
Gene by environment interaction mouse model reveals a functional role for 5-hydroxymethylcytosine in neurodevelopmental disorders
- Ligia A. Papale1,5,
- Andy Madrid1,2,5,
- Qi Zhang3,5,
- Kailei Chen4,
- Lara Sak1,
- Sündüz Keleş4 and
- Reid S. Alisch1
- 1Department of Neurological Surgery, University of Wisconsin, Madison, Wisconsin 53719, USA;
- 2Neuroscience Training Program, University of Wisconsin, Madison, Wisconsin 53719, USA;
- 3Department Mathematics and Statistics, University of New Hampshire, Durham, New Hampshire 03824, USA;
- 4Department of Statistics, Biostatistics, and Medical Informatics, University of Wisconsin, Madison, Wisconsin 53719, USA
-
↵5 These authors contributed equally to this work.
Abstract
Mouse knockouts of Cntnap2 show altered neurodevelopmental behavior, deficits in striatal GABAergic signaling, and a genome-wide disruption of an environmentally sensitive DNA methylation modification (5-hydroxymethylcytosine [5hmC]) in the orthologs of a significant number of genes implicated in human neurodevelopmental disorders. We tested adult Cntnap2 heterozygous mice (Cntnap2+/−; lacking behavioral or neuropathological abnormalities) subjected to a prenatal stress and found that prenatally stressed Cntnap2+/− female mice show repetitive behaviors and altered sociability, similar to the homozygote phenotype. Genomic profiling revealed disruptions in hippocampal and striatal 5hmC levels that are correlated to altered transcript levels of genes linked to these phenotypes (e.g., Reln, Dst, Trio, and Epha5). Chromatin immunoprecipitation coupled with high-throughput sequencing and hippocampal nuclear lysate pull-down data indicated that 5hmC abundance alters the binding of the transcription factor CLOCK near the promoters of these genes (e.g., Palld, Gigyf1, and Fry), providing a mechanistic role for 5hmC in gene regulation. Together, these data support gene-by-environment hypotheses for the origins of mental illness and provide a means to identify the elusive factors contributing to complex human diseases.
Developmental brain disorders usually present themselves years after birth; however, the molecular pathogenesis is thought to arise earlier, either during pregnancy or just after birth. Genetic studies have revealed hundreds of rare risk genes that cumulatively account for <25% of neurodevelopmental disorder cases (Sebat et al. 2007; Glessner et al. 2009; Weiss et al. 2009). The search for environmental factors contributing to the origins of neurodevelopmental disorders has been motivated by their multifactorial genetic heredity and incomplete concordance in monozygotic twins (Tordjman et al. 2014). There is clear evidence that individual differences in the developmental timing and the severity of early-life stressors, including in utero, can predict emotional dispositions later in life and influence the risk for the development of psychopathology (O'Connor et al. 2003; Heim et al. 2008; Ceschi et al. 2014). This process of “fetal programming” may be mediated, in part, by the impact of the early-life experience on the developing hypothalamic-pituitary-adrenal (HPA) axis (Weinstock 1997; Van den Hove et al. 2006). The HPA axis responds to stressors in early embryonic development and is highly sensitive to early life adversities (Meaney 2001).
5-Hydroxymethylcytosine (5hmC) is an environmentally sensitive epigenetic mark that is highly enriched in neuronal cells (Kriaucionis and Heintz 2009; Song et al. 2011; Szulwach et al. 2011a,b) and is associated with the regulation of neuronal activity, suggesting that 5hmC plays an important role in coordinating transcriptional activity in the brain, where it has been linked to developmental brain disorders (e.g., fragile X syndrome, Rett syndrome, and autism) (Mellen et al. 2012; Wang et al. 2012; Yao and Jin 2014; Zhubi et al. 2014). Recently we reported that the Cntnap2 knockout mouse model of autism shows a genome-wide disruption of 5hmC in a significant number of orthologous genes (N = 57/232) and pathways common to human neurodevelopmental disorders (Papale et al. 2015), supporting an interaction between Cntnap2 and 5hmC in developmental brain disorders.
The Cntnap2 (also known as Caspr2) gene encodes a neuronal transmembrane protein member of the neurexin superfamily involved in neuron–glia interactions and clustering of potassium channels in myelinated axons (Poliak et al. 1999, 2003). Humans harboring a homozygous loss of CNTNAP2 function show severe developmental delays and intellectual disabilities, contributing to a variety of neurodevelopmental disorders such as Gilles de la Tourette syndrome, obsessive-compulsive disorder, cortical dysplasia–focal epilepsy syndrome, autism, schizophrenia, Pitt–Hopkins syndrome, and attention deficit hyperactivity disorder (Poot 2015). Mice harboring a homozygous loss of Cntnap2 function show parallels to the major neuropathological features in autism spectrum disorders, including abnormal social behavior, deficits in communication, stereotypic repetitive behaviors, cortical dysplasia, and focal epilepsy (Penagarikano et al. 2011). In addition, these mice show defects in neuronal migration of cortical projection neurons and a reduction in the number of striatal GABAergic interneurons. In contrast, heterozygous Cntnap2 mutant mice lack any of the behavioral or neuropathological abnormalities found in the homozygous mutant (Penagarikano et al. 2011).
Recent evidence in humans does not support a relationship between heterozygous CNTNAP2 deletions and neurodevelopmental disorders (Toma et al. 2018); however, it remains possible that the combination of heterozygous CNTNAP2 deletions in a genomic background of increased risk may lead to mental illness (Toma et al. 2018). With the onset of Cntnap2 expression in mice beginning on embryonic day (E) 14, its expression may be sensitive to prenatal stressors that alter the neuronal epigenome. Thus, we hypothesized that Cntnap2 heterozygote mutants exposed to prenatal stress would have genome-wide disruptions in 5hmC levels and behavioral deficits, similar to the homozygote mutant.
