
Somatic structural variant formation is guided by and influences genome architecture
- Nikos Sidiropoulos1,2,9,
- Balca R. Mardin3,4,9,
- F. Germán Rodríguez-González1,2,
- Ivan D. Bochkov5,
- Shilpa Garg6,
- Adrian M. Stütz3,
- Jan O. Korbel3,
- Erez Lieberman Aiden5,7 and
- Joachim Weischenfeldt1,2,8
- 1Biotech Research and Innovation Centre (BRIC), University of Copenhagen, Copenhagen 2200, Denmark;
- 2The Finsen Laboratory, Rigshospitalet, Copenhagen 2200, Denmark;
- 3European Molecular Biology Laboratory (EMBL), Genome Biology Unit, 69117 Heidelberg, Germany;
- 4BioMed X Institute, 69120 Heidelberg, Germany;
- 5The Center for Genome Architecture, Baylor College of Medicine, Houston, Texas 77030, USA;
- 6Department of Biology, University of Copenhagen, Copenhagen 2200, Denmark;
- 7Center for Theoretical Biological Physics, Rice University, Houston, Texas 77030, USA;
- 8Department of Urology, Charité-Universitätsmedizin Berlin, 10117 Berlin, Germany
-
↵9 These authors contributed equally to this work.
Abstract
The occurrence and formation of genomic structural variants (SVs) is known to be influenced by the 3D chromatin architecture, but the extent and magnitude have been challenging to study. Here, we apply Hi-C to study chromatin organization before and after induction of chromothripsis in human cells. We use Hi-C to manually assemble the derivative chromosomes following the occurrence of massive complex rearrangements, which allows us to study the sources of SV formation and their consequences on gene regulation. We observe an action–reaction interplay whereby the 3D chromatin architecture directly impacts the location and formation of SVs. In turn, the SVs reshape the chromatin organization to alter the local topologies, replication timing, and gene regulation in cis. We show that SVs have a strong tendency to occur between similar chromatin compartments and replication timing regions. Moreover, we find that SVs frequently occur at 3D loop anchors, that SVs can cause a switch in chromatin compartments and replication timing, and that this is a major source of SV-mediated effects on nearby gene expression changes. Finally, we provide evidence for a general mechanistic bias of the 3D chromatin on SV occurrence using data from more than 2700 patient-derived cancer genomes.
Understanding the mechanistic forces that shape somatic aberrations in cancer genomes can provide important basic and clinically relevant information on the causes of the disease. Mutations are caused by stochastic, mechanistic, and selective forces, and the sources of single-nucleotide variants (SNVs) have been explored extensively (Lawrence et al. 2014; Gonzalez-Perez et al. 2019). These studies have shown a strong association between SNV occurrence and one-dimensional features of the genome, including local chromatin, replication timing, and transcription factor binding.
In contrast to SNVs, structural variants (SVs) can impact multiple genes through gene loss, disruption, gene–gene fusion formation, amplification, or dysregulation of the cis-regulatory landscape, in some cases through reorganizing contact domains, also known as topology-associated domains (TADs), which can lead to enhancer hijacking (Northcott et al. 2014; Weischenfeldt et al. 2017). A pan-cancer study found relatively few TAD-affecting SVs to be associated with changes in nearby gene expression (Akdemir et al. 2020), which is in line with a general realization that changes in gene expression do not necessarily depend on changes in contact domains (Despang et al. 2019; Ghavi-Helm et al. 2019). The sources of SVs in cancer have also proved challenging to disentangle, partly due to their complex nature and orders-of-magnitude lower occurrence frequency compared to SNVs (Drier et al. 2013; Rheinbay et al. 2020). SVs can be simple (e.g., deletions, duplications, translocations) or highly complex, as in the case of chromothripsis (CT) (Stephens et al. 2011; Rausch et al. 2012), which leads to chromosome shattering and reordering of genomic segments.
The 3D nuclear organization is known to be important in gene regulation through bringing genomically distant loci in close physical proximity (Nora et al. 2012; Le Dily et al. 2014). Several lines of evidence have suggested that SV occurrence is linked with chromatin architecture. Early cancer studies using fluorescence in situ hybridization (FISH) found that the proximity in 3D genome space influences the probability of forming fusion gene products (Lukášová et al. 1997; Roix et al. 2003). On a more global scale, the effects of genome architecture on SVs in cancer genomes have been inferred (De and Michor 2011; Fudenberg et al. 2011; Harewood et al. 2017) using reference maps of genome organization. Moreover, SVs can alter the 3D organization, which can have direct consequences on gene regulation (Lupiáñez et al. 2015; Northcott et al. 2017; Weischenfeldt et al. 2017; Dixon et al. 2018; Akdemir et al. 2020). However, it remains unsolved to what extent prior 3D contacts in the same cells impact on somatic SVs formation in human cells and how these acquired SVs impact on gene expression in cis.
The lack of model systems to study the relationship between genome organization before and after SV occurrence in the same population of cells and the lack of information on the physical composition of the chromosomes in the cell after multiple rearrangements presents an obstacle to appreciate the consequences of SVs on genome architecture and gene regulation. It is therefore desirable to construct a “derivative chromosome” that represents the order in which genomic regions have been stitched together following complex SV formation.
