oxidase (lmo0722) and phosphotransacetylase (lmo2103), two alternatives for formation of acetyl phosphate exist, explaining why both genes are dispensable despite the necessity of ackA1 (Fig. 3). Likewise, ldh, encoding lactate dehydrogenase, and its three paralogs were dispensable, as were the alsS and alsD genes required for acetoin formation. Pyruvate carboxylase PycA generates oxaloacetate for biosynthesis of several amino acids, and consequently, the pycA gene was required for growth.
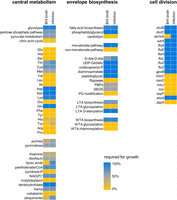
Necessity of genes acting in central metabolism, envelope biosynthesis, and cell division for L. monocytogenes growth. Heat maps illustrating the necessity of selected genes and pathways for growth of L. monocytogenes. Where genes were aggregated according to their pathways, their necessity for growth is expressed as the number of genes required for growth divided by the number of all genes in this particular pathway. Necessity for growth is shown for standard growth conditions (BHI broth) and during growth in J774 macrophages (infection).
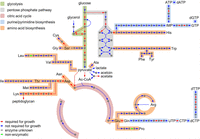
Necessity of genes acting in central carbon and nitrogen metabolism for L. monocytogenes growth in BHI broth. Scheme illustrating the metabolic pathways in central carbon metabolism, in biosynthesis of nucleobases and their corresponding nucleotides and biosynthesis of amino acids according to the KEGG database (https://www.genome.jp/kegg-bin/show_organism?org=T00066). Reactions are colored according to the necessity of their corresponding genes for growth of L. monocytogenes in BHI broth.
The tricarboxylic acid (TCA) cycle is incomplete in L. monocytogenes (Trivett and Meyer 1971), and in line with this, all TCA cycle genes were dispensable under the tested conditions (Figs. 2, 3). In contrast, selected genes for the upper oxidative part of the pentose phosphate pathway (PPP) were not found to be disrupted by transposons, whereas the genes in the lower nonoxidative part could largely be inactivated (Fig. 3). The only enzyme in this lower part of the pathway that is required for growth is lmo1818, which encodes one out of six paralogous ribulose-phosphate 3-epimerase genes and was the only paralog expressed under standard growth conditions in a previous transcriptome analysis (Hauf et al. 2019).
All steps in the biosynthesis of fatty acids from acetyl-CoA are mediated by enzymes that were required for growth (Fig. 2; Supplemental Fig. S4). One exception is enoyl-acyl carrier protein reductase, which occurs in four isoforms (FabI, FabL, FabK1, and FabK2) (Yao et al. 2016). The fabI and fabL genes were required for growth, whereas fabK1 and fabK2 are individually dispensable. Taken together, glycolytic glucose conversion to acetyl-CoA for fatty acid biosynthesis or to acetate for energy conservation represents the essential backbone of L. monocytogenes carbon catabolism (Figs. 2, 3).
Biosynthesis and acquisition of amino acids, purines, and pyrimidines
The biosynthetic pathways for all 20 proteinogenic amino acids are complete, but only some genes were required for growth in BHI broth according to Tn-Seq. Among them were the genes for aspartate amino transferase AspB (lmo1897), asparagine synthase AnsB (lmo1663), glutamine synthetase GlnA (lmo1299), and serine acetyltransferase CysE (lmo0238) (Figs. 2, 3). All genes required for lysine biosynthesis except the last gene in this pathway (lysA) were also required (Fig. 3). LysA catalyzes lysine production from diaminopimelate. This pattern of gene necessity shows that lysine can be acquired from external resources, whereas diaminopimelate is essential for peptidoglycan (PG) biosynthesis and must be synthesized. The components of two peptide transport systems were indispensable for growth: the ctaP (lmo0135)-lmo0136-lmo0137 cysteine transporter and the oppD and oppF ATPase subunits of the oppABCDF oligopeptide transporter (Borezee et al. 2000). This suggests that uptake is the preferred mode of amino acid acquisition in rich medium, as previously suggested by metabolomics (Kutzner et al. 2016). L. monocytogenes encodes all genes necessary for de novo biosynthesis of purines and pyrimidines (Fig. 3; Glaser et al. 2001) and additionally contains several genes for nucleobase uptake (lmo0456, lmo0573, lmo1839/pyrP, lmo1845, lmo1884, lmo2254). Because biosynthesis and uptake can mutually replace each other, the genes for these transporters and all genes required for nucleobase biosynthesis were dispensable. In contrast, many of the genes required for generation and interconversion of nucleotides are indispensable under the condition analyzed here (Figs. 2, 3).
Cofactors and vitamins
L. monocytogenes requires riboflavin, thiamine, biotin, and lipoic acid to grow in minimal medium (Premaratne et al. 1991; Tsai and Hodgson 2003). Consequently, their biosynthetic pathways are incomplete (Glaser et al. 2001), and the few remaining genes were not required for growth. The riboflavin/FMN/FAD transporter gene ribU (lmo1945) (Matern et al. 2016; Rivera-Lugo et al. 2022) was not found to be disrupted by transposons, implying the absence of alternative uptake systems in EGD-e (Supplemental Fig. S3). In contrast, the only known thiamine (lmo1429) (Schauer et al. 2009) and biotin uptake genes (lmo0598) (Dowd et al. 2011) were not required for growth. The pdxST genes encoding pyridoxal phosphate synthase were also not required for growth (Fig. 2), suggesting the presence of alternative routes for acquisition of these latter compounds.
The entire gene set for the de novo formation of NAD(P)+ from aspartate and nicotinate is present, but only nadD, nadE, and nadF (lmo0968) for the terminal reactions were required for growth (Supplemental Fig. S5).