Results
Prenatal stressed Cntnap2 heterozygous mice show altered social and repetitive behaviors
To test the long-lasting effects of a prenatal environmental stress on the development of altered behaviors, we subjected wild-type (WT) pregnant mice (carrying WT and Cntnap2+/− pups) to 7 d of variable prenatal stress from E12 to E18, which overlaps the onset of Cntnap2 expression (E14; Methods) (Mueller and Bale 2008; Penagarikano et al. 2011). These mild stressors were previously published and were selected because they do not include pain or directly influence maternal food intake or weight gain (Mueller and Bale 2006, 2007). Both WT and HET mice were taken from the same litter, providing an ideal internal control for our findings. Offspring were monitored for consistent maternal/offspring interactions and left undisturbed until weaning day (postnatal day 18) (Supplemental Methods). At 3 mo of age, prenatal stressed (PS) heterozygous Cntnap2 (PS-HET) and PS wild-type (PS-WT), as well as non-PS HET (control-HET) and non-PS WT (control-WT), offspring were subjected to behavioral testing or sacrificed for molecular analysis. All behavioral groups (PS and controls for both genotypes and sex; N = 9–12 per genotype and for each sex) were assessed using a variety of behavioral tests for general locomotor activity, anxiety and depression-like levels, and social aptitude (Supplemental Methods). Only female PS-HET mice showed altered social and repetitive behaviors in two independent social behavioral tests, the three-chamber social test, and a 10-min reciprocal social interaction test, respectively. As expected for the highly social strain C57BL/6J, female controls (control-WT, PS-WT, and control-HET) showed a significant preference to be in the chamber and interacting with the cup containing an unfamiliar mouse in the three-chamber social test. In contrast, female PS-HET mice mirror the homozygous Cntnap2 mutants by not showing a significant preference (differential in mean time spent in each chamber was 65.3 sec for control-WT, 80.3 sec for PS-WT, 102.4 sec for control-HET, and 27.7 sec for PS-HET; two-way ANOVA, P-value < 0.05 for controls only) (Fig. 1A,B; Supplemental Fig. S1; Penagarikano et al. 2011). Similarly, the time spent interacting with the unfamiliar mouse instead of the empty cup during the three-chamber test was significant for the controls but not for the PS-HET female mice (differential in mean time spent interacting in each chamber was 50.2 sec for control-WT, 53.6 sec for PS-WT, 65.4 sec for control-HET, 16.1 sec for PS-HET; two-way ANOVA, P-value < 0.05 for controls only) (Fig. 1C,D; Supplemental Fig. S1F). The mice were then subjected to an additional social interaction test that confirmed the female PS-HET mice spent significantly more time grooming/digging than interacting with an unfamiliar mouse compared with all controls (total time spent grooming/digging among pairs of mice matched for treatment, genotype, and sex was 96.4 sec for control-WT, 74.4 sec for PS-WT, 101.2 sec for control-HET, and 160.8 sec for PS-HET; one-way ANOVA, P-value < 0.05) (Fig. 1E). This repetitive grooming/digging behavior phenotype was only present in a social setting, as it was not observed in mice allowed to explore a cage alone for 10 min (total time spent grooming/digging during a 10-min cage exploratory test was 64.5 sec for control-WT, 52.2 sec for PS-WT, 59.3 sec for control-HET, and 44.2 sec for PS-HET; two-way ANOVA, P-value > 0.05) (Table 1). Deficits in these three social-related measures were reproducible in an independent cohort of female mice (N = 8–10 per treatment, sex, and genotype) (Supplemental Fig. S1). Male mice (control-WT, control-HET, PS-WT, and PS-HET) did not show any altered social or repetitive behavior phenotypes (Supplemental Fig. S1). In addition, both sexes and all experimental groups showed no significant differences in general locomotor activity and in anxiety and depression-like levels (P-value > 0.05) (Table 1; Supplemental Table S1). Together, these data support altered social and repetitive behaviors in female PS-HET mice, revealing a sex-specific gene by environment (GxE) interaction effect on behavior. Because homozygous mutants show similar altered social and repetitive-like behaviors (Penagarikano et al. 2011) and genome-wide disruptions in 5hmC (Papale et al. 2015), we next sought to examine the 5hmC profile in this GxE interaction model.
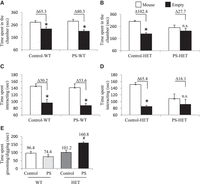
Results from social behavioral tests in female offspring. (A–D) Three-chamber social interaction test: (A,B) time spent in the chamber with either an unfamiliar mouse inside a cup (mouse) or with an empty cup (empty). Data analysis details: (A) two-way ANOVA for control-WT versus PS-WT groups—effect of time spent in either chamber, F(1,42) = 28.5, P-value < 0.001; and (B) two-way ANOVA for control-HET versus PS-HET groups—effect of time spent in either chamber, F(1,32) = 16.2, P-value < 0.001, and interaction between time spent in either chamber and treatment, F(1,32) = 5.3, P-value = 0.002. (C,D) Time spent interacting with either an unfamiliar mouse inside a cup (mouse) or with an empty cup (empty). Data analysis details: (C) two-way ANOVA for control-WT versus PS-WT groups—effect of time spent interacting, F(1,42) = 29, P-value < 0.001; and (D) two-way ANOVA for control-HET versus PS-HET groups—effect of time spent interacting in either chamber, F(1,32) = 9.9, P-value = 0.003. All asterisks denote P-value ≤ 0.05, two-way ANOVA with Bonferroni multiple comparison test–determined post hoc significance. Δ denotes the difference in time spent. (E) Ten-minute reciprocal social interaction test in female offspring. One-way ANOVA detected effect of groups on time spent grooming/digging during a 10-min reciprocal social interaction test (F(3,16) = 10.9, P-value < 0.001). Hashtag (#) denotes significance for time spent grooming/digging between PS-HET in comparison to Control-WT, Control-HET, and PS-WT (P-value < 0.05). N = 9–10 per treatment for each genotype.
Summary of behavioral tests in female offspring
Long-lasting disruption of 5hmC in the female GxE interaction model
To determine the effect of this GxE interaction on the genome-wide distribution of 5hmC, we used an established chemical labeling and affinity purification method, coupled with high-throughput sequencing technology (Szulwach et al. 2011b; Papale et al. 2015, 2016; Li et al. 2016). Age-matched PS-HET and control HET, as well as PS-WT and control-WT female, offspring (N = 3/group) were sacrificed at 3 mo of age as independent biological replicates. 5hmC-containing DNA sequences were enriched from pools (N = 2) of hippocampal tissues, a brain region involved in the pathology of social behavior with high expression of the Cntnap2 gene since E14 and previously shown to be correlated with altered sociability in the Cntnap2−/− mice (Penagarikano et al. 2015). High-throughput sequencing resulted in a range of approximately 25 million to 43 million uniquely mapped reads from each pool of biological replicates (Supplemental Table S2, Supplemental Methods). These data showed no visible differences among the chromosomes, except depletion on Chromosome X, which is consistent with previous observations (Szulwach et al. 2011b; Papale et al. 2015, 2016), and an overall equal distribution of 5hmC levels in both genotypes and in both conditions on defined genomic structures and repetitive elements (Supplemental Fig. S2).
Identification and characterization of differentially hydroxymethylated regions in the female GxE interaction model
To identify distinct 5hmC distribution patterns following prenatal stress throughout the genome of both Cntnap2 heterozygous and WT female mice, we characterized differentially hydroxymethylated regions (DhMRs) between (1) PS-HET and control-HET groups, to investigate the long-lasting effects of the GxE interaction on 5hmC levels (HET-DhMRs), and (2) PS-WT and control-WT groups, to determine the long-lasting effects of prenatal stress on 5hmC levels (WT-DhMRs). A total of 1381 PS-HET increases in hydroxymethylation (PS-HET-specific 5hmC) and 1231 PS-HET decreases in hydroxymethylation (control-HET-specific 5hmC) were found, and these loci were distributed across all chromosomes (Fig. 2A; Supplemental Data Set S1; Supplemental Methods). In contrast, only a total of 735 significant PS-WT increases in hydroxymethylation (PS-WT-specific 5hmC) and 710 significant PS-WT decreases in hydroxymethylation (control-WT-specific 5hmC) were found, and these loci were distributed across all chromosomes (Supplemental Fig. S3A; Supplemental Data Set S1; Supplemental Methods). These data indicate that the GxE interaction results in nearly twice the number of changes in 5hmC levels, but also suggest that prenatal stress alone is sufficient to cause stable changes in 5hmC levels. Because specific regions of the genome are differentially methylated based on the biological functions of the genes contained within each region, we next determined if GxE- and PS-induced DhMRs are enriched or depleted on certain chromosomes using a binomial test of all detected 5hmC peaks as the background (Supplemental Methods). This analysis revealed that DhMRs were not significantly enriched or depleted on any chromosomes (binomial P-value < 0.05) (Fig. 2A; Supplemental Fig. S3A).