We have previously developed a model system that uses doxorubicin (dox) treatment to induce CT in human cells, characterized by massive genomic rearrangements in a single cell cycle (Mardin et al. 2015). Dox, a widely used chemotherapeutic, traps Topoisomerase II (TOP2) on DNA and acts as a poison for TOP2 cleavage complexes by inhibiting DNA religation, causing TOP2-linked double-strand breaks (DSBs), which can result in the formation of massive, genome-wide SVs (Fornari et al. 1994). Our model uses retinal pigment epithelial cell line RPE-1, a near diploid cell line widely used in genomic instability studies (Passerini et al. 2016; Santaguida et al. 2017). The cells are TP53-deficient—previous studies revealed that mutations of TP53 are strongly associated with chromothripsis in several subtypes of cancer (Rausch et al. 2012; Korbel and Campbell 2013).
To shed light on the interplay between SVs, genome organization, and gene regulation, we applied Hi-C to our human cell line model system before and after induction of CT. We use this to compare and assemble chromosomes before and after CT, which enabled us to resolve complex SVs in an allele-specific manner from Hi-C (Lieberman-Aiden et al. 2009). We leverage this to investigate key properties of the 3D genome folding conformation and replication timing and how it impacts SV formation. Finally, we validate the generalization of these properties across 2700 cancer genomes.
Results
Reconstructing the derivative chromosomes from highly rearranged genomes
To investigate the 3D chromatin before and after CT, we performed Hi-C sequencing on the wild-type (C93), two TP53-deficient single clones (C29 and DCB2; maternal), and four treated (BM175, BM178, BM838, and BM780, daughter) clones showing signs of massive rearrangements (Fig. 1A; Supplemental Fig. S1A; Supplemental Tables S1, S2). Hi-C maps can be leveraged to provide information on SVs because physical proximity mediated by SVs is reflected in novel contacts in the Hi-C (Harewood et al. 2017; Dixon et al. 2018) map (Fig. 1). We used Hi-C analysis to identify 306 SVs, the majority (93.4%) of which were also identified by mate-pair sequencing. A subset of these (48%) (Supplemental Fig. S2B–D) were identified by hic_breakfinder (Dixon et al. 2018). The 306 SVs were explicitly occurring in the daughter cells, the majority of which were clustered on a few chromosomes (Supplemental Fig. S1A; Supplemental Tables S3–S5), a typical feature of chromothripsis.
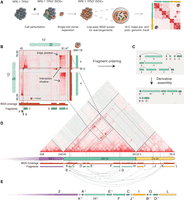
Hi-C-based assembly of a highly rearranged human cell line model system. (A) Schematic overview of Complex Alterations after Selection and Transformation (CAST) coupled with in situ Hi-C. TP53-deficient RPE-1 cells are perturbed with doxorubicin (DOX), followed by single-cell sorting and expansion. Surviving populations are screened for genomic rearrangements with low-depth WGS. Hi-C libraries from control (maternal) and treated (daughter) clones are sequenced to identify acquired and drug-induced rearrangements. (B) Annotation of fragment size and edge positions. The latter is annotated at peaks of ectopic contacts between two distal sites. Duplicated regions, highlighted at fragments F and G, exhibit distinct interaction patterns and produce nonorthogonal ectopic interactions, as revealed by the interaction profile between fragments C-F and B-G, respectively. The genomic regions depicted on the Hi-C maps correspond to the highlighted regions on the chromosome ideograms. (C, upper) Following the above-described annotation strategy, we create a graph of nodes (fragments) and edges (rearrangements) for the ectopic interactions of panel B. (Lower) Traversing the graph produces the derivative assembly. (−1) denotes fragment inversion. Duplicated regions that are shared between fragments A-B and F-G are colored in pink. (D) The Hi-C map depicts rearrangements within and across Chromosomes 2, 12, and 22. Straight diagonal lines on the Hi-C map trace the position of the SV calls back to the reference genome. Following the graph method in B–D, we annotated 12 rearranged DNA fragments (A–L) and their juxtapositions, represented as green and yellow rectangles and directed arches, respectively. The telomere of Chromosome 2q is connected to 12 and 22, observed as stripes protruding from the 2q telomere to the q-arm of 12 and 22. (E) Using Chromosome 2 as a starting point, we walk along the annotated edges and segments to produce the assembly of the derivative 2-12-22 chromosome.
In agreement with the CT model (Korbel and Campbell 2013), we found that the SVs occurred on a single haplotype, had a minimal positional recurrence of breakpoints, and random orientation of SVs (see Supplemental Text; Supplemental Fig. S1).
We next sought to reconstruct the derivative chromosomes as they appeared in the genomes of the rearranged cells. This is challenging due to the highly rearranged nature and heterozygous state of the genomes, and no automated methodologies are currently able to reconstruct the derivative chromosomes from such genomes. Thus, we used our Hi-C data to perform reference-guided manual assembly on the chromothriptic daughter cells to obtain the derivative chromosomes. Hi-C is particularly tractable due to the long-range genomic contact information that can be utilized to order genomic fragments and assign them to whole derivative chromosomes (Dudchenko et al. 2017; Ghavi-Helm et al. 2019). The long-range information can assist in connecting even repeat-rich sequences, which are impossible to assemble with standard short-read paired-end sequencing. Although Hi-C data can detect all SV types, we note that the method has limitations in detecting small-sized SVs (Supplemental Fig. S2D), which can affect the assembly.