Two pathways feed pantothenate biosynthesis, one starting from pyruvate (ilvB, ilvH, ilvC, ilvD) and the second from valine (lmo0978). Genes for both pathways were dispensable up to the point where they merge, and the genes downstream were required for growth (panB, lmo2046, panC, panD), as were the genes necessary for coenzyme A formation from pantothenate (lmo1825, coaD, coaE, acpS, acpP). The lmo0221 and lmo0922 encoding two alternative pantothenate kinases were a notable exception (Supplemental Fig. S5).
The eight genes for molybdopterin biosynthesis from GTP are complete in EGD-e (lmo1038, lmo1042, lmo1043, lmo1044, lmo1045, moaC, moaA, lmo1048), but none of them was required for growth, suggesting alternative routes for molybdopterin acquisition (Supplemental Fig. S5). In contrast, all genes for biosynthesis of tetrahydrofolate (THF) from GTP (folE, folA, folK, sul, folC, lmo1873) and THF regeneration (fmt, folD, thyA) were required for growth (Supplemental Fig. S5).
The genes for heme, vitamin B12, and menaquinone biosynthesis are completely present. A significant number of heme biosynthesis genes was required for growth (Supplemental Fig. S6), despite the presence of heme transporters (Klebba et al. 2012). In contrast, genes for vitamin B12 and menaquinone biosynthesis were largely dispensable for growth in BHI broth (Fig. 2; Supplemental Fig. S6).
Taken together, essential enzymes acting in biosynthesis of pantothenate/CoA, THF, or NAD(P)+ could be the promising targets for identification of novel antilisterial compounds.
Envelope biosynthesis and cell division
More stringent essentiality patterns are observed in the pathways for biosynthesis of the cellular envelope (Fig. 2). Genes for the cytosolic steps of PG biosynthesis up to lipid II (glmSMU, murABCDEF, mraY, murG) were collectively required for growth, as well as the genes for diaminopimelate production from aspartate and generation of D-Ala-D-Ala from D-alanine (Fig. 2; Supplemental Fig. S4). The two enzymes for generation of undecaprenyl-P from farnesyl-PP were also required for growth (lmo1315, lmo2017), but from the two pathways for isoprenoid biosynthesis, only the mevalonate pathway genes were not hit by transposons, whereas all MEP/DOXP pathway genes could be inactivated (Fig. 2; Supplemental Fig. S4). Listeria encodes two potential MurJ-like flippases for lipid II transport (lmo1624, lmo1625), and both could be disrupted. Moreover, only one (ftsW1) out of six SEDS-type transglycosylases and two (pbpB1, pbpB2) out of five high-molecular-weight penicillin-binding proteins were required for growth (Supplemental Fig. S4), as shown in previous gene deletion experiments (Rismondo et al. 2015; Rismondo et al. 2019).
Similarly, the genes participating in phospholipid biosynthesis were generally required for growth, but cardiolipin synthase was not (Supplemental Fig. S4). Also, the genes contributing to formation of wall teichoic acids (WTAs) were not inactivated by Tn insertions, with the exception of the two tagO genes (lmo0959 and lmo2519), two paralogs for the first enzymatic step that can compensate each other (Eugster and Loessner 2012). The dltABCD genes for WTA decoration with D-alanine were all required for growth under our conditions, whereas the rmlTACBD and lmo2550, gtcA, and lmo1079 genes for rhamnosylation (Carvalho et al. 2015) and glycosylation of WTAs (Rismondo et al. 2018), respectively, as well as several key enzymes in lipoteichoic acid biosynthesis were dispensable (Supplemental Fig. S4). Likewise, most core divisome components were required for growth. However, genes with auxiliary function in divisome assembly and turnover (divIVA, minCD, minJ, sepF, and zapA) were found to be inactivated (Fig. 2). The lmo2472 gene, encoding a homolog of the B. subtilis auxiliary cell division gene whiA, was an interesting exception. This gene was found to be required for growth in L. monocytogenes even though its deletion only gave a mild phenotype in B. subtilis (Surdova et al. 2013). An L. monocytogenes mutant lacking this gene could be generated, and this mutant was viable at 37°C but had a growth defect at 42°C (Supplemental Fig. S7). This further supports our assumption that heat-sensitive mutants are counterselected in our approach.
Implications for PASTA kinase signaling
We previously showed that prkA, encoding a eukaryotic-like serine/threonine protein kinase important for regulation of PG biosynthesis, is essential in L. monocytogenes EGD-e (Wamp et al. 2020, 2022). Among the substrates of this kinase are YvcK and ReoM, both contributing to PG biosynthesis (Pensinger et al. 2016; Wamp et al. 2020, 2022; Kelliher et al. 2021). The precise role of YvcK is not clear, but ReoM controls ClpCP-dependent degradation of MurA, which catalyzes the first committed step in PG biosynthesis (Wamp et al. 2020). In contrast to our previous results, prkA was hit by Tn insertions. However, the vast majority of Tn insertions in prkA clustered in the C-terminal half of the gene and thus separated the extracellular PASTA domains from the cytoplasmic kinase domain. In contrast, only few insertions not exceeding background signals were seen in the kinase domain (Fig. 4A; Supplemental Fig. S1). To confirm that the prkA kinase domain and not the PASTA domains is required for viability, we tried to remove the prkA C terminus from the chromosome. A viable mutant lacking the PASTA domains (prkAΔC) could be generated and grew with wild-type kinetics under standard laboratory conditions, whereas PrkA depletion prevented growth (Fig. 4B). The prkAΔC strain was susceptible to ceftriaxone (Fig. 4C), suggesting perturbation of PG biosynthesis. MurA levels were not reduced as previously shown for PrkA-depleted cells (Wamp et al. 2020). Instead, the MurA level in the prkAΔC mutant accumulated slightly above the wild-type level (Fig. 4D). Because situations in which ReoM cannot be phosphorylated lead to rapid MurA degradation and thus are lethal (Wamp et al. 2020), this suggests that the isolated PrkA kinase domain is active and phosphorylates ReoM (Wamp et al. 2020). Apparently, the kinase domain provides the enzyme function required for growth, whereas the PASTA domains are dispensable for growth under standard cultivation conditions. In contrast, the prkAΔC strain shows a significant delay during intracellular replication (Fig. 4E), indicating that the PASTA domains become critical for growth during infection.