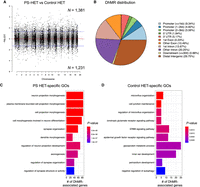
Distribution of hippocampal HET DhMRs. (A) A Manhattan plot shows the distribution of 5hmC sequenced data across the genome (x-axis; alternating black/gray for alternating cromosomes) and the level of significance [–log10(P-value)] along the y-axis. Dots above the top blue line represent PS-HET-specific (hyper)-DhMRs, whereas dots below the bottom blue line represent Control-HET-specific (hypo)-DhMRs (P-value < 0.05). (B) A pie chart displays the proportion of DhMRs across standard genomic structures. (C,D) Bar plot showing the top 10 significantly overrepresented GO terms (y-axis) based on GO analysis of PS-HET-specific DhMR-associated genes (C) and Control-HET-specific DhMR-associated genes (D). The number of DhMR-associated genes linked to the GO terms are displayed on the x-axis. Bar color is based on P-value.
We proceeded to define the genomic features associated with the GxE interaction by annotating HET-DhMRs to overlapping gene structures or to intergenic regions if >10 kb away from any gene structure. The overall distribution of these data indicated that the largest fractions of HET-DhMRs were intronic (∼34%) or were in distal intergenic regions (∼30%), which is not a significant change in distribution compared with distribution of the 5hmC data, suggesting that DhMR are not targeted to specific gene structures (Fig. 2B). Binomial testing revealed that the HET-DhMRs were significantly depleted in exons and promoter regions (binomial P-value < 0.05) (Supplemental Fig. S4A). Although the distribution of WT-DhMRs also revealed structures with significant fluctuations, the genomic location of the WT-DhMR fluctuations was unique to those found for the HET-DhMRs (Supplemental Figs. S3B, S4B). Together, these data indicate that the GxE interaction–induced and stress alone–induced DhMRs are unique and not randomly distributed throughout the genome.
Finally, DNA hypermethylation on repetitive elements is believed to play a critical role in maintaining genomic stability (Coufal et al. 2009). To investigate the genome-wide disruptions of 5hmC on repetitive elements, we aligned the total 5hmC reads and DhMRs to RepeatMasker and the segmental duplication tracks of NCBI37v1/mm9 and found that the largest fraction of HET-DhMRs (∼70%) was in the SINE and simple repeat regions of the genome; however, they were only enriched in LINEs (Supplemental Fig. S4C). In contrast, the WT-DhMRs were enriched in LTR repeats and were specifically enriched in DNA repeats and SINEs (PS-WT-specific and Control-WT-specific, respectively; binomial P-value < 0.05) (Supplemental Fig. S4D). These data suggest a long-term stability of 5hmC at the majority of repeat sequences in female mice following prenatal stress.
Annotation of GxE interaction DhMRs to genes reveals known and potentially novel stress- and neurodevelopmental disorder–related genes
To determine the genes and pathways affected by the long-lasting molecular effects of a GxE interaction on female mice, we annotated the HET-DhMRs to genes and found 895 and 770 genes that contained PS-HET-specific and control-HET-specific 5hmC levels, respectively. One-hundred ninety-five genes were found to have more than one DhMR, leading to a total of 1600 unique genes with disruptions in 5hmC levels (Supplemental Data Set S1; Supplemental Fig. S5). Although 65 genes were identified to have both PS-HET-specific and control-HET-specific 5hmC, DhMRs residing on the same gene were at distinct genomic loci (average >50 kb away from each other), suggesting they have unique molecular roles (e.g., gene regulation) following a GxE interaction. A significant number of HET-DhMR-associated genes were found in well-known stress genes such as Homer1 (Wagner et al. 2015) and Ncam1 (524/1600 χ2 test P-value < 0.0001) (Bisaz et al. 2009). Moreover, several of the HET-DhMR-associated genes were previously linked with developmental brain disorders, including genes that encode synaptic proteins/receptors (e.g., glutamate, GABAergic, calcium channels, potassium channels, sodium channels, as well as neurexin and neuroligin transmembrane proteins). To examine whether the DhMR-associated genes were enriched for neurodevelopmental disorder-related genes, we compared them to a list of developmental brain disorder genes (N = 232 genes) (Supplemental Methods; Gonzalez-Mantilla et al. 2016). This comparison revealed that a significant number of orthologs (N = 56 of 232; hypergeometric test P-value < 0.01) harbor HET-DhMRs, suggesting that 5hmC has a molecular role in the disruption of neurodevelopment following a GxE interaction. Indeed, this finding is similar to the DhMR-associated genes found in the Cntnap2 homozygous mouse (Papale et al. 2015). The HET-DhMR-associated genes also included several genes known to function in epigenetic pathways, including Dnmt3a, Tet2, and Hdac4. To further examine the biological significance of the HET DhMRs-associated genes, we performed separate Gene Ontology (GO) analyses of the PS- and control-specific HET DhMR-associated genes (N = 895 and 770, respectively) and found a significant enrichment of neuronal GO terms only among the PS-specific HET DhMR-associated genes, which included regulation of synaptic organization, axon development, and neuron projection morphogenesis (P-value < 0.05 and FC > 1.5 for GO enrichment tests) (Fig. 2C; Supplemental Data Set S2). In contrast, the control-specific HET DhMR-associated genes were related to organ-related developmental and morphological processes (Fig. 2D; Supplemental Data Set S2). Together, increased 5hmC in the GxE model results in disruptions of genes that encode synaptic proteins/receptors and signaling pathways, supporting a link to brain-related disorders.
Genomic annotation of the WT-DhMRs to genes revealed that 510 and 450 genes contained PS-WT-specific and control-WT-specific 5hmC, respectively. Sixty genes were found to have more than one DhMR, leading to a total of 931 unique genes with disruptions in 5hmC levels (Supplemental Data Set S1). Although 28 genes were identified to have both PS-WT-specific and control-WT-specific 5hmC, WT-DhMRs residing on the same gene were at distinct genomic loci (average >50 kb away from each other), suggesting that when found proximal to the same gene WT-DhMRs likely have unique molecular roles (e.g., gene regulation) following prenatal stress. Nearly 80% of WT-DhMR-associated genes were unique from the HET-DhMR-associated genes (Supplemental Fig. S3C), indicating that the majority of hydroxymethylation changes were specific to genotype. Moreover, to gain insight into 5hmC changes that may predispose heterozygous mice to show repetitive behaviors and altered sociability when exposed to prenatal stress, we compared control-HET and control-WT groups and found 1430 DhMR-associated genes. Overlap of these genes with PS-HET-associated and PS-WT DhMR-associated genes revealed a significant overlap among all three groups (N = 50) (Supplemental Fig. S3D; Supplemental Data Set S1). The overlapping DhMR-associated genes may indicate top candidates contributing to the GxE behavioral outcomes.
