
Genetic exchange with an outcrossing sister species causes severe genome-wide dysregulation in a selfing Caenorhabditis nematode
- 1Department of Biology, Hong Kong Baptist University, Hong Kong, China;
- 2College of Life Sciences, Capital Normal University, Beijing, 100048, China;
- 3Department of Biochemistry, University of Oxford, Oxford, OX1 4BH, United Kingdom;
- 4State Key Laboratory of Environmental and Biological Analysis, Hong Kong Baptist University, Hong Kong, China
Abstract
Different modes of reproduction evolve rapidly, with important consequences for genome composition. Selfing species often occupy a similar niche as their outcrossing sister species with which they are able to mate and produce viable hybrid progeny, raising the question of how they maintain genomic identity. Here, we investigate this issue by using the nematode Caenorhabditis briggsae, which reproduces as a hermaphrodite, and its outcrossing sister species Caenorhabditis nigoni. We hypothesize that selfing species might develop some barriers to prevent gene intrusions through gene regulation. We therefore examined gene regulation in the hybrid F2 embryos resulting from reciprocal backcrosses between F1 hybrid progeny and C. nigoni or C. briggsae. F2 hybrid embryos with ∼75% of their genome derived from C. briggsae (termed as bB2) were inviable, whereas those with ∼75% of their genome derived from C. nigoni (termed as nB2) were viable. Misregulation of transposable elements, coding genes, and small regulatory RNAs was more widespread in the bB2 compared with the nB2 hybrids, which is a plausible explanation for the differential phenotypes between the two hybrids. Our results show that regulation of the C. briggsae genome is strongly affected by genetic exchanges with its outcrossing sister species, C. nigoni, whereas regulation of the C. nigoni genome is more robust on genetic exchange with C. briggsae. The results provide new insights into how selfing species might maintain their identity despite genetic exchanges with closely related outcrossing species.
Eukaryotic species frequently evolve a selfing reproduction mode from their outcrossing ancestral species. It is well known that the reproduction mode of a species plays a crucial role in shaping its genome size (Lynch and Conery 2003). For example, highly selfing plant species have significantly smaller genome sizes than those of their most closely related outcrossing relatives, which appears to be driven by reduced activity and spread of repetitive elements in selfing plants (Brandvain and Haig 2005). Genome size reduction is also observed in selfing animal species. For example, the nematode, Caenorhabditis elegans, a well-established model organism, mostly reproduces through selfing as a hermaphrodite, albeit with a rare chance of outcrossing with males in the wild (Brenner 1974). It has a significantly smaller genome than its most closely related outcrossing species (The C. elegans Sequencing Consortium 1998; Li et al. 2015; Kanzaki et al. 2018; Yoshimura et al. 2019). In addition to C. elegans, other Caenorhabditis species, including Caenorhabditis briggsae and Caenorhabditis tropicalis, have independently evolved hermaphroditism, which has also resulted in a significantly reduced genome size relative to the most-closely related outcrossing species (Kiontke et al. 2004; Ren et al. 2018; Yin et al. 2018; Cutter et al. 2019; Woodruff and Teterina 2020; Noble et al. 2021). These studies provide strong evidence that the genomes of selfing species rapidly reduce their size once speciation starts from its outcrossing ancestral species.