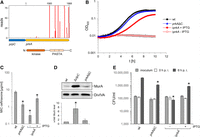
Dispensability of the prkA PASTA domains for L. monocytogenes viability. (A) Tn insertions at the prkA locus. Numbers refer to nucleotides in the prkA gene. The arrangement of PrkA domains is given below. (B) Viability of an L. monocytogenes prkAΔC mutant. L. monocytogenes strains EGD-e (wt), LMSW84 (iprkA), and LMS278 (prkAΔC) were grown in BHI broth (containing IPTG where indicated) at 37°C. Average values and standard deviations calculated from technical replicates (n = 3) are shown. (C) Increased cephalosporin sensitivity of an L. monocytogenes prkAΔC mutant. Susceptibility of the same set of strains as in B against ceftriaxone was determined using E-tests. Average values and standard deviations were calculated from three independent repetitions. Asterisks mark statistically significant differences (P < 0.01, t-test with Bonferroni–Holm correction). (D) MurA level in an L. monocytogenes prkAΔC mutant. Western blots showing signals specific for MurA (top) in L. monocytogenes strains EGD-e (wt), LMJR138 (ΔclpC), and LMS278 (prkAΔC). A parallel western blotting showing DivIVA-specific signals was included for a control (middle). MurA signals were quantified by densitometry, and average values and standard deviations calculated from three independent repetitions are shown (bottom). The asterisk indicates significance level (P < 0.05, t-test with Bonferroni–Holm correction, n = 3). (E) Reduced intracellular multiplication of the prkAΔC mutant. The same set of strains as above was used to infect J774 mouse macrophages, and bacterial cell numbers were determined right after (0 h post infection [p.i.]) and 6 h p.i. The experiment was performed as a triplicate from which average values and standard deviations were calculated. Statistical significance is indicated by asterisks (P < 0.001, t-test with Bonferroni–Holm correction).
Tn-Seq-based identification of genes important during macrophage infection
Next, Tn-Seq was used to identify genes that become essential during infection. For this, J774 mouse macrophages were infected with the Tn insertion library; samples were collected before and 24 h after infection; and the number of Tn insertions in each gene before and after infection was determined. All genes with a significant (P ≤ 0.05) reduction of Tn insertions during infection of at least twofold in two independent experiments were considered as important during intracellular stages. We identified 42 genes that were required for intracellular growth using this strategy (Table 1). First of all, four of the seven virulence genes that form the Listeria pathogenicity island LIPI-1 (prfA, plcA, hly, plcB) were identified, which shows the suitability of our approach (Fig. 5A; Table 1). The two LIPI-1 genes actA and mpl were not among the identified genes. However, a ΔactA mutant was able to replicate in J774 macrophages with wild-type kinetics in a separate experiment (Fig. 5B), suggesting that mutants in genuine spreading genes are not counter-selected in our J774 setup. Besides this, a number of other genes were identified that also had been described previously as important during infection, including genes acting in purine/pyrimidine (purA, purB, pyrD) (Faith et al. 2012) or menaquinone biosynthesis (aroE, aroF, menB, menC, menF, menI) (Stritzker et al. 2004; Chen et al. 2017; Smith et al. 2021), utilization of branched-chain amino acids (bkdAA, bkdAB) (Sun and O'Riordan 2010), regulation of flagellar motility (mogR) (Gründling et al. 2004), daughter cell separation (divIVA, secA2) (Lenz et al. 2003; Halbedel et al. 2012), lipoate metabolism (lipL, lplA1) (O'Riordan et al. 2003; Dowd et al. 2016), or RNA degradation (nrnA) (Table 1; Gall et al. 2022).
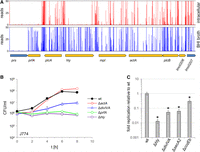
Requirement of selected virulence genes for intracellular growth in macrophages. (A) Diagrams visualizing the necessity of LIPI-1 genes during growth under standard growth conditions (BHI broth; blue) and during intracellular growth in J774 mouse macrophages (intracellular; red). Tn insertions are mapped on the chromosomal region carrying the LIPI-1 genes (prfA-lmo0206). Tn insertions are not found in genes that are required for growth under a certain condition. The prs gene preceding LIPI-1 is required for growth even under normal growth conditions. (B) Intracellular multiplication of L. monocytogenes strains EGD-e (wt), LMS250 (Δhly), LMS251 (ΔactA), and BUG2214 (ΔprfA) inside J774 mouse macrophages over a time course of 6 h. Strain LMS2 (ΔdivIVA) was included for comparison. The experiment was performed in triplicate. Average values and standard deviations are shown. (C) Intracellular replication of L. monocytogenes strains EGD-e (wt), LMS250 (Δhly), LMS2 (ΔdivIVA), LMS81 (ΔsecA2), and LMJR87 (ΔcozEb) mutants in J774 mouse macrophages. Ratio of bacterial titers right after and 6 h after infection is shown. The wild type replicated 35-fold during this time and was arbitrarily set to one. Average values and standard deviations were calculated from experiments performed in triplicate. Statistical significance is indicated by asterisks (P < 0.01, t-test with Bonferroni–Holm correction).