A significant number of WT DhMR-associated genes were previously linked to stress, including Dnmt1, Pten, and Fkbp5 (312/931; hypergeometric test P-value < 0.01) (Unterberger et al. 2006; Bassi et al. 2013; Zannas et al. 2016). Moreover, a significant number of orthologs, best known for their role in human neurodevelopmental disorders, have disruptions in 5hmC levels (35/232; hypergeometric P-value < 0.01; e.g., Aut2, Nckap5, Setbp1) (Papale et al. 2015; Gonzalez-Mantilla et al. 2016), further implicating stress alone in the pathogenesis of neurodevelopmental disorders. Finally, WT DhMR-associated genes included Dnmt1 from the epigenetic pathway. Annotation of the PS- and control-specific WT DhMR-associated genes (N = 510 and 450 genes, respectively) to GO terms found a significant overrepresentation of terms connected with metabolism/catabolism and limb development/morphogenesis (Supplemental Figs. S3E,F; Supplemental Data Set S2). Together, these data indicate that prenatal stress alone disrupts 5hmC on biologically relevant genes and pathways.
Confirmation of long-lasting disruptions of 5hmC in the GxE interaction model
As an initial confirmation of genome-wide disruption of 5hmC following prenatal stress, we profiled 5hmC levels in an independent cohort of mice and another brain region associated with social and stress-related behaviors (striatum) that may contribute to pathways of risk in female autism spectrum disorders (Jack et al. 2021). Age-matched PS-HET and control-HET, as well as PS-WT and control-WT female mice (N = 3/group), were sacrificed at 3 mo of age as independent biological replicates. 5hmC containing DNA sequences were enriched from striatal total DNA, and high-throughput sequencing resulted in a range of approximately 20 million to 70 million uniquely mapped reads from each biological replicate (Supplemental Table S3; Supplemental Methods). Similar to the hippocampal 5hmC data, sequence read density mapping showed no visible differences among the chromosomes, except depletion on Chromosome X, and an overall equal distribution of 5hmC levels in both genotypes and in both conditions on defined genomic structures and repetitive elements (Supplemental Fig. S6). To compare the long-lasting molecular effects of a GxE interaction on female mice in the striatum and hippocampus, we annotated the striatal HET-DhMRs to genes (N = 2224) and found a significant overlap of differentially hydroxymethylated genes between both tissues (N = 395 genes; P-value < 0.05), including genes well known to play a role in neurodevelopmental disorders (e.g., Nrxn1, Nlgn1, and Grip1/Ncoa2) (Supplemental Fig. S7A). Again, similar to the hippocampal findings, the Gene Ontologies of the striatal DhMR-associated gene contained a significant neuronal enrichment of terms (e.g., synapse organization/transmission [glutamatergic] and negative regulation of axonogenesis; P-value < 0.05 and FC > 1.5 for GO enrichment tests) (Supplemental Data Set S2). The DNA hydroxymethylation levels of several regions also were validated using an alternative method (Supplemental Fig. S7B; Supplemental Methods). Together, these striatal 5hmC data provide a validation that 5hmC levels are stably disrupted following a GxE interaction in brain regions linked to stress response and social behavior.
Because the prenatal stressed Cntnap2+/− female mice showed repetitive behaviors and altered sociability, similar to the homozygote phenotype, we compared the DhMR-associated genes between these two models (e.g., PS-HET and female homozygotes) and found a significant overlap of genes in the striatum (N = 443; P-value < 0.05) (Supplemental Fig. S7C). In addition, the DhMR-associated genes from the homozygote striatum also had a significant overlap of genes compared with the PS-HET genes from the hippocampus (N = 346; P-value < 0.05) (Supplemental Fig. S7C). Together, these data further validate the finding that 5hmC disruptions are linked to neurodevelopment-related disorders and support GxE hypotheses for complex disorders.
Candidate functional DhMRs in the female GxE interaction model
To gain insight into the potential molecular mechanism(s) for long-lasting GxE interaction-induced DhMRs, we used RNA sequencing (RNA-seq) to profile gene expression in the hippocampus of the same mice surveyed for 5hmC (see Supplemental Methods). Comparison of transcript levels in PS-HET and control-HET mice revealed 524 differentially expressed genes, 162 up-regulated and 363 down-regulated (FDR P-value < 0.1) (Fig. 3A; Supplemental Data Set S3). Separate Gene Ontologies of these 162 up-regulated and 363 down-regulated genes found terms related to metabolic-related processes (up-regulated genes) (Fig. 3B; Supplemental Data Set S4) and axonal development and histone modifications (down-regulated genes) (Fig. 3C; Supplemental Data Set S4; Supplemental Methods). DhMRs associated with differentially expressed genes represent candidate functional DhMRs that may have a direct role in gene regulation. An overlay of the HET-DhMR data with these HET-RNA-seq data revealed that 72 differentially expressed genes harbor a HET-DhMR, which included neurodevelopmental disorder genes (e.g., Reln, Dst, and Trio) (Gonzalez-Mantilla et al. 2016), known stress-related genes (e.g., Ncoa2) (Barko et al. 2019), and epigenetic genes (Dnmt3a and Hdac4) (Fig. 3D; Supplemental Data Set S3). The Pearson's correlation between DNA methylation and gene expression across all genes was similar to the correlations found in previous studies (r = 0.267) (Zeng et al. 2012; Alisch et al. 2014). Although general relationships between 5hmC abundance and gene expression levels were not observed in these genes, there was a significant enrichment of DhMRs located in the introns of differentially expressed genes (P-value < 0.05). These results indicate that the GxE interaction induces long-lasting effects in the regulation of genes functioning in neuronal development and synaptic plasticity, and support a role for 5hmC in this dysregulation that is associated with altered social behaviors in adult GxE mice.
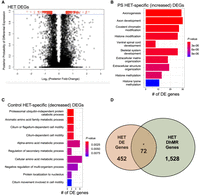
Characterization of differentially expressed genes (DEGs) between PS-HET and Control-HET. (A) A modified volcano plot depicts the log2(posterior fold change; x-axis) versus the posterior probability of differential expression (y-axis). Open circles (red/black) represent each gene examined, and differentially expressed genes (red) are shown above the significance line (blue; FDR P-value < 0.1). (B,C) A bar plot showing the top significantly overrepresented GO terms based on GO analysis of PS-HET-specific (B) differentially expressed genes and Control-HET-specific (C) differentially expressed genes. The number of DEGs linked to the GO terms are displayed on the x-axis. Bar color is based on P-value. (D) A Venn diagram depicts the overlap of DhMR-associated genes (green circle) and differentially expressed genes (orange circle) from the PS-HET versus Control-HET comparison. The asterisk (*) denotes a significant overlap (hypergeometric enrichment P-value < 0.05).