The rapid divergence in genome size between selfing species and outcrossing species descended from a common ancestor is challenging to understand in situations in which the two species have a chance to occupy the same habitat and interbreed. In particular, crossing studies in plants with differing mating systems led to the weak inbreeder/strong outbreeder (WISO) hypothesis, which claims that in the crosses between species with different mating systems, outcrossing parent species will “overpower” selfing parents such that their genomes have a higher “effective ploidy” in hybrid offspring (Brandvain and Haig 2005). Frequent introgressions from outcrossing species into selfing species would therefore be expected to counter the diminution in genome size. The WISO hypothesis was formulated in the context of plant development in which there is a natural asymmetry in the contribution of parental and maternal genomes to the endosperm and the seed. Nevertheless, recent studies suggest that the WISO hypothesis may apply in animals although effective ploidy is irrelevant in animal hybrids for the lack of triploidy as seen in plants. Interspecies mating between Caenorhabditis nematodes sterilizes maternal individuals, and the hermaphrodites are more vulnerable to such detrimental effects than the female of outcrossing species (Ting et al. 2014). A similar situation applies in the case of the androdiecious C. briggsae, which mostly reproduces as a hermaphrodite, and its dioecious sister species, C. nigoni, in which mating is always between male and female. Hybrid progeny between C. nigoni mothers and C. briggsae fathers produces fertile males and females and sterile males, whereas the reciprocal cross produces fertile females only but no viable males (Fig. 1A). In other words, there is an apparent asymmetry in terms of mutually introducing genomic fragments from one species into the genome of the other species; that is, it is straightforward to introduce the genomic fragments from C. briggsae into the genome of C. nigoni, but not feasible in the opposite direction. Furthermore, fertile hybrid F1 females from either crossing direction produce viable hybrid F2 females when mated with C. nigoni males, but no viable progeny when mated with C. briggsae males (Fig. 1C; Woodruff et al. 2010; Bundus et al. 2015), which is consistent with the WISO hypothesis. Notably, many C. nigoni strains carrying an introgression fragment from C. briggsae genome show a compromised fitness relative to C. nigoni wild isolate in the laboratory (Bi et al. 2015, 2019). It is worth noting that the F2 animals are expected to carry ∼75% of the genome of the backcrossing male and ∼25% of the genome from the other species (Fig. 1A). Despite the apparent asymmetry in genetic exchange in backcrossing progeny, the molecular mechanism underlying it is poorly defined. Expression analysis of the F2 hybrid populations presents an excellent opportunity to investigate the consequences of this asymmetry for gene regulation.
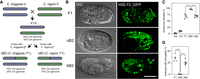
Crossing direction–dependent lethality in the backcross hybrid embryos between Caenorhabditis briggsae and Caenorhabditis nigoni. (A) Schematic diagram illustrating crossing strategy and expected genome composition of the hybrid progeny between C. briggsae (Cbr, blue) and C. nigoni (Cni, green) as indicated, including their hybrid F1 embryos (F1 female), the backcrossing embryos, that is, bB2 and nB2, which are the crossing progeny between the F1 females and C. briggsae or C. nigoni males, respectively. The coloring scheme is used throughout the paper. (B) DIC (left) and fluorescent micrographs (right) showing the typical phenotypes of arrested hybrid embryos. The fluorescent embryo nuclei were ubiquitously labeled by a HIS-72::GFP marker. An unusually large nucleus in the bB2 embryo is indicated by arrowhead. Differentiated head is indicated with dashed line. Scale bar represents 20 µm. Note a more severe phenotype in the bB2 than in the F1 and nB2 embryos. (C) Quantification of the ratio of embryonic lethality. Shown are the data from five replicates with n = 1718, 1709, 1549, 2800, 2636 for Cbr, Cni, F1, bB2, and nB2, respectively. The error bar represents the standard error of mean. (***) Adjusted P < 0.001 (one-way ANOVA followed by multiple pairwise t-test, FDR adjusted). (D) Percentage of dead embryos with elongation. Shown are the data from five replicates with n = 483, 820, 521 for F1, bB2, and nB2, respectively. The error bar represents the standard error of mean. (****) Adjusted P < 0.0001 (one-way ANOVA followed by multiple pairwise t-test, FDR adjusted). Note that nearly all bB2 embryos arrested before the elongation stage without obvious morphogenesis.
It is well established that transposable elements (TEs) are involved in both pre- and postzygotic isolation (Serrato-Capuchina and Matute 2018). For example, a wide range of TEs have been shown to be reactivated in the lethal hybrid males of two Drosophila species, Drosophila melanogaster and Drosophila simulans, but not in their viable intraspecies hybrids (Kelleher et al. 2012). Further studies have identified an association between the mutation of two extensively studied hybrid incompatibility (HI) genes, Hmr and Lhr, and the dysregulation of TEs possibly because of their malfunctioning in heterochromatin binding (Satyaki et al. 2014). More TE dysregulation was observed in hybrid F2 and subsequent crossing progeny. For instance, an increase in the number of dysregulated TEs was identified in the backcrossing hybrids of two lake whitefish lineages compared with the parents or relatively healthy F1 hybrids (Dion-Côté et al. 2014). In addition to TEs, the extensive dysregulation of protein-coding genes (referred to as coding genes hereafter) has been frequently observed in hybrids (Wei et al. 2014; Lu et al. 2018; Moran et al. 2021). Notably, our previous study of hybrid sterile males between C. briggsae and C. nigoni, resulting from extensive backcrossing with C. nigoni, also revealed the dysregulation of spermatogenesis genes (Li et al. 2016b).