L. monocytogenes genes required for intracellular growth during macrophage infection
Among the genes so far not linked to intracellular survival were gcvPA and gcvPB, encoding two components of the glycine cleavage system (GCS); the thrC gene encoding threonine synthase; and the lmo2214-2215 genes, coding for the components of an ABC transporter of unknown function. Several genes with putative functions in PG biosynthesis (cozEb, ylaN, yvcK, walJ) and cell division (divIVA, ftsK, parA, secA2) also showed up in this screen (Table 1), out of which some (divIVA, secA2, yvcK) had been identified in previous work (Lenz et al. 2003; Halbedel et al. 2012; Pensinger et al. 2016).
Novel genes required for intracellular replication
To validate the Tn-Seq results, we created clean deletion mutants in cozEb, walJ, ftsK, parA, gcvPA-gcvPB, thrC, and lmo2214-2215 by allelic replacement. First, replication of the ΔcozEb mutant was analyzed in J774 macrophages. The ΔcozEb mutant generated only 31 ± 16% of the bacterial titer obtained with the wild type after 6 h of intracellular growth (Fig. 5C), even though no general fitness effect was associated with the cozEb deletion in broth culture (Fig. 6A). Although this degree of attenuation was clearly less pronounced than that observed with a Δhly mutant (1.2 ± 0.5%), it confirmed the importance of cozEb for growth inside macrophages. The cozEb gene is located downstream from secA2 in the chromosome of different Listeria species, which together with divIVA is crucial for autolysin secretion and virulence (Halbedel et al. 2012). The divIVA and secA2 genes were also identified in the screen (Table 1), but ΔdivIVA and ΔsecA2 mutants showed a stronger degree of attenuation (3.7 ± 2.5% and 4.9 ± 3.1% of wild-type CFU numbers, respectively) than the ΔcozEb mutant (Fig. 5C), suggesting a distinct function for cozEb.
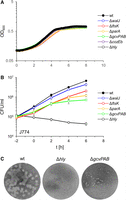
Novel genes contributing to L. monocytogenes growth inside macrophages. (A) Growth of L. monocytogenes strains EGD-e (wt), LMS250 (Δhly), LMJR87 (ΔcozEb), LMS283 (ΔwalJ), LMS284 (ΔftsK), LMS290 (ΔparA), and LMS305 (ΔgcvPAB) in BHI broth. (B) Intracellular growth of the same set of strains in J774 mouse macrophages. Average values and standard deviations were calculated from triplicates. (C) Plaque formation assay testing cell-to-cell spread of the ΔgcvPAB mutant (strain LMS305) in 3T3 mouse embryo fibroblasts. L. monocytogenes strains EGD-e (wt) and LMS250 (Δhly) were included for control.
Intracellular replication of the remaining mutants was tested in a separate experiment. Although mutants lacking the lmo2214-2215 ABC transporter and thrC did not show a delayed intracellular replication, the mutant lacking walJ showed a modest growth retardation intracellularly (37 ± 27% of wild-type CFU numbers after 6 h, P < 0.05) (Fig. 6B). In contrast, intracellular growth of mutants lacking either ftsK (32 ± 24%, P < 0.01), parA (25 ± 19%, P < 0.005), or gcvPAB (20 ± 16%, P < 0.001) was clearly delayed (Fig. 6B), despite the absence of growth defects in BHI broth (Fig. 6A). Finally, the same strains were tested in plaque formation assays for possible defects in cell-to-cell spread. This revealed a strong defect for the ΔgcvPAB mutant (plaque area: 8 ± 3% of wild-type level) (Fig. 6C), whereas no effects were found for the remaining mutants. Taken together, the cozEb, walJ, ftsK, parA, and gcvPAB genes are required for optimal replication in the cytosol of macrophages, and gcvPAB is additionally essential for cell-to-cell spread.
Discussion
We here have identified the complement of genes that is required to support growth of L. monocytogenes under standard conditions and during growth in macrophages. With 394 genes, almost 14% of all genes are required for growth in rich medium. Similar numbers of genes required for growth were discovered by Tn-Seq in other bacteria, such as Staphylococcus aureus (420), Streptococcus pyogenes (298), Enterococcus faecalis (349), Escherichia coli (358), or Pseudomonas aeruginosa (352) (Valentino et al. 2014; Le Breton et al. 2015; Lee et al. 2015; Goodall et al. 2018; Gilmore et al. 2020). In Mycoplasma genitalium, which contains one of the smallest genomes of self-replicating organisms, 382 out of 482 protein encoding genes (79%) are essential (Glass et al. 2006). We observed a large overlap between the L. monocytogenes set of genes required for growth and the 258 essential genes determined by stepwise gene inactivation in B. subtilis (Kobayashi et al. 2003; Commichau et al. 2013). Moreover, naturally occurring premature stop codons were underrepresented in the 394 L. monocytogenes genes required for growth. Thus, our assignment represents a first estimation of the essential L. monocytogenes genome, even though several limitations must be considered. First, a transient incubation at 40°C was required during library generation, and this has probably counterselected heat-sensitive mutants, for example, in whiA (this work), gpsB, clpP, dnaK, or lmo1450 (Hanawa et al. 1999; Gaillot et al. 2000; Markkula et al. 2012a; Rismondo et al. 2016). Second, we found several genes as required for growth by Tn-Seq, even though their inactivation had been reported by others. Heat sensitivity and different strain backgrounds could be reasons for this. Also, some reported mutants in genes that we consider as required for growth showed growth defects (ackA, ltaS, oppD, oppF, sipZ, sod, smpB, lmo2487) (Bonnemain et al. 2004; Gravesen et al. 2004; Archambaud et al. 2006; Gueriri et al. 2008; Webb et al. 2009; Mraheil et al. 2017; Krypotou et al. 2019), longer lag phases (dltA, ptsI) (Abachin et al. 2002), genetic instability (spoVG1) (Burke and Portnoy 2016), or sensitivities against oxidative stress (menA, sod) or aerobic conditions (spxA1) (Chen et al. 2017; Whiteley et al. 2017). Most likely, Tn insertions in such genes were counterselected because of their detrimental effects on growth at the chosen selection conditions. In other instances, insertional disruptions were reported for genes that we classify as required for growth, but deletion of these genes (hprK, ptsH, pycA) turned also out to be impossible (Mertins et al. 2007; Schär et al. 2010). Probably, these genes are in fact essential, because insertional disruptions are genetically unstable and may lead to erroneous conclusions because transient merodiploidy with one wild-type and one mutant allele in the same cell may arise during chromosome replication. Furthermore, suppressor mutations might also have rescued otherwise lethal gene deletions right during strain construction. Thus, it could be interesting to re-examine mutants in genes that we found to be required for growth but whose deletions were without adverse effects on growth in other studies (i.e., the genes shown in Supplemental Fig. S3), for the possible presence of suppressor mutations, and for their heat sensitivity or to recreate these deletions in a clean background. One of the genes in this category, ribU, was required for growth in BHI broth in our hands but could be deleted in the background of strain 10403S (Rivera-Lugo et al. 2022). RibU is the flavin binding protein of an energy coupling factor (ECF) transporter and required for uptake of flavins, for which L. monocytogenes is auxotrophic (Rivera-Lugo et al. 2022). Thus, 10403S must have alternative routes of flavin uptake that, however, are absent in EGD-e.