Comparison of transcript levels in PS-WT and control-WT mice revealed only five genes that were altered by prenatal stress, all down-regulated (FDR P-value < 0.1) (Supplemental Fig. S8; Supplemental Data Set S3). An overlay of the WT-DhMR data with the WT-RNA-seq data did not reveal any potentially functional DhMRs. These results suggest that WT-DhMRs represent stable molecular alterations that may have affected gene expression at earlier developmental time points, suggesting that the PS-WT mice may have earlier phenotypes related to the prenatal stress.
The GxE interaction alters transcription factor binding in DhMRs
Syndromic forms of neurodevelopmental disorders can involve the disruption of transcription factor function (Gharani et al. 2004; Bienvenu and Chelly 2006); thus, we investigated the potential for the HET-DhMRs to alter DNA binding of transcription factors by testing for enrichments of known transcription factor sequence motifs among the DhMRs (Supplemental Methods). Several transcription factor sequence motifs were significantly enriched in the hippocampal HET-DhMRs. Many of the transcription factors that bind these motifs have links to neurodevelopmental behaviors and disorders, such as CLOCK, NPAS2, HIF1A, ARNT (also known as HIF1B), and USF1 (Table 2; Supplemental Data Set S5; Schmidt-Kastner et al. 2006; Nicholas et al. 2007; Shi et al. 2016; Hamilton et al. 2018). Together, these findings suggest that 5hmC influences transcription factor binding, which may explain the observed correlated disruptions in gene expression. An initial test of this hypothesis examined transcription factor sequence motif enrichments among only the potentially functional DhMRs (i.e., genes with correlated disruptions in 5hmC and expression; N = 72) (Supplemental Methods). Five transcription factors sequence motifs were significantly enriched among the potentially functional HET-DhMRs (BHLHE40, MYC, CLOCK, ARNT, and USF1). The CLOCK transcription factor binds E-box enhancer elements, its consensus binding sequence contains a CpG dinucleotide (CACGTG) that can be hydroxymethylated, and its expression/dysregulation is associated with autism (Hu et al. 2013; Schuch et al. 2018). RNA sequence data and Western blot analysis revealed that Clock is not differentially expressed at the transcript and protein levels due genotype and/or condition (i.e., PS-HET, control-HET, PS-WT, and control-WT) (Fig. 4A; Supplemental Data Set S3). To determine the effect of this GxE interaction on the genome-wide distribution of CLOCK binding, we used chromatin affinity purification coupled with high-throughput sequencing technology (ChIP-seq). Age-matched PS-HET and control HET, as well as PS-WT and control-WT female offspring, (N = 3/group) were sacrificed at 3 mo of age as independent biological replicates. DNA sequences containing CLOCK binding sites were enriched from the hippocampus total DNA and high-throughput sequencing resulted in approximately 10 million uniquely mapped, nonduplicated, reads from each biological replicate (Methods; Supplemental Data Set S6). A differential analysis of these data filtered to only DhMRs containing a putative CLOCK binding site (N = 577; CACGTG) identified 19 DhMRs with differential CLOCK binding (Fig. 4B; Supplemental Data Set S6). Three of these DhMRs were associated with genes that also were differentially expressed as a result of the GxE interaction (Gigyf1 ∼1.8× and both Palld and Fry ∼1.5×) (Supplemental Data Set S3). The DhMR containing CLOCK binding sequences reside near the promoters of each of these genes (Fig. 4C), and all three genes have links to autism (Garbett et al. 2008; Voineagu and Eapen 2013; Paulraj et al. 2019; Satterstrom et al. 2020). Together, these data suggest that differential hydroxymethylation levels may mediate CLOCK binding, supporting a mechanism for 5hmC in the regulation of gene expression of genes associated with the observed phenotype.
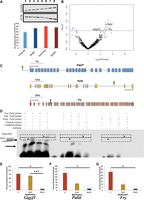
Functional assays reveal that 5hmC represses CLOCK binding. (A) Western blot analysis identified no gross differences in CLOCK protein expression between Control-WT (lanes 1,2), PS-WT (lanes 3,4), Control-HET (lanes 5,6), or PS-HET (lanes 7,8; top panel) or in ACTB (middle panel), used as a loading control in all lanes. A bar plot (bottom panel) depicts the quantified levels of CLOCK proteins normalized to ACTB from each respective lane. Error bars show the SEM (N = 2). (B) A volcano plot displays the –log2(fold-change) of PS-HET compared with Control-HET (x-axis) and the significance (y-axis) from differential analysis of ChIP-seq data. Open circles (blue/black) and red triangles represent DhMRs containing a canonical CLOCK binding site. DhMRs with significant differential binding of CLOCK (blue open circles and red triangles) are located above the significance line (P-value < 0.05). Red triangle DhMRs also show differential gene expression (Gigyf1, Palld, and Fry). (C) Gene schematics of Gigyf1 (blue), Palld (yellow), and Fry (orange) are shown and indicate the location of the DhMRs containing the differential CLOCK binding (green triangle). (D) Electromobility shift assays with hippocampal lysates. Lysate shift (lanes 2,6,10), cold competition (lanes 3,7,11), and supershift (lanes 4,8,12) of DNA probes corresponding to a CLOCK E-box motif in the DhMR of Palld that showed differential CLOCK binding. Biotinylated DNA probes contained either an unmodified E-box motif (lanes 1–4), a methylated E-box motif (lanes 5–8), or a hydroxymethylated E-box motif (lanes 9–12). Regions containing bands of interest are highlighted (dashed boxes); DNA::protein complexes that are abrogated with cold competition are indicated (black arrowheads) and supershift complexes are shown (carrot). (E–G) Quantifications of biotinylated DNA probe intensities. The E-box modifications (x-axis) and mean biotinylated DNA probe intensity (y-axis) are depicted ±SEM for Gigyf1 (E), Palld (F), and Fry (G) probes. The asterisk (*) denotes a significant decrease in CLOCK binding box: (*) P-value < 0.05; (***) P-value < 0.001.