Insight into the molecular mechanisms underlying gene expression dysregulation in hybrids may come from the fact that dysregulation of small regulatory RNAs is also commonly observed (Kelleher et al. 2012; Kotov et al. 2019). For example, in Drosophila hybrids, regulatory changes in PIWI-interacting RNAs (piRNAs) are frequently associated with the failure of maternally distributed piRNAs to repress the expression of paternally inherited TEs (Pilar and Guerreiro 2014; Kotov et al. 2019). Likewise, rampant gene expression divergence has been linked to the dysregulation of microRNAs (miRNAs) or piRNAs in rapeseed and tomato (Shivaprasad et al. 2012; Shen et al. 2017). In nematodes, the most abundant class of small regulatory RNAs linked to TE and gene regulation are known as 22G small RNAs (22G RNAs). 22G RNAs are made by RNA-dependent RNA polymerases, and target RNAs are used as templates to produce 22G RNAs that are antisense. 22G-RNA formation may be initiated by target recognition by other classes of small RNAs, in particular piRNAs or 26G RNAs, but once synthesis begins it can be maintained independently of these small RNAs, even over multiple generations (Almeida et al. 2019). We previously showed that the up-regulation of a specific group of 22G RNAs was associated with the down-regulation of spermatogenesis genes in hybrid sterile C. nigoni males carrying an introgression fragment from the C. briggsae genome (Li et al. 2016b), supporting a role of small RNAs in mediating hybrid sterility. Here, we investigated the mechanism of asymmetric genetic exchange between two nematodes with differential reproduction modes by sequencing the transcriptomes and small RNAs of the parents, that is, C. briggsae and C. nigoni, and their hybrid F1 and F2 progeny. We focused on contrasting the expression between two types of F2 hybrids resulting from reciprocal backcrossing, which carry an expected 75% and 25% of the genomes from either parent (Fig. 1A). The results of this study are expected to provide explanations to the observed asymmetry in gene regulation between selfing C. briggsae and outcrossing C. nigoni on genetic exchange between each other.
Results
Hybrid F2 embryos carrying 75% of the C. briggsae genome arrested with more severe defects than those carrying 75% of the C. nigoni genome
A previous study showed the crossing direction–dependent hybrid F2 breakdown between C. briggsae and C. nigoni (Woodruff et al. 2010), that is, all F2 progeny resulting from backcrossing with C. briggsae arrested as embryos, whereas the F2 progeny resulting from backcrossing with C. nigoni were partially viable (Fig. 1B,C). To determine whether the arrested embryos show comparable phenotypes between the two F2 populations, we first determined the embryonic lethality for both parents, that is, C. briggsae (AF16) and C. nigoni (JU1421), in their F1 hybrids fathered by C. briggsae (F1), and in their backcrossing F2 hybrids in both directions, which we designated as bB2 (C. briggsae father and F1 mother) and nB2 (C. nigoni father and F1 mother) (Fig. 1A). Consistent with previously reported results (Woodruff et al. 2010; Kozlowska et al. 2012; Bi et al. 2015), we observed almost 100% embryonic lethality in the bB2 embryos, which was significantly higher than that in the nB2 and F1 embryos (One-way ANOVA, P < 0.0001, multiple pairwise t-test, FDR adjusted P < 0.0001) (Fig. 1C). We observed comparable lethality rates between the F1 and nB2 embryos, both of which showed a significantly higher embryonic lethality rate (∼70%, multiple pairwise t-test, FDR adjusted P < 0.0001) than the parental species. In addition, we observed a low rate of embryonic lethality (1%–3%) in wild-type C. briggsae embryos, and a significantly elevated lethality rate (8%–10%) in the wild-type C. nigoni embryos (multiple pairwise t-test, adjusted P < 0.001). The elevated embryonic lethality in C. nigoni could be caused by inbreeding of the isolate in the laboratory, leading to homozygosity of some lethal alleles.