Ribosomal genes, genes acting in protein biosynthesis, and genes with functions in replication, segregation, and maintenance of the chromosome constitute the three largest groups within the 394 genes. Furthermore, stringent patterns of gene essentiality were also observed along central metabolic pathways, first of all glycolysis, and the pathways for biosynthesis of the cell envelope. In contrast, the majority of genes for biosynthesis of amino acids and nucleotides was not required for growth, which reflects the presence of efficient uptake systems. Most of these genes will become indispensable in minimal medium, in which all amino acids (except cysteine, for which EGD-e is auxotrophic) need to be synthesized de novo using ammonium as the nitrogen source (Tsai and Hodgson 2003). The need of lysine and aspartate biosynthesis genes for growth probably reflects the role of the intermediate/descendant diaminopimelate as a constituent of the PG peptide bridges. PG accounts for one-fifth of the weight of a Gram-positive cell (Reith and Mayer 2011), and the high number of diaminopimelate molecules needed for this might be difficult to satisfy based on aspartate uptake alone.
The pathways for thiamine, riboflavin, lipoic acid, and biotin are incomplete (Glaser et al. 2001; Tsai and Hodgson 2003), and their few remaining genes were mostly found disrupted by transposons. Consequently, these cofactors must be acquired through essential transporters. In contrast, the genes for biosynthesis of the other cofactors are present, but their necessity for growth varies. There is a high essentiality ratio in the pathways for coenzyme A, NAD(P)+, and THF biosynthesis, ruling out the existence of uptake systems but also disclosing the potential of their enzymes as drug target candidates. The genes for biosynthesis of pyridoxal-phosphate, molybdopterin, porphyrins, and ubiquinones, although invariably present, are mostly or partially dispensable during growth in BHI broth. Thus, these compounds or precursors thereof must be taken up via known (porphyrins) or yet-to-be-identified transporters (pyridoxal-phosphate) or could be dispensable during standard growth (ubiquinones and molybdopterin). A hemin transporter is known in L. monocytogenes (Xiao et al. 2011), and an ABC transporter for pyridoxal-phosphate was recently discovered in the γ-proteobacterium Actinobacillus pleuropneumoniae (Pan et al. 2021). A homolog of this transporter is not present in L. monocytogenes, but one of the many other ABC transporters could execute this function.
Only 42 genes become additionally necessary when L. monocytogenes grows inside mouse macrophages. Possibly more genes would be identified in the analysis if other time points after infection are sampled or if other types of macrophages (or even nonphagocytic cell lines) are used as an infection host. Only half of the genes that we identify act in uptake or biosynthesis of cellular building blocks, suggesting a decent nutrient supply inside the host cell. Among this latter group of genes are such for biosynthesis of certain nucleobases, menaquinones, and D-alanine, as well as for acquisition of lipoic acid. This is concordant with previous studies (Thompson et al. 1998; O'Riordan et al. 2003; Stritzker et al. 2004; Faith et al. 2012; Chen et al. 2017), but it raises the question as to how these compounds contribute to intracellular growth. Scarcity of lipoate and absence of D-alanine explain conditional essentiality of lplA1, lipL, and daaA (Thompson et al. 1998; O'Riordan et al. 2003). Likewise, a shortage of nucleotide precursors, primarily uracil and xanthine, for the uptake of which many putative transporter genes are present in the genome (Elbourne et al. 2017), could explain the necessity of the nucleotide biosynthesis genes purA, purB, and pyrD during intracellular growth. The aro and men genes are collectively needed for menaquinone biosynthesis, and although menaquinones are not required for growth in macrophages (Chen et al. 2017), one of the intermediates in this pathway, 1,4-dihydroxy-2-naphthoate (DHNA), was shown to be required for intracellular survival. The specific reason is not clear yet, but DHNA possibly facilitates redox reactions as a cofactor (Smith et al. 2021).
The GCS components GcvPA/B were required for intracellular growth and for cell-to-cell spread. GCS is a lipoate-dependent enzyme system similar to the branched-chain alpha-keto acid dehydrogenase (BKD) enzyme complex, whose genes were also needed for growth inside macrophages. BKD is involved in formation of branched-chain fatty acids, which become necessary during infection (Sun and O'Riordan 2010), whereas GCS transfers a methyl group to THF, the biosynthesis of which is strictly essential. Intracellular necessity of BKD and GCS would also provide an explanation for the importance of lipoate transfer enzymes for intracellular growth. GCS might be required intracellularly because methylated THF is a methyl group donor in other essential reactions (e.g., during pantothenate biosynthesis) or because NADH2 generated during glycine degradation is useful as source of energy and electrons.