A subset of the transcription factors with binding motifs associated with DhMRs in hippocampus PS-HET × Control-HET
5hmC alters CLOCK binding
To formally test whether 5hmC can alter CLOCK binding, hippocampal nuclear lysates were incubated with oligonucleotide probes containing sequences from the differentially bound CLOCK sites in the DhMRs associated with Gigyf1, Palld, and Fry genes. The CpG dinucleotide in the E-box motif of CLOCK located in each probe contained either cytosine, 5-methylcytosine, or 5hmC (Methods). Electromobility shift assays using unmodified cytosine probes from each of these genes revealed successful binding of CLOCK from hippocampal nuclear lysates (Fig. 4D–G; Supplemental Fig. S9). These DNA::protein complexes were abrogated in cold completion assays and super-shifted with the addition of antibodies against CLOCK (Fig. 4D, left panel; Supplemental Fig. S9). In contrast, hydroxymethylated probes significantly reduced the binding of CLOCK (Fig. 4D–G; Supplemental Fig. S9), indicating that 5hmC prevents CLOCK from binding to the E-box motifs located near the promoters of the differentially expressed genes Gigyf1, Palld, and Fry. Positive controls for both 5mC (Kcnk4) and 5hmC (Camk2a) confirm that these DNA modifications show unique binding efficiencies for specific transcription factors (Supplemental Fig. S10). Together, these findings suggest a working model for 5hmC regulation of developmentally important genes that when altered can result in social-related disorders. In the example presented here, the GxE interaction results in a loss of 5hmC near the promoters of Gigyf1, Palld, and Fry that subsequently improves the binding of CLOCK to act as a repressor of Gigyf1, Palld, and Fry expression (Fig. 5). Thus, Gigyf1, Palld, and Fry are top candidate genes contributing to the adult sex-specific phenotype found in female Cntnap2 heterozygous mice exposed to prenatal stress.
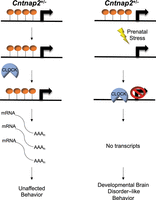
A working model of a functional role for 5hmC in developmental brain disorder–like behaviors. Control Cntnap2+/− mice (left panel) 5hmC levels (orange lollipops) remain abundant and prevent CLOCK (blue semicircle) from binding to genes implicated in developmental brain disorders (e.g., Gigyf1), which results in adequate gene expression levels (black curved lines) and unaffected behaviors. Cntnap2+/− mice exposed to prenatal stress (PS; lightning bolt; right panel) show a loss of 5hmC abundance in genes implicated in developmental brain disorders (e.g., Gigyf1). This reduction in 5hmC levels allows CLOCK to bind to DNA and repress transcription, which results in developmental brain disorder–like phenotypes.
Discussion
Here we provide evidence of a functional role for 5hmC in a gene (partial loss of Cntnap2) by environment (prenatal stress; GxE) interaction model that results in sex-specific alterations of neurodevelopmental behaviors. These 5hmC disruptions were found in the orthologs of a significant number of genes implicated in human developmental brain disorders, significantly overlapping with those found in both male and female Cntnap2 homozygous mouse mutants (Papale et al. 2015). Integration of epigenetic and gene expression data from the same mice revealed relations with genes (e.g., Nrxn1, Reln, Grip1, Dlg2, and Sdk1) known to contribute to the behavioral deficits of neurodevelopmental disorders. Finally, we focused on an enrichment of transcription factors sequence motifs among the potentially functional PS-HET DhMRs and found that the loss of 5hmC in these regions resulted in the loss of CLOCK binding. Together these data show a molecular mechanism for 5hmC in the regulation of developmentally important genes that when altered can result in social-related disorders. A GxE model and 5hmC disruptions may represent a common etiology for developmental brain disorders in humans.
Having data from both striatal and hippocampal tissues provides a unique opportunity to examine the combined and differential contributions of these tissues to neurodevelopment-related behaviors, which both supported that the epigenome plays a role in the long-lasting effects of prenatal environmental exposures on brain and behavior. For example, both tissues carried GxE DhMRs in orthologous genes common to human neurodevelopmental disorders, such as nuclear receptor genes, glucocorticoid receptor interacting protein 1 (Grip1 also called Ncoa2) (Barko et al. 2019), as well as several genes associated with neurotransmission and synaptic plasticity (Nrxn1, Nlgn1, Dlg2, and Negr1) (Gonzalez-Mantilla et al. 2016; Szczurkowska et al. 2018). The hippocampus-specific GxE DhMRs also included genes related to human neurodevelopmental disorders, including Homer1, which forms high-order complexes with Shank1 that are necessary for the structural and functional integrity of dendritic spines (Orlowski et al. 2012). In addition, the hippocampus-specific GxE DhMRs contained links to circadian function (e.g., Clock, Homer1, and Shank2); the prenatal stress administered here is sleep disruptive, supporting a hypothesis that circadian rhythms contribute to the pathogenesis of neurodevelopmental disorders (Geoffray et al. 2016). Finally, investigation of the hippocampus-specific GxE DhMRs with correlated disruptions in gene expression revealed links to the observed adult behavioral deficits, including specific links to the limbic system and synaptic plasticity (e.g., Dst, Reln, Lpp, Trio, Notch1 Col4a1, and Utrn) (Sibbe et al. 2009; Bhandare et al. 2010; Wong et al. 2014; Wang et al. 2015). Together, these data suggest insights into the molecular pathogenesis of GxE interactions that result in neurobehavioral alterations. Although prenatal stress alone disruptions in 5hmC (WT-DhMRs) were largely unique from the GxE DhMRs (HET-DhMRs), WT-DhMR-associated genes also included previous links to neurodevelopmental disorders (e.g., Auts2, Cacna1c, and Setd5) (Gonzalez-Mantilla et al. 2016). Despite the fact that many of these genes have been implicated in disorders with social deficits, 5hmC disruption on these genes was not correlated with altered transcript levels and was not sufficient to cause a behavioral phenotype.
Mechanistically, it is intriguing that the binding motifs of several transcription factors with known roles in neuronal development were found in the GxE DhMRs. These findings included binding motifs for transcription factors best known for their roles in circadian rhythm, immune response, learning and memory, and hypoxia. For example, CLOCK, ARNTL, BHLHE40, and BHLHE41 all regulate the circadian clock; in addition, they are also linked to neurodevelopmental disorders, such as major depressive disorders, bipolar disorders, and autism spectrum disorders (Nicholas et al. 2007; O'Roak et al. 2012; Shi et al. 2016; Hamilton et al. 2018); again, consistent with the prenatal stress paradigm used here being sleep disruptive. CLOCK functions as a pioneer-like transcription factor (Menet et al. 2014) while also displaying histone acetyltransferase activity (Doi et al. 2006), corroborating CLOCK as a regulator of chromatin accessibility and contributor to the epigenetic framework in cells. Although CLOCK generally acts as a transcriptional activator, it also shows repressive activity that is dependent on the assembly of larger protein complexes (Michael et al. 2017; Yang et al. 2020; Cao et al. 2021). Paired box protein 5 (Pax5) and aryl-hydrocarbon receptor (Ahr) have defined roles in immune response and also are connected to autism spectrum disorders and major depressive disorders (Wong et al. 2008; O'Roak et al. 2012; Neavin et al. 2018). The stress paradigm used in this study increases placental inflammation (Bronson and Bale 2014); indeed, immune activation history in the mother is associated to increased symptom severity in children with mental illness (Patel et al. 2018). It is notable that the GxE interaction resulted in distinct differences between PS-HET and PS-WT enriched transcription factor sequence motifs, perhaps suggesting an interaction between CNTNAP2 and 5hmC in transcription factor binding and function.