Two key sources of information encoded in the ectopic Hi-C contact maps formed the main guidelines of our manual assembly. First, as it has been previously described (Harewood et al. 2017; Dixon et al. 2018), the breakpoint location of SVs can be identified as peaks of high-intensity ectopic interactions, which we term the edge position (Fig. 1B). Second, and most importantly, the length and type of SV can be identified from the decay in contact frequency originating at the breakpoint site, which we term interaction shadow. The orientation of the interaction shadow can be used to identify the particular SV type, such as deletion, duplication, and inversion-type (Fig. 1B; Supplemental Fig. S2A). We initially focused on the highly rearranged Chromosomes 12 and 22 of one of the daughter clones, BM178, to map all juxtapositions of the rearranged fragments in the derivative chromosome. We found that the interaction shadow is also helping to resolve conflicts across overlapping signals, as seen in the highlighted region between fragments F and G in Figure 1B. Fragment F has enriched contacts with fragment C, observed as high-intensity regions on the Hi-C map, and depleted with B, whereas the opposite is true for G. Only the highlighted region has contact enrichment across all four fragments. This can only occur if the region was duplicated with a copy existing in both fragments F and G, each one with its distinct interaction signature. The predicted duplication is confirmed by a copy-number gain in the WGS coverage track. A similar scenario is observed between fragments A and B, and their interactions with fragments G and H.
We used these two features, Hi-C edge position and interaction shadows, to create an assembly graph composed of genomic segments, represented as nodes, and SVs represented as edges. Traversing the graph produces the derivative chromosome (Fig. 1C). We identified 13 genomic segments of Chromosome 12q stitched in seemingly random orientations with four segments from Chromosome 22q (Fig. 1D,E). The SVs and segment annotations are in alignment with copy-number switches and loss-of-heterozygosity identified by WGS (Fig. 1B; Supplemental Fig. S3D). Moreover, we found evidence that the derivative chromosome resides at the telomere of Chromosome 2 (Supplemental Fig. S2E), supported by contact frequency enrichment between fragments from Chromosomes 12 and 22, and the q arm of Chromosome 2 (Fig. 1D; Supplemental Fig. S2F,G; Supplemental Text).
Sites of structural variants are linked with pre-existing genomic contacts
After assembling the derivative chromosome, we turned to the 3D genome organization prior to CT. We generated WGS and Hi-C data from the maternal (before CT) and derivative cell lines (after CT), and we examined the impact of CT on genomic contacts in 3D and SV formation. WGS on the derivative cell lines was performed on an early passage after CT induction, whereas Hi-C was performed approximately 20 passages later. This difference in passage timing between the library preparation of WGS and Hi-C allowed us to dissect SVs that occurred as a direct result of dox treatment or due to subsequent genomic instability (from hereon termed “induced” and “spontaneous,” respectively) (see Supplemental Fig. S4; Supplemental Text). We used these data to ask whether and to what extent linear and 3D features of the genome were associated with juxtaposition of distal genomic regions and the formation of SVs.
The genome can be partitioned into A and B compartments, associated with open and closed chromatin, respectively (Lieberman-Aiden et al. 2009; Dixon et al. 2015). Although 95% of all compartments were conserved across different tissues (Fig. 2A,B), we found tissue-specific compartments to be enriched for both induced and spontaneous SV (Fig. 2B), and enrichment of induced SVs in A compartments (P = 3.5 × 10−13, Fisher's exact test) (Fig. 2C, left).
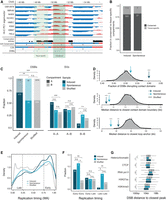
Genome architecture features at SV sites. (A) DSBs associated with conserved and tissue-specific compartments. PC1 eigenvectors from eight human cell lines (Rao et al. 2014), with signs adjusted so that positive values correspond to A (red) and negative values to B (blue) compartments. A/B conservation track annotates every 100-kb bin to a compartment it was found to be in the same state in more than half of the cell lines. Compartment entropy reflects the compartment concordance across the cell lines, with higher values representing less conserved compartments. DSBs in tissue-specific compartments overlap nonconserved regions or regions where the C29 compartment annotation disagrees with the A/B conservation track. (NC) Not conserved. (B) Distribution of DSBs in conserved and tissue-specific compartments between induced and spontaneous rearrangements; 114 out of 614 (18.6%) and seven out of 37 (18.9%) occurred in tissue-specific compartments. (C, left) Induced DSBs (n = 610) are highly enriched within A compartments compared to the spontaneous (n = 44) and shuffled (n = 588,388) set. Fisher's exact test. (Right) SVs are enriched in A to A and depleted in B to B in the induced SV set compared to the shuffled, whereas spontaneous SVs are evenly distributed across A and B compartments. Post-hoc Fisher's exact test, FDR corrected. (D) Observed and expected distribution of DSBs across features that reflect genome organization. (Top) Induced DSBs are enriched inside contact domains (n = 6165), whereas spontaneous are enriched in interdomain loci. Both of them occur significantly closer to insulating factors, such as contact domain boundaries (middle, n = 12,326) and chromatin loop anchors (bottom, n = 17,374) than the shuffled sets. (E) Replication timing weighted average (WA) values at DSBs show enrichment of induced (dark blue, n = 609) and spontaneous (light blue, n = 44) breaks during early replication compared to the permuted set (gray, n = 586,044). (F) SVs occur significantly more frequently between early-to-early replication domains for both induced and spontaneous rearrangements. In contrast, late-to-late replication timing SVs are depleted for induced and enriched for spontaneous SVs. Post-hoc Fisher's exact test. (G) Distance from DSBs to the closest ChIP-seq peak for H3K4me3 (n = 122,258), H3K27ac (n = 103,673), RNA pol II (n = 59,021), and CTCF (n = 43,285) peaks compared to the permuted ones (t-test). Box plots show the median, first, and third quartiles, and outliers are shown if outside the 1.5× interquartile range. (*) P-value < 0.05, (**) P-value < 0.01, (***) P-value < 0.001.