Our study also highlights the importance of cell division, PG biosynthesis, and chromosome segregation for extra- and intracellular growth. First of all, several cell division genes are indispensable for standard growth (ezrA, ftsA, ftsEX) or growth at high temperature (whiA) even though their homologs in B. subtilis are not (Beall and Lutkenhaus 1989; Levin et al. 1999; Garti-Levi et al. 2008; Surdova et al. 2013). The essentiality of genes like ezrA (Considine et al. 2011) may be masked in B. subtilis owing to a higher degree of gene redundancy; however, essentiality in L. monocytogenes may open up new starting points to further investigate the function of these genes in bacterial cell division. Second, several new genes (cozEb, ftsK, parA, walJ) from this functional category have been identified as crucial inside the host cell, even though their function is not understood in all cases. FtsK and ParA are both ATPases needed for chromosome segregation (Bouet et al. 2014), and beyond this, an L. monocytogenes ΔftsK mutant was impaired in cell division with a tendency to form chains of unsegregated cells (Chang et al. 2013). The role of L. monocytogenes walJ remains to be studied, but its B. subtilis homolog contributes to coordination of cell division with cell wall biosynthesis and chromosome segregation (Biller et al. 2011). CozEb is a homolog of S. pneumoniae CozE, which directs the activity of the penicillin binding protein PBP1a to the MreCD complex to ensure normal PG biosynthesis (Fenton et al. 2017). S. pneumoniae cozE is essential, but L. monocytogenes encodes two cozE-like genes. Possibly, their simultaneous deletion is not tolerated as reported for S. aureus (Stamsås et al. 2018). Third, our analysis confirms the special role of the two cell division genes divIVA and secA2 for intracellular growth. Deletion of either gene strongly impaired daughter cell separation after septum closure, which leads to formation of long chains of unseparated cells that cannot multiply in macrophages (Lenz et al. 2003; Halbedel et al. 2012). Lastly, we found that the PASTA domains of the PrkA kinase can be deleted. This was surprising because the full-length prkA gene could not be removed in previous attempts, and depletion of PrkA was lethal in L. monocytogenes EGD-e (Wamp et al. 2020, 2022). Thus, the kinase domain contributes the functionality required for growth in BHI broth. PrkA regulates PG biosynthesis through phosphorylation of ReoM, a small cytosolic protein that controls proteolytic stability of MurA, which is essential itself (Wamp et al. 2020, 2022; Kelliher et al. 2021). ReoM needs to be phosphorylated to prevent degradation of MurA (Wamp et al. 2020). Deletion of PASTA domains has lowered the activity of StkP, the PrkA homolog of S. pneumoniae (Zucchini et al. 2018). As MurA can be detected in cells lacking the PASTA domains (Fig. 4D), we assume that the activity of the isolated kinase domain of PrkA is sufficient for ReoM∼P formation. A viable prkA mutant with reduced kinase activity would be very useful to further study the role of this protein in L. monocytogenes growth and virulence.
Methods
Bacterial strains and growth conditions
All strains and plasmids used in this study are listed in Supplemental Table S3. Strains of L. monocytogenes were cultivated in BHI broth or on BHI agar plates at 37°C. Antibiotics and supplements were added when required at the following concentrations: erythromycin (5 µg/mL), X-Gal (100 µg/mL), and IPTG (as indicated). E. coli TOP10 was used as host for all cloning procedures (Sambrook et al. 1989). Ceftriaxone minimal inhibitory concentrations were determined using E-test strips with a ceftriaxone concentration range of 0.016–256 µg/mL (bestbiondx).
General methods, manipulation of DNA, and oligonucleotide primers
Standard methods were used for transformation of E. coli and isolation of plasmid DNA (Sambrook et al. 1989). Transformation of L. monocytogenes was performed as previously described (Monk et al. 2008). Restriction and ligation of DNA was performed according to the manufacturer's instructions. All primer sequences are listed in Supplemental Table S4.
Transposon library construction
The Tn-delivery vector pKRMIT was transformed into L. monocytogenes EGD-e. Transformants were selected on BHI agar plates containing 25 mg/L kanamycin and 5 mg/L spectinomycin after incubation for 24 h at 30°C. One clone was selected and grown overnight at 30°C in BHI broth containing both antibiotics. This culture was used to inoculate 25 mL BHI broth containing 25 mg/L kanamycin to a starting OD600 of 0.01 and then further incubated at 30°C until an OD600 of 0.1 was reached. Plasmid replication was stopped by increasing the cultivation temperature to 40°C followed by further incubation until an OD600 of 0.5. Aliquots were harvested, mixed with glycerol (50% final concentration), and stored at −80°C. These aliquots were used to inoculate 25 mL of BHI broth containing kanamycin (25 mg/L) to an OD of 0.1, which was grown at 37°C to stationary phase. This culture was diluted back and grown to stationary phase two more times, a procedure that was needed to effectively deplete clones that retained the plasmid or have lost the plasmid without transposition. At the end of the experiment, bacterial titers were determined on BHI agar plates and on BHI agar plates containing either kanamycin or spectinomycin to calculate the transposition frequency and the ratio of plasmid loss. Five hundred microliters of the culture was centrifuged, and genomic DNA was extracted for Tn insertion sequencing.