Focusing more specifically on significant enrichments of transcription factor sequence motifs in potentially functional DhMRs, especially enriched motifs located in the promoter regions, revealed known and novel genes contributing to the adult phenotype. Gigyf1, Palld, and Fry were the examples reported here with differential CLOCK binding and links to autism (Garbett et al. 2008; Voineagu and Eapen 2013; Paulraj et al. 2019; Satterstrom et al. 2020). In addition, catenin (cadherin associated protein), alpha 2 (Ctnna2) is an example of a gene that is differentially expressed and contains a DhMR located in its promoter harboring a different enriched transcription factor sequence motif (USF1). Rodent Ctnna2 encodes a protein that regulates morphological plasticity of synapses, and its loss in neurons leads to defects in neurite stability and migration, as well as cortical dysplasia (Schaffer et al. 2018). Indeed, cortical dysplasia is a feature in humans and mice lacking expression of CNTNAP2, and mice show defects in neuronal migration of cortical projection neurons and a reduction in the number of striatal GABAergic interneurons (Penagarikano et al. 2011; Poot 2015). It will be important to characterize the role of 5hmC on the binding of other transcription factors (e.g., USF1) in promoters and enhancers of genes containing potentially functional DhMRs. Although only the 5hmC probes containing the transcription factor binding sites had statistically significant different binding efficiencies compared with the 5C probes, it will be important for future studies to characterize the potential role of 5mC in altered transcription factor binding associated with GxE interactions resulting in neurodevelopmental disorders. Despite the fact that enriched motifs in DhMRs located in promoter regions of differentially expressed genes represent top candidate genes directly contributing to the adult phenotype, enriched motifs in DhMR-associated genes that are not differentially expressed in the adult may identify genes contributing to the origins of the adult behavioral deficits. Further studies at earlier developmental time points are needed to determine the role of these DhMRs in the development of the observed adult behavioral deficits.
Exposure to stress during different gestational periods has unique effects on epigenetic programming of the developing embryo. During early gestation, prenatal stress-induced epigenetic changes in neuronal precursor cells are predominantly mediated by hormonal mechanisms, which in part can be transmitted via placental pathways (Bock et al. 2015). Stress during this time period predominantly affects behavioral development in males (Li et al. 2008; Mueller and Bale 2008). However, in late gestational stages, the mechanisms underlying prenatal stress-induced epigenetic changes become more complex, involving the activation of the excitatory and inhibitory endocrine modulatory systems, which may interfere with stress-induced synaptic reorganization (Bock et al. 2015). Stress during this time period predominantly affects behavioral development in females (Li et al. 2008; Mueller and Bale 2008); thus, there is a sex-specific effect on behavior depending on the gestational timing of the stress exposure. Consistent with these reports, late gestational stress in this present study results in long-lasting behavioral and molecular alterations in females on genes known to be associated with synaptic plasticity and mental disorders. The repetitive behaviors and social deficits render the PS-HET mice an attractive model for other psychiatric disorders in humans. Because both insults (i.e., genetic and environment) are required for the expression of an altered behavioral phenotype, this model (i.e., a two-hit model) directs studies to consider environmentally sensitive molecular mechanisms contributing to stress-induced behavioral outcomes. Moreover, it supports the study of other “multihit” hypotheses in the origins of mental illness to identify elusive factors that could contribute to the complexity of psychiatric disorders in humans. Although there is precedence for multihit models disrupting brain development (van den Hove et al. 2011; Schraut et al. 2014; Bell et al. 2016; Schaafsma et al. 2017), it will be important to examine the generalizability of these findings using other developmentally important genes in the brain that also may contribute to the vulnerability/resilience toward mental illness.
CNTNAP2 has been implicated in numerous neurodevelopmental disorders and heterozygous disruptions have been found in humans with intellectual disability (ID), seizures, and signs of autism spectrum disorders (e.g., repetitive behaviors, and social deficits). However, heterozygous losses of CNTNAP2 also have been inherited from healthy parents, suggesting that heterozygous disruptions of CNTNAP2 by themselves are not sufficient to elicit cellular or organismal phenotypes. Aside from striatum and hippocampus, high levels of CNTNAP2 expression have been found in the olfactory bulb, ventricular zone, and thalamus (Abrahams et al. 2007; Penagarikano et al. 2011), warranting future examinations of 5hmC in other brain-related behaviors that are linked to these additional brain regions. CNTNAP2 also has an organizing function to assemble neurons into neural circuits (Anderson et al. 2012), suggesting future studies should examine the electrophysiology of the excitatory and inhibitory synaptic transmissions in the PS-HET mice. Finally, prenatal stress may have revealed a molecular connection between 5hmC and Cntnap2 involving the binding of a transcription-enhancing transcription factor TCF4, which can transactivate the CNTNAP2 and NRXN1 promoters. Heterozygous deletions of TCF4 causes Pitt–Hopkins syndrome: a syndrome that also is linked to the loss of either CNTNAP2 or NRXN1 function (Amiel et al. 2007; Brockschmidt et al. 2007; Zweier et al. 2007). Thus, TCF4 may modulate the expression of CNTNAP2 and NRXN1 in the regulatory network involved in Pitt–Hopkins syndrome (Forrest et al. 2012). We find several DhMRs associated with Tcf4 and Nrxn1 following GxE interaction, suggesting that the regulatory network involved in Pitt–Hopkins syndrome may be similarly disrupted in the PS-HET mice. Together, these findings potentially provide novel insight into the molecular connection between 5hmC, Cntnap2, Tcf4, and Nrxn1 in neurodevelopmental disorders, such as Pitt–Hopkins syndrome.
Molecular factors influencing vulnerability and resilience to environmental stress clearly involve environmentally sensitive epigenetic mechanisms such as DNA methylation. Thus, perturbations of GxE interactions may underlie common pathways for many mental illness–related disorders in humans. The identification and characterization of a potentially modifiable substrate (e.g., 5hmC) contributing to GxE interaction–induced sex-specific neurologic behaviors are significant and comes at a time when there is great interest to harness the diagnostic and therapeutic power of these substrates toward healthy outcomes.
Methods
Prenatal stress paradigm
All experiments were approved by the University of Wisconsin–Madison Institutional Animal Care and Use Committee (M02529). To minimize the stress of animal handling, all of the following were conducted by a single researcher: animal colony maintenance, breeding, prenatal stress, and behavioral tests. Pregnant mice assigned to the variable stress group experienced a different daily stressor on each of the 7 d during late pregnancy (E12 to E18; the timing of which was chosen because it overlaps with the onset of Cntnap2 expression [E14]). These variable stressors included 36 h of constant light, 15 min of fox odor (catalog no. W332518) beginning 2 h after lights on, overnight exposure to novel objects (eight marbles), 5 min of restraint stress (beginning 2 h after lights on), overnight novel noise (white noise, nature sound-sleep machine, Brookstone), 12 cage changes during the light period, and overnight saturated bedding (700 mL, 23°C water) (Mueller and Bale 2006, 2007). These mild stressors were previously published and were selected because they do not include pain or directly influence maternal food intake or weight gain. Both WT and HET sibling pairs from multiple litters were used for both behavior and molecular studies, providing an ideal internal control for our findings and negates the need for cross-fostering of the prenatally stressed pups to nonstressed moms to find effects of prenatal exposure to stress.