Because the frequency of interactions between similar compartments is known to be high (Lieberman-Aiden et al. 2009), we hypothesized that pre-existing long-range contacts between distant compartments would increase the probability of SV formation between these compartments. To this end, we quantified the proportion of SVs that occurred within (|A-to-A| or |B-to-B|) and between (|A-to-B|) compartments. Compared to a random background model, we found within-compartment SVs to be strongly enriched (Fig. 2C, right)—more than half of all dox-induced SVs occurred in |A-to-A| compartments (P = 0.0002 compared to random set, post-hoc Fisher's exact test) and fewer than 10% in |B-to-B| compartments. Spontaneous SVs displayed a more uniform distribution with a relatively higher proportion of within than between compartment SVs, although this did not reach statistical significance when compared to the random background set, potentially due to the relatively low number of spontaneous SVs in our set.
The genomic folding principles can be reconciled at the level of contact domains, which are genomic loci with a high within-contact frequency, associated with CTCF and cohesin binding (Hadjur et al. 2009; Sanborn et al. 2015). Contact domains are similar to, but more well-defined than, TADs and are thought to play an important role in gene regulation. Given the enrichment of SVs occurring between similar compartments, we next explored to what extent these SVs were expected to disrupt contact domains. We found a significant enrichment of doxorubicin-induced SVs associated with contact domain disruption and both induced and spontaneous SVs to occur in close proximity to contact domain boundaries and loop anchors (Fig. 2D).
We next asked whether the observed patterns of compartment-associated SVs in our model system could be recapitulated in patient-derived cancer genomes and to what extent this varied across different tumor types. Whereas patient-derived cancer genomes are strongly influenced by both positive and negative selective pressure, we hypothesized that the mechanistic biases of the 3D chromatin on SV formation would be comparable to our model system. To examine the association between chromatin conformation and SVs identified in cancer genomes, we analyzed SVs from almost 2700 tumor samples across 30 different tumor types (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020; Li et al. 2020). We also undertook a separate analysis of three common cancer types with high SV burden—breast, prostate, and uterine cancer. In these cancer genomes, we found overall a more even distribution of SVs occurring in A and B compartments (Supplemental Fig. S5B). In agreement with the findings from our model system, a significant majority of SVs occurred within compartments (|A-to-A| and |B-to-B|, P < 0.01 for all cancer types compared to our background model, Fisher's exact test). Uterine cancer showed a striking enrichment of |A-to-A| SVs, the highest proportion across all 30 tumor types and 24% higher compared to pan-cancer SVs and 60% higher compared to our background model (P < 10−16, Fisher's exact test), suggesting that SVs in uterine cancers are particularly associated with prior 3D folding and proximity of A compartments.
Faulty DNA replication is thought to underlie many complex SVs in cancer genomes through, for example, replication fork stalling and template switching or replication fork collapse (Yang et al. 2013; Canela et al. 2017). Replication timing is also strongly linked with genome architecture and contact domains. Early replicating regions often overlap with A compartments, whereas late-replicating regions coincide with B compartments (Ryba et al. 2010; Pope et al. 2014). We performed Repli-seq on C29, BM175, and BM178 (Supplemental Table S1) to explore the influence of replication timing on SV formation and the changes in chromatin conformation. In agreement with earlier findings, we find a robust correlation between early and late replication timing and A and B compartments, respectively (Pearson's correlation 0.77, P < 2.2 × 10−16) (Supplemental Fig. S5C,D). Whereas induced SVs formed primarily between early-replication domains, spontaneous SV formation was enriched between the same replication timing domain (Fig. 2E–G), suggesting that spontaneous formed SVs are highly influenced by replication timing per se and to a lesser extent by whether it is an early- or late-replication domain. We next performed a parallel analysis across the 2700 cancer genomes to examine whether the strong preference for SVs to occur between similar replication timing domains can be recapitulated in cancer genomes. Indeed, we found that the cancer genomes displayed a distribution similar to the spontaneous SVs with a significantly higher fraction of SVs occurring between similar replication timing domains (high within-replication timing frequency) compared to our background model (P < 0.01, Fisher's exact test). In addition to the enrichment of uterine cancer within compartment juxtapositions, we also observed a particularly pronounced enrichment of SVs associated with the juxtaposition between early-replication domains in this tumor type, 60% higher compared to pan-cancer and, conversely, 40% depleted for SVs between early- and late-replication domains compared to pan-cancer analysis (P-value < 2.2 × 10−16, Fisher's exact test) (Supplemental Fig. S5E,F).
In summary, our findings suggest a strong mechanistic bias for SVs to occur between sites of identical chromatin compartments and replication timing domains and that this is a common phenomenon across both cell lines and in patient-derived cancer genomes.
SVs in regulatory regions cause expression dysregulation of nearby genes
Our findings of enrichment of SVs intersecting the transcriptionally active A compartments prompted us to investigate the influence of mRNA transcription levels on SV formation. We compared allele-specific mRNA transcription between the maternal cells and two daughter cells, C29 and DCB2, and found that actively transcribed genes were associated with a fourfold increased risk of SV occurrence and that highly expressed genes (above and below median expression of active genes) were more strongly associated with SV occurrence (OR = 1.91; P = 0.001, and OR = 1.35; P = 0.217 for C29 and DCB2, respectively) (Fig. 3A). We also found a significant correlation between breakpoint proximity and altered gene expression (P = 0.01, Pearson correlation coefficient = −0.13) (Fig. 3B), with 10% of breakpoint regions associated with differentially expressed genes in cis and 63% of genes displaying a significant change in gene expression within 25 kb (P = 0.018) (Fig. 3C). This is in line with a recent study, showing a distance relationship between the disrupted enhancer and the observed changes in gene expression (Fulco et al. 2019).