Generation and sequencing of Tn-Seq amplicon libraries
Transposon library sample aliquots were used for DNA extraction by a phenol-chloroform isoamyl alcohol procedure (Sambrook et al. 1989). DNA pellets were resuspended in 200 µL 10 mM Tris/HCl (pH 7.5), and DNA concentration was determined using the Qubit dsDNA HS assay. Three micrograms of the isolated DNA was digested using 2 U MmeI for 6 h at 37°C followed by heat inactivation for 10 min at 80°C. Digestion was confirmed by agarose gel electrophoresis. Five units of quick CIP calf intestinal phosphatase was directly added to the digestion mix and incubated for 1 h at 37°C. The reaction mix was then purified using a column-based PCR clean-up kit. Double-stranded DNA adapters were obtained by mixing 40 µL of the oligonucleotides MF38 and the 5′ and 3′ phosphorylated MF52 (both 100 µM) followed by denaturation for 10 min at 99°C. Three microliters of these adapters was added to the cleaned-up transposon library DNA and ligated using 5 U T4 DNA ligase. The produced fragments were purified using the Qiagen size-selection kit and eluted in 20 µL 10 mM Tris/HCl (pH 7.5). Sequencing adapters were attached to the fragments via PCR. For this purpose, 8 µL of the eluate was used as template in an 80 µL PCR reaction with MF38 and MF53 as the primers. The size of the PCR products was controlled using agarose gel electrophoresis, and the products were purified from the gel. The adapter-linked PCR products were used as a template to introduce multiplex identifiers for pooled sequencing. PCR was performed with the following conditions: 2.5 µL template (3 ng/µL), 2.5 µL Nextera XT index primer (each N7 and S5, Illumina), and 12.5 µL 2x KAPA HiFi HotStart ReadyMix (Roche) and 5 µL H2O using a PCR program with these settings: 3 min at 95°C; eight cycles (each 30 sec) of 5 min at 95°C, 55°C, 72°C, 72°C; and storage at 4°C until bead-clean up. Sequencing was performed in a 2 × 150-bp paired-end run on a NextSeq550 instrument (Illumina) using v2.5 midout chemistry.
Bioinformatic processing of sequencing reads and gene essentiality prediction
Only raw reads containing the terminal fraction of the transposon were accepted for further analysis. The sequence region of the inverted repeat was removed afterward using cutadapt (Martin 2011), and 13 bases following the inverted repeat were kept for determination of the insertion locus. The insertion locus was determined by back-mapping of the trimmed sequences against the L. monocytogenes EGD-e genome (Toledo-Arana et al. 2009) using Bowtie (Langmead et al. 2009) with the options “-S -v 0 -n 0 -p 20 -l 13 -m 1,” allowing no mismatch within the alignment and only unique insertion sites to be counted. The resulting SAM file was used for the analysis in Tn-Seq Explorer version 1.5 (Solaimanpour et al. 2015). For the determination of zbar values, the SAM file was further transformed to wiggle file format using an in-house script (Supplemental File S1) and analyzed using the software TRANSIT version 3.1.0 (DeJesus et al. 2015).
Tn-Seq Explorer and TRANSIT were used for the prediction of essentiality and determination of prediction accuracy (zbar). For Tn-Seq Explorer analysis, sequencing run data in the form of the SAM file and the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) file of L. monocytogenes EGD-e (accessed under accession number NC_003210.1) were uploaded to TnSeq-Explorer. Insertion loci were excluded if they were covered by fewer than five reads mapping onto them. Repetitive hits of one insertion site were counted as one unique insertion, so that loci potentially preferred during PCR amplification would not compromise further analysis. Genes with an insertion density lower than 0.01 were defined as genes required for growth. The insertion density is defined as the number of unique insertions divided by the number of bases of the gene (Solaimanpour et al. 2015). Identified genes were visually confirmed using the Artemis comparison tool (Carver et al. 2012).
Premature stop codon analysis
Assembled genome sequences of all L. monocytogenes isolates that were available at the NCBI Pathogen Detection pipeline server (https://www.ncbi.nlm.nih.gov/pathogens) in December 2019 (n = 27,118) were downloaded. All FASTA files were imported in SeqSphere+ (Ridom) to perform core genome and accessory genome multi locus sequence typing (cgMLST/agMLST) by automated allele submission to cgMLST.org (Ruppitsch et al. 2015). Afterward, all allele sequences for all targets of the L. monocytogenes cgMLST (n = 1701) and agMLST schemes (n = 1158) deposited at cgMLST.org were downloaded, and alleles with premature stop codons between 5% and 80% relative to the start of the ORF were determined for each target using an in-house script (https://gitlab.com/s.fuchs/lmo-essential-genes). The frequency of occurrence of such alleles among the 27,118 genomes was quantified.
Construction of plasmids and strains
Plasmid pSH541 was generated for deletion of hly. For its construction, hly up- and downstream fragments were amplified using the primers SHW858/SHW860 and SHW861/859, respectively, fused together by overlapping extension (SOE)-PCR, and inserted into pMAD by restriction-free (RF) cloning (van den Ent and Löwe 2006).
Plasmid pSH542 was generated for deletion of actA. Fragments upstream of and downstream from actA were amplified by PCR using the oligonucleotides SHW838/SHW841 and SHW840/SHW839, respectively. Both fragments were spliced together by SOE-PCR and then inserted into pMAD using RF cloning.
Plasmid pSH554, facilitating deletion of the prkA C terminus, was obtained by amplification of fragments upstream of and downstream from the prkA region to be deleted using the primer pairs SHW899/SHW902 and SHW901/SHW900, respectively, which were then fused together by SOE-PCR and inserted into pMAD by RF cloning.
A chromosomal fragment containing whiA together with its up- and downstream regions was amplified using the primers MF93/MF94 and inserted into pMAD by RF cloning, which resulted in plasmid pSH553. The whiA gene of this plasmid was then removed in a PCR with MF95/MF96 as the primers, yielding the whiA deletion plasmid pSH556.