Statistics for behavioral tests
Data are reported as mean ± S.E.M. Homogeneity of variance was assessed using the Brown–Forsythe and the normal distribution of the data was tested by the Shapiro–Wilk test. Two-way ANOVA also was used to detect differences between genotype (WT × HET) and treatment (Control × PS) for time spent in the inner circle and number of entries into that circle (open field test); time spent in closed arm, time spent in open arm, and time spent in center (elevated plus maze); time spent in light side (light–dark box test); and time spent floating (forced swim test). Two-way ANOVA was used to detect differences between treatment (Control × PS) for time spent in the chamber (three-chamber social test) and interacting with the novel mouse and time spent in the chamber with the empty cup. One-way ANOVA was used to detect differences between groups for time spent engaged in repetitive behaviors (grooming and digging). Post hoc analysis was performed using a Bonferroni correction. Statistics were performed using SigmaPlot version 14.
5hmC enrichment of genomic DNA, library preparation, and high-throughput sequencing
Chemical labeling-based 5hmC enrichment was described previously (Song et al. 2011; Papale et al. 2015). 5hmC-enriched libraries were generated using the NEBNext ChIP-seq library prep reagent set for Illumina sequencing, according to the manufacturer's protocol. Fifty-cycle single-end (striatum) or paired-end (hippocampus) sequencing was performed by Beckman Coulter Genomics or the University of Wisconsin Biotechnology Center, respectively. Paired-end sequencing for hippocampal tissue was chosen for reasons unrelated to this project. Image processing and sequence extraction were performed using the standard Illumina Pipeline.
Identification of DhMRs
We mapped the sequence reads to mouse NCBI37v1/mm9 reference genome using Bowtie 0.12.7 (Langmead et al. 2009) and called peaks using the MACS2 algorithm v2.1.2 (Zhang et al. 2008). Newer versions of the genome always add more sequence data, but because we achieved >90% alignment of the reads to mm9, realigning to mm10 or beyond would only nominally increase the alignment efficiency, not altering the main findings of these analyses. Summits from both striatal and hippocampal data were extracted for each peak for each sample and extended ±500 bp for downstream analysis. For each genotype, the stress and control groups were pooled and merged to form the candidate differential regions within each genotype. The Bioconductor package edgeR was used to test each candidate region (P-value < 0.05) (Robinson et al. 2010).
Analysis of RNA-seq data
Read alignment and calculation of transcript expression levels: We used the mm9 assembly as our reference genome and the GENCODE M1 (NCBIM37) as our gene annotation library (the Y Chromosome was deleted for the alignment of female samples). We ran RSEM to calculate the expression at both gene level and isoform level (Li and Dewey 2011). EBSeq was used to detect the DE genes and transcripts in each of the pairwise comparisons of PS-WT versus control WT, and PS-HET versus control HET, with an FDR < 0.1.
ChIP-sequencing and analysis
High-throughput sequence libraries were generated from immunoprecipitated and IgG eluted DNA using the NEBNext ChIP-seq library prep reagent set for Illumina sequencing, according to the manufacturer's instructions. Fifty-cycle paired-end sequencing was performed by the University of Wisconsin–Madison Biotechnology Center. Image processing and sequence extraction were performed using the standard Illumina Pipeline. Raw paired-end sequencing files were assessed for quality using FastQC (https://www.bioinformatics.babraham.ac.uk/), trimmed using the Trim Galore! package in R (https://github.com/FelixKrueger/TrimGalore), and aligned to the mm9 genome using Bowtie 2 v2.3.4 (Langmead et al. 2009), local alignment. Following alignment, SAMtools to filter multimapping reads (Li et al. 2009) and PCR duplicates. Uniquely mapped and filtered sequence reads were mapped to DhMRs found to contain a putative CLOCK binding site (CACGTG) by motif enrichment analysis (N = 577). Only data from these sites were used for differential binding analyses (R package edgeR) (Robinson et al. 2010), after controlling for background through normalization to sequenced IgG reads. A P-value threshold of 0.05 was used to determine significant differential binding of CLOCK between groups.
Electrophoretic mobility shift assays
Complementary 5′-biotinylated (0.5 µM) and unlabeled oligonucleotides (Integrated DNA Technologies) (Supplemental Methods) were annealed by heating equal concentrations of sense and antisense oligonucleotides to 95°C and lowering the temperature by 2°C in 3-min intervals until reaching 23°C. Mouse hippocampal nuclear extracts were isolated from adult female mice (P90) using a nuclear and cytoplasmic extraction kit per the manufacturer's instructions (Thermo Fisher Scientific 78833). Shift assays were performed using the LightShift chemilumiescent EMSA Kit (Thermo Fisher Scientific 20148). Reaction products were separated by electrophoresis, and protein::DNA complexes were transferred onto a positively charged nylon membrane (Thermo Fisher Scientific 77016). Protein::DNA complexes were detected using the nucleic acid detection module kit (Thermo Fisher Scientific 89880) per the manufacturer's instructions, and imaged using an Odyssey Fc imaging system (Li-Cor). A two-sided t-test (R [R Core Team 2021] environment) was used to determine significant alterations in binding affinity to methylated and hydroxymethylated oligonucleotides, using three independent EMSA replicates.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE87032.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Sergio Tufik for his endless support, the UW biotechnology center, and Dr. Sisi Li for her assistance with brain dissections. This work was supported in part by the University of Wisconsin-Madison departments of Neurosurgery and Psychiatry, University of Wisconsin Vilas Cycle Professorships #133AAA2989 and University of Wisconsin Graduate School MSN184352 (all to R.S.A.), National Alliance for Research on Schizophrenia and Depression Young Investigator Grant from the Brain & Behavioral Research Foundation 22669 and 25212 (L.A.P.), Ruth L. Kirschstein National Research Service Award MH113351-02 (A.M.), National Institutes of Health grants HG003747 and U54AI117924 (all to S.K.), HG007019 (S.K. and Q.Z.), and the University of Nebraska Lincoln department of Statistics (Q.Z.). Part of Q.Z.'s research was performed when he was a postdoc in the statistical genomics group at UW Madison led by S.K.
Author contributions: Conceptualization was by L.A.P., A.M., and R.S.A. Methodology was by L.A.P., A.M., Q.Z., K.C., L.S., S.K., and R.S.A. Validation was by L.A.P., A.M., L.S., and R.S.A. Software was by A.M., Q.Z., K.C., S.K., and R.S.A. Formal analysis was by L.A.P., A.M., Q.Z., K.C., and R.S.A. Writing of the original draft was by L.A.P., A.M., and R.S.A. Review/editing was by L.A.P., A.M., Q.Z., K.C., L.S., S.K., and R.S.A.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276137.121.
- Received August 23, 2021.
- Accepted December 22, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