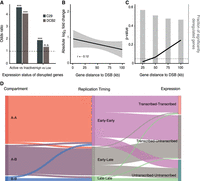
SV-mediated effects on gene expression in cis. (A) DSBs are associated with highly transcribed genes. (Left) Out of 414 DSBs in C29-derived clones, 198 were located within gene bodies; 116 in active, 82 in inactive. The DCB2-derived clone had 137 out of 198 DSBs occurring within gene regions; 80 active and 57 inactive. Genes were defined as active if they had TPM > 1 across all replicates. (Right) Further subdivision of active genes links highly (>50%) transcribed genes with the formation of DSBs. (***) P-value < × 10−5, (n.s.) nonsignificant, Fisher's exact test. (B) Absolute log2 fold change as a function of distance from the closest DSB cluster in BM175 cells. We analyzed in total 274 breakpoints in 182 clustered breakpoint loci. For every DSB cluster, we considered the gene with the highest absolute log2 fold change in a 10-kb sliding window with no overlap. The mean fold change effect at 1 kb versus 100 kb was 0.95 ± 0.35, 95% confidence interval and 0.53 ± 0.33, 95% confidence interval. (C) P-value estimation of the number of significantly deregulated genes as a function of genomic distance from the most proximal TOP2-linked DSB. Background distribution was estimated by randomly shuffling the gene status (deregulated, non-deregulated; see Methods section). Bar plots depict the fraction of deregulated genes within the marked genomic window. (D) Effects of active chromatin, replication timing and gene expression on the formation of TOP2-linked, induced SVs (n = 90). Intermediate replication timing states (n = 214) are excluded for visual simplicity. (See also Supplemental Fig. S6.)
These results collectively demonstrate that SV formation following doxorubicin treatment is both associated with prior active transcription and has a subsequent impact on nearby gene expression (Fig. 3D).
SV-mediated chromatin reorganization affects both gene regulation and DNA replication in cis
Genomic compartments disappear during mitosis and reassemble during G1 and S phase (Nagano et al. 2017). Although many replication domain boundaries coincide with genomic compartments, replication is a dynamic process that occurs during the S phase of the cell cycle. We next hypothesized that SVs could impact chromatin organization, for example, by causing compartment and replication domain switches. Indeed, we found several loci associated with such changes in compartment and replication timing between maternal and daughter cells. A representative example is shown in Figure 4A from the q-arm of Chromosome 15 in BM175, which was associated with chromothriptic SVs, including translocations to Chromosomes 2, 8, 11, and 12 (Supplemental Fig. S1B,C). Whereas the majority of loci were constant (Fig. 4A), we noticed a locus with late-replication and B compartmentalization in the C29 maternal clone (Fig. 4A, red shade), where several DSBs occurred in the BM175 daughter clone (Fig. 4A, bottom panel). This was associated with a switch in replication from late to early (measured by delta WA) (Fig. 4A) and a compartment switch from B to A. The SV-associated compartment and replication timing switch was linked with up-regulation of both genes within this locus (WDR72, fold-change = 7.5, and UNC13C, fold-change = 7.8) (Fig. 4B) and the emergence of new loop domains (upper right square, shown as open boxes on the Hi-C map) (Fig. 4B). Most of these new looping contacts coincided directly with sites of DSBs, strongly suggesting that they were a direct consequence of the SVs.
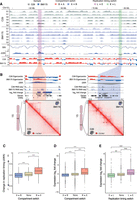
Interplay between compartment switches, replication timing switches, and gene expression in BM175 (daughter clone) compared to C29 (maternal clone). (A) Representative examples of regions with replication timing switches from early → late (green) and late → early (pink) coupled with compartment switches from A → B (blue) and B → A (red), respectively. Percent-normalized density values (PNDVs) for C29 (gray, maternal) and BM175 (blue, daughter) are shown in the top panels. Weighted average values and their difference (ΔWA) pinpoint switches in replication timing, similar to the Hi-C eigenvector tracks below for compartment switches. The switches were evenly distributed genome-wide, irrespective of the presence of DSBs. (B) Alterations in chromatin structure and expression in regions with a combined compartment and replication switch. (Left) B → A and late → early switch accompanied with up-regulation of WDR72 (log2fc = 7.5, P-value = 5 × 10−11) and UNC13C (log2fc = 7.8, P-value = 3 × 10−13), and formation of chromatin loops. (Right) In contrast, an A → B and early → late switch is coupled with down-regulation of ADAMTS7 (log2fc = −3.9, P-value = 1 × 10−15), CTSH (log2fc = −2.2, P-value = 6 × 10−17), and RASGRF1 (log2fc = −2.24, P-value = 0.0002), and loss of chromatin looping. (16/5 kb2) corresponds to 16 normalized counts per 5-kb width. (C) Genome-wide association of compartment switches and changes in replication timing (ΔWA). A → B (n = 298,916) switches have significantly lower ΔWA (E → L) values than regions with no switch (n = 1,912,662), and B → A (n = 317,686) significantly higher (L → E). (***) P-value < 2.2 × 10−16, two-sided t-test. (D,E) A → B (n = 1798) and E → L (n = 327) switches are significantly down-regulating genes compared to regions with no switch, whereas B → A (n = 1063) and L → E (n = 255) up-regulate them significantly. (**) P-value = 3.9 × 10−7, (***) P-value < 7.4 × 10−14, two-sided t-test. Box plots show the median, first, and third quartiles, and outliers are shown if outside the 1.5× interquartile range.
To quantify these observations genome-wide, we computed the extent to which A-to-B and B-to-A compartment domain switches were accompanied by early-to-late and late-to-early replication timing-switches. In agreement with our observations, A-to-B and B-to-A switches were significantly associated with early-to-late and late-to-early switches and with decreased and increased gene expression, respectively (P << 2.2 × 10−16) (Fig. 4C–E; Supplemental Fig. S6).