Plasmid pJR57 was constructed for deletion of cozEb. To this end, chromosomal regions upstream of and downstream from cozEb were amplified using JR153/KK104 and KK103/JR154, respectively, as the primers and spliced together by SOE-PCR. The resulting fragment was then cloned into pMAD using EcoRI/BamHI.
Likewise, plasmid pSH559 was constructed for deletion of walJ. Regions flanking the walJ gene were amplified using the oligonucleotides SHW919/SHW922 and SHW921/SHW920 and spliced together by SOE-PCR. Finally, the resulting fragment was inserted into pMAD by RF cloning.
For construction of plasmid pSH563, fragments upstream of and downstream from ftsK were amplified using SHW915/SHW917 and SHW916/SHW918, respectively, as the primers. Both fragments were spliced together by SOE-PCR and inserted into pMAD by RF cloning.
Fragments upstream of and downstream from parA were amplified by PCR using oligonucleotides SHW911/SHW914 and SHW913/SHW912, respectively, and then fused together by SOE-PCR. The resulting fragment was inserted into pMAD by RF cloning, yielding plasmid pSH566.
For construction of plasmid pTE11, fragments upstream of and downstream from lmo2214-lmo2215 were amplified by PCR using TE8/TE85 and TE84/TE7, respectively, fused together by SOE-PCR and inserted into pMAD by RF cloning.
Plasmid pTE51 was designed for deletion of thrC. Fragments upstream of and downstream from thrC were amplified by PCR using TE203/TE204 and TE205/TE202, respectively, as the primers. The two fragments were spliced together in a SOE-PCR and then inserted into pMAD by RF cloning.
Plasmid pTE57, constructed for deletion of gcvPAB, was generated similarly. First, regions upstream of and downstream from gcvPAB were amplified by PCR using the oligonucleotides SHW942/SHW944 and SHW945/SHW943, respectively. In a second step, both fragments were joined by SOE-PCR, and the resulting fragment was inserted into pMAD by RF cloning.
Derivatives of pMAD plasmids were transformed into the L. monocytogenes recipients, and genes were deleted as described elsewhere (Arnaud et al. 2004). All gene deletions were confirmed by PCR.
Isolation of cellular proteins and western blotting
Cells were grown in BHI broth at 37°C to an optical density of OD600 = 1.0 and harvested by centrifugation. Cell pellets were washed with ZAP buffer (10 mM Tris/HCl at pH 7.5, 200 mM NaCl), resuspended in 1 mL ZAP buffer to which 1 mM PMSF was added and disrupted by sonication. Cellular debris was removed by centrifugation, and the supernatant was considered to contain total cellular proteins. Sample aliquots were separated by standard SDS polyacrylamide gel electrophoresis. Gels were transferred onto positively charged polyvinylidene fluoride membranes by semidry transfer. DivIVA and MurA were immune-stained using polyclonal rabbit antisera originally generated against B. subtilis DivIVA (Marston et al. 1998) and MurAA (Kock et al. 2004) as the primary antibodies and an anti-rabbit immunoglobulin G conjugated to horseradish peroxidase (Sigma-Aldrich A0545) as the secondary one. The ECL chemiluminescence detection system (Thermo Fisher Scientific) was used for detection of the peroxidase conjugates on the PVDF membrane in a chemiluminescence imager.
Infection experiments
Infection experiments with the transposon library were performed in J774.A1 mouse ascites macrophages (ATCC TIB-67) similar to a previously published protocol (Halbedel et al. 2014) but with some modifications; 107 macrophages were seeded in 500-mL coated cultivation flasks (VWR) containing 75 mL DMEM + 10% FCS and incubated for 48 h at 37°C and 5% CO2. For bacterial infection, an overnight culture of the transposon library (incubated at 37°C at 250 rpm in BHI) was adjusted to an OD600 of 0.001 in DMEM, and before infection, a sample aliquot was stored at −20°C for DNA extraction to serve as a control. J774.A1 macrophages were infected with 106 bacterial cells, resulting in a MOI = 0.1. After 1 h of incubation, the medium was removed from the cell culture, and cells were briefly washed with 10 mL room temperature DMEM + 10% FCS and further incubated for 1 h in 50 mL DMEM + 10% FCS + 40 mg/L gentamicin to eliminate free bacterial cells. After 1 h, the medium was removed, and 75 mL of DMEM + 10% FCS + 10 mg/L gentamicin was added to the cell culture, which was then further incubated for 24 h. At the end of the incubation, cells were harvested in 10 mL cold Triton X-100 in PBS, 50 µL of which was used for dilution series on BHI agar plates to determine the number of viable bacterial cells recovered from the infected macrophages, whereas the rest of the cells were centrifuged at 5000g for 15 min at 4°C, decanted, and stored for DNA extraction at −20°C.
J774.A1 macrophage infection experiments and plaque formation assays with 3T3 mouse fibroblasts for analysis of individual deletion mutants were essentially performed as described earlier (Halbedel et al. 2014).
Data access
All raw and processed sequencing data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under project accession number PRJEB51140 as samples ERS10768787–ERS10768791, each containing the sequencing raw data as well as the used BAM files.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Birgitt Hahn for excellent technical assistance and Jeanine Rismondo for help with construction of strain LMJR87. We also thank Kevin S. McIver for kindly sharing plasmid pKRMIT and Pascale Cossart for sharing the prfA mutant. This work was supported by the German Federal Ministry of Health/National Research Platform for Zoonoses (LISMORES to S.H.), by the Robert Koch Institute (Geno2Pheno to S.H.), and by the German Research Foundation (HA6830/2-1, HA6830/4-1 to S.H.).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276747.122.
- Received March 11, 2022.
- Accepted August 4, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