Having established how SVs can change compartments and replication timing to alter gene expression on average, we noticed a striking set of SVs at Chromosome 11, which led to novel chromatin looping and a series of gene expression changes in cis. The locus involved a 90-Mb deletion and a 35-kb inversion involving BCL9L and CXCR5 (Fig. 5). Our Hi-C-based assembly showed an exchange of the regulatory regions between these two genes and a long-range (>900 kb) ectopic loop to BDNF (Fig. 5A–C). Integrating allele-specific expression demonstrated simultaneous up-regulation of both CXCR5 and BCL9L (>10-fold and twofold, respectively) (Fig. 5D). Conversely, the long-range loop was associated with down-regulation of BDNF (twofold), suggesting the emergence of looping with a repressive element or disruption of existing enhancer-promoter interactions at the locus. In short, this locus exemplified how a few SVs can cause altered chromatin organization and deregulation of several genes, one of them being BCL9L, a known oncogene (Zatula et al. 2014).

Complex SVs form ectopic loops to simultaneously deregulate BDNF, CXCR5, and BCL9L in cis. (A) Hi-C map highlighting a complex SV on Chromosome 11 in BM175. The SV results in the juxtaposition of the BCL9L/CXCR5 locus (yellow) 90 Mb upstream and the formation of an ectopic loop with BDNF (purple). The genomic regions depicted on the Hi-C maps correspond to the highlighted (purple and yellow) regions on the chromosome ideogram. (B) Gene expression tracks stratified by plus/minus strand (bulk RNA-seq) and A/B allele (allele-specific RNA-seq). SVs affect the B allele only. Dashed vertical lines track the breakpoint positions. (C) Schematic representation of the resolved assembly and the SV-mediated deregulation of BDNF, BCL9L, and CXCR5 in cis, as inferred by Hi-C and RNA-seq data. (D) Allele-specific gene expression of BDNF, CXCR5, and BCL9L. (*) P-value < 0.05, (***) P-value < 0.001.
In summary, our integrative results using derived assemblies integrated with chromatin organization and replication information implicate a direct and quantitative functional consequence of SVs on genomic architecture, where SVs not only change the chromatin structure but can also have direct consequences on replication and nearby gene expression.
Discussion
To gain mechanistic insight into SV formation and their consequences on gene regulation, we employed an approach that leverages the power of Hi-C to detect complex SVs and to infer the position and orientation of the rearranged genomic fragments in the derivative chromosomes (Fig. 6). Our findings provide evidence that the folding architecture of the genome is an important determinant for SV formation—and that SVs can have a direct impact on the folding architecture. We find that SVs can cause domain-wide switches, from, for example, A-to-B compartment and early-to-late replication timing, and repression of gene expression. This resembles the recently discovered cis-elements termed “early replication control elements” (ERCEs), which were shown to form long-range CTCF-independent loops to other ERCEs (Sima et al. 2019) and may constitute key replication control elements disrupted by the SVs. The 3D genome impacts directly on SV formation, which in turn influences the 3D genome organization and gene expression in cis. Such “action-reaction” effects between chromatin organization and SVs imply that studies of disease-associated rearrangements should be evaluated in the context of the 3D genome architecture.
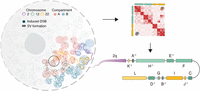
Schematic illustration of genome conformation during double-strand break induction. The Hi-C map allows us to track SV formation and assemble the derivative chromosomes, which enables us to study how SVs affect chromatin organization and gene expression.
In this study, we used cells subjected to dox, a TOP2 inhibitor, to induce SVs. Together with micronuclei formation and telomere attrition, generation of double-strand breaks by TOP2 inhibition can lead to CT in cell-based models (Koltsova et al. 2019). We demonstrate that the induced SVs are strongly associated with chromatin architecture, replication timing, and transcription. Transcription activity has been found to correlate with TOP2 activity (Uusküla-Reimand et al. 2016; Gothe et al. 2019), suggesting a role for active transcription in SV formation. The spontaneous SVs, formed generations after the induced SVs, displayed a similar though less pronounced pattern, with a noticeable preference for SVs to occur between the same compartment and replication timing domain, suggesting that compartment proximity is more important than the identity of the compartment.
We extended our analysis to cancer samples across 30 different tumor types to evaluate our findings from the model system in the context of samples that are expected to have undergone many series of rearrangements over time. We found a consistent pattern of higher A-A compartment SVs across the tumor types. Uterine cancers displayed an extreme pattern with high A-A and, in particular, high levels of early–early replication timing SVs. The nature of this preference for early replication timing and A compartments in uterine cancers is unclear, but it is tempting to speculate that mutations in factors involved in replication timing could play a role in shaping this pattern. Indeed, uterine cancers are associated with frequent mutations in replication timing components, such as RIF1 (Mei et al. 2017) and POLE (Hussein et al. 2015). We note that the uterine tumors are all treatment naive, suggesting that the effects are endogenous to the tumor type.
We also find a strong influence of active transcription on DSB formation and locus partner selection. This process could involve key factors in the enhancer-promoter interaction such as transcription factors and bromodomain proteins, Mediator as well as RNA pol II. A previous study using the balancer chromosome in Drosophila melanogaster revealed a small but significant proportion of SVs to affect nearby gene expression (Ghavi-Helm et al. 2019). Here, we extend these observations to mammalian cells and show a significant distance relationship, with 63% of genes in close proximity of breakpoints to be differentially expressed.
Contact domains have been implicated to play an important role in regulating gene expression, and contact domain-disrupting SVs can lead to dysregulation of developmental genes (Lupiáñez et al. 2015; Nagano et al. 2017) and activation of oncogenes (Weischenfeldt et al. 2017). We find that approximately half of all SVs occur in regions of the genome associated with A compartments, active transcription, and early replication and that SVs are highly enriched at loop anchors and sites bound by chromatin-associated factors. The molecular factors involved in genomic proximity-mediated SV formation, as well as the subsequent consequences on the genome architecture, will need further studies, including, for example, depletion and mutation studies. Cohesin, CTCF, and WAPL are important factors in generating the genome topology by, for example, loop extrusion in mammalian cells (Sanborn et al. 2015; Haarhuis et al. 2017; Rao et al. 2017), and perturbing these could provide insights into their relevance in SV formation. Moving forward, cell type–specific chromatin maps will be needed to advance our understanding, and several large studies are under way to profile cell type–specific chromatin organization such as the 4DN (Dekker et al. 2017), which will provide a deeper understanding of the cell type–specific mechanistic biases of the 3D chromatin and how these impact the occurrence of SVs in diseases such as cancer.
Methods
Preparation of WGS, mate-pair, and RNA-seq libraries
Cell lines were treated as previously described (Mardin et al. 2015). See Supplemental Methods for details. Unless stated otherwise, the NCBI GRCh38 (hg38) human genome reference was used for the alignment of all sequenced reads and the GENCODE v30 reference annotation for all human gene related assessments (Frankish et al. 2019). Comparative sequencing metrics are provided in Supplemental Table S2.
Preparation of Repli-seq libraries
Repli-seq libraries were prepared according to Hansen et al. (2010) based on a previously described STS replication assay (Hansen et al. 1993). See Supplemental Methods.
Preparation of in-situ Hi-C libraries and data processing
All in-situ Hi-C libraries were prepared based on Rao et al. (2014). See Supplemental Methods for details.
All Hi-C data sets were processed using Juicer (Durand et al. 2016) and visualized with Juicebox (Durand et al. 2016). We used HiCCUPS to call loops in C29 with -m 2048 -r 5000,10000 -k KR -f .1,.1 -p 4,2 -i 7,5 -t 0.02,1.5,1.75,2 -d 20000,20000 ‐‐ignore_sparsity parameters which produced a set of 8730 loops at 5-kb and 10-kb resolution. We performed domain calling on C29 Hi-C maps using arrowhead at 5-kb resolution with default parameters and identified 6163 contact domains with 170-kb median size.
Pearson's correlation eigenvectors were computed by the eigenvector module on Knight-Ruiz balanced matrices at 100-kb resolution. For every chromosome, the eigenvector sign with the highest association with gene expression, H3K4me3 and H3K27ac, was assigned to the A compartment.
Allele-specific maps
RPE-1 phased SNPs were obtained from a recent study of RPE-1 cell haplotyping from long-range sequencing (Tourdot et al. 2021). We examined Hi-C read-pairs with MAPQ ≥ 10 where at least one read was overlapping one or more informative SNPs and annotated each informative position to the A or B allele according to the phased information. Reads with varying allele assignments were discarded. Intrachromosomal read-pairs having only one of the reads phased were inferred to the same allele as the phased read. For interchromosomal events, we required both reads to span at least one informative SNP each. Applying these filters yielded allele-specific Hi-C maps with ∼12% of the full set of valid Hi-C interactions.
Derivative chromosome assembly
The derivative chromosome assemblies, illustrated in Figure 1B–E, were produced manually, using the interaction shadow and edge position information derived from the Hi-C map of BM178 (see main text and Fig. 1B,C).
Allele-specific assembly
Allele-specific merged_nodups.txt files were processed with the 3D-DNA pipeline (Dudchenko et al. 2017) to lift chromosomal coordinates to assembly ones and produce Hi-C maps compatible with Juicebox Assembly Tools (JBAT), using the hg38 genome reference as the draft genome.
Compartment conservation
We used the same process as with the RPE-1 cells to obtain A/B compartment annotations from eight human cell lines (Rao et al. 2014). Compartment conservation was measured by the concordance of the compartment annotation across the cell lines in 100-kb
bins. Bins with <5 compartment concordance were annotated as nonconserved (NC). Conservation entropy S was calculated as in
Xiong and Ma (2019) and given by the formula
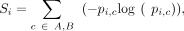 where pi,c is the fraction of cell lines with annotation c for a given 100-kb region i.
where pi,c is the fraction of cell lines with annotation c for a given 100-kb region i.
Copy number alterations and ploidy estimates
WGS data were aligned with BWA-MEM v0.7.15 (Li and Durbin 2009). Copy number alterations (CNAs) in BM175 and BM178 were identified by Sequenza v3.0.0 (Favero et al. 2015), using WGS from the wild-type C93 as normal. Regions with low mappability scores were removed from the analysis. Ploidy estimates were selected based on the Scaled Log Posterior Probability (SLPP).
Structural variant calling
SVs were annotated on the Hi-C map at 5-kb resolution in regions of ectopic contacts with high interaction frequency. The exact breakpoint position was identified as the pixel with the highest intensity, located at the origin of the interaction shadow. Ties within the same rearranged fragment were resolved by choosing the position with the highest contact frequency, requiring at least five supporting read-pairs with MAPQ ≥ 30 in the given 5 × 5-kb pixel.
The derivative assembly of BM178 was produced using SV calls identified exclusively by Hi-C. For the analysis of DSBs and juxtaposition mechanisms, we used SV calls identified by both mate-pair and Hi-C data, except for spontaneous SVs which were only present in the Hi-C maps. See Supplemental Table S2 for comparative sequencing metrics.
Disrupted contact domains are defined as SVs where one breakpoint is inside the contact domain and the other breakpoint is outside of the contact domain.
See Supplemental Methods for details on mate-pair SV calling.
PCAWG data
SV calls from 2693 tumor samples were generated by consensus calling from four SV calling pipelines (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020; Rheinbay et al. 2020).
Replication timing
Repli-seq reads were processed according to Hansen et al. (2010) guidelines. The sequenced tag density of each cell-cycle fraction was calculated in 50-kb sliding windows at 1-kb intervals
and normalized to tags per million, excluding intervals with Ns. To account for sequence, mapping, and copy-number variation
biases the TPM values were converted to percent-normalized density values (PNDVs), representing the percentage of total replication
occurring on a given 1-kb bin within the examined cell-cycle fraction. PNDVs across all six cell-cycle fractions were combined
into a single weighted average (WA) score using the following ENCODE formula:
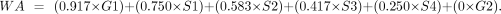 WA reflects replication timing, with high values representing early replication and low values late. Regions with WA values
lower than the first quartile (WA < 0.22) and higher than the third (WA > 0.74) were classified as late- and early-replication,
respectively.
WA reflects replication timing, with high values representing early replication and low values late. Regions with WA values
lower than the first quartile (WA < 0.22) and higher than the third (WA > 0.74) were classified as late- and early-replication,
respectively.
Repli-seq was performed on C29 on BM175 cells, and WA scores were computed independently for each sample. WA was highly correlated between C29 and BM175 (Pearson's correlation 0.81, P < 2 × 10−16). To identify regions with a replication timing switch, we calculated the difference in replication timing (ΔWA) by subtracting the WA of C29 from BM175 so that positive values represent a transition to earlier replication timing. We defined regions below the 10% quantile (ΔWA < −0.11) and above the 90% quantile (ΔWA > 0.29) as early-to-late and late-to-early, respectively.
ChIP-seq
Single-end H3K4me3 ChIP-seq reads generated in this study and publicly available RPE-1 H3K27ac, RNA polymerase II, CTCF, and whole-cell extract (WCE) ChIP-seq reads (NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] accession number GSE60024) were we processed by the ENCODE ChIP-seq (https://github.com/ENCODE-DCC/chip-seq-pipeline2), with ‐‐species hg38 ‐‐type histone ‐‐se. With the exception of H3K4me3, we used WCE as a control.
RNA-seq
Single-end (C29, DCB2) and paired-end (C29, BM175) RNA-seq reads were aligned using STAR v2.5.3 (Dobin et al. 2013). See Supplemental Methods for details.
Cis effects/P-value estimation
To assess the effects of SVs on nearby gene expression, we measured the frequency of significantly differentiated genes (q < 0.05) that are proximal to an SV, setting the distance cut off to 25, 50, 75, and 100 kb. To obtain an empirical P-value, we estimated the background frequency model by randomly shuffling the gene status to differentiated/nondifferentiated 10,000 times and evaluated the P-value by counting the number of random shuffled genes with an effect larger than the observed.
Significance testing
Odds ratio scores and P-values were estimated using Fisher's exact test unless stated otherwise. Empirical distributions of SVs were estimated by shuffling the induced SVs on the same chromosome as the observed set of SVs using BEDTools (Quinlan and Hall 2010), excluding telomeres and centromeres, and preserving the size of intrachromosomal SVs. Each shuffle was performed 1000 times.
Visualization
Hi-C maps were produced by Juicebox v2.0.0 (Durand et al. 2016) and genome browser tracks by the Integrative Genomics Viewer, IGV v2.5.3 (Robinson et al. 2011). All other plots were produced in R v3.6.0 (R Core Team 2019) using the base, ggplot2 (Wickham 2016), circlize (Gu et al. 2014), and flipPlots packages.
Data access
All raw and processed sequencing data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/) under accession number PRJEB40747. Code used in this study is available at Bitbucket (https://bitbucket.org/weischenfeldt/sv_3dchromatin_paper) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Olga Duchenko for helpful comments on the manuscript and Cheng-Zhong Zhang for providing SNP-phased RPE-1 genome data. J.W. and N.S. were partly funded by a grant from the Independent Research Fund Denmark (0134-00265B). Members of the Korbel group received funding from the European Research Council (ERC, 336045).
Author contributions: Conceptualization: J.W. and N.S.; Hi-C-based assembly methodology: N.S., S.G., I.D.B., E.L.A., and J.W.; Edge-and-Shadow concept: N.S. and E.L.A.; RPE-1 cell line generation and perturbation: B.R.M. and J.O.K.; Hi-C experiment: B.R.M. and F.G.R.-G.; Hi-C data analysis: N.S.; ChIP-seq experiment: B.R.M. and A.M.S.; ChIP-seq data analysis: N.S.; RNA-seq experiment: B.R.M. and F.G.R.-G.; RNA-seq data analysis: N.S.; Repli-seq experiment: B.R.M.; Repli-seq data analysis: N.S.; Hi-C-based assembly: N.S.; integrative data analysis: N.S.; visualization: N.S.; manuscript, first draft: J.W. and N.S.; manuscript, revisions: J.W., N.S., B.R.M., A.M.S., and J.O.K.; project administration: J.W.; funding acquisition: J.W. and J.O.K.; supervision: J.W. and E.L.A.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275790.121.
-
Freely available online through the Genome Research Open Access option.
- Received May 18, 2021.
- Accepted February 11, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