To further evaluate whether F1, bB2, and nB2 hybrid embryos arrested at similar or different stages, we repeated the above crossings using a transgenic C. briggsae strain that ubiquitously expresses a histone-green fluorescent protein (GFP) fusion (HIS-72::GFP) in the nuclei (Zhao et al. 2010). The dominant GFP marker allowed the direct visualization of all nuclei in arrested embryos. We found that ∼35% of the F1 (169 out of 483) and 25% of the nB2 (130 out of 521) embryos were able to develop up to the elongation stage, respectively, as judged by the presence of a pharynx and nucleus positioning (Fig. 1B,D). The remaining dead embryos arrested at stages ranging from as early as approximately the 150-cell stage to the bean stage. In contrast, we barely observed any bB2 embryos that were able to develop to the elongation stage, usually without obvious gastrulation (Fig. 1D, One-way ANOVA, P < 0.0001, multiple pairwise t-test, adjusted P < 0.0001), suggesting more severe dysregulation of gene expression in bB2 embryos than in other hybrid embryos. In addition, unusually large nuclei, which are characteristic of failures in cell nucleus division (e.g., chromosome segregation failure) or gastrulation (e.g., mislocalized intestine precursors), were observed frequently only in bB2 embryos (Fig. 1B). In summary, we observed more severe phenotypes in arrested bB2 embryos than in hybrid F1 and nB2 arrested embryos in addition to their differential lethality rate. We reasoned that the phenotypic differences between bB2 and nB2 embryos may be explained, at least in part, by their different genomic compositions and subsequent difference in their transcriptome. We tested this hypothesis by examining the expression profiles of coding genes, TEs, and small RNAs by RNA sequencing (RNA-seq) analyses of the staged hybrid embryos and wild-type embryos (Supplemental Fig. S1; Supplemental Table S1).
TEs show a disproportionately higher level of derepression in bB2 embryos than in nB2 embryos
Derepression of TEs is frequently observed in the plant and animal hybrids (Kelleher et al. 2012; Laporte et al. 2019). We performed RNA-seq analysis of poly(A)-tailed RNAs to quantify the abundances of TE transcripts in the embryos of C. briggsae (Cbr) and C. nigoni (Cni), their F1 hybrids fathered by C. briggsae, and their F2 hybrids fathered by C. briggsae (bB2) and C. nigoni (nB2) (Supplemental Figs. S1, S2). We generated approximately 165, 174, 182, 145, and 188 million reads for the embryos of C. briggsae, C. nigoni, their F1 hybrids, and bB2 and nB2 hybrids, respectively, with three replicates each (Supplemental Table S1). To evaluate the quality of our sequencing results, we performed principal components analysis of all sequencing reads mapped to the consensus sequences of TEs or to the one-to-one orthologous coding genes between C. briggsae and C. nigoni in each replicate (see Methods). The results showed a clear separation between different samples with replicates from the same sample clustering together (Supplemental Fig. S2A,B). To further evaluate the reproducibility of our sequencing data, we generated a heatmap to contrast the abundance of reads mapped to TEs and the one-to-one orthologs using row Z-score normalization as previously described (Li et al. 2016b). The replicates of the same sample clustered together (Supplemental Fig. S2C,D), demonstrating a reasonable quality of our sequencing data.
We compared TE expression between F1 or F2 hybrids and their parent C. briggsae or C. nigoni species. We observed that the TE expression levels in the F1 embryos were much more similar to that in the C. nigoni embryos than in the C. briggsae embryos (Supplemental Fig. S3). The biased similarity may reflect a maternal effect because C. nigoni served as mother in producing the F1 hybrids. In F2 hybrids, the TE expression levels in the bB2 embryos showed comparable similarity to that in both C. briggsae and C. nigoni embryos, with Pearson's correlation coefficients of 0.813 (95% confidence interval: 0.681–0.939) and 0.794 (95% confidence interval: 0.713–0.878), respectively (Fig. 2A). This is unexpected because bB2 embryos carried about 75% of the C. briggsae genome but only 25% of the C. nigoni genome. In contrast, as expected, the TE expression level in the nB2 embryos was highly similar to that in C. nigoni but not C. briggsae, with Pearson's correlation coefficients of 0.953 (95% confidence interval: 0.931–0.977) and 0.558 (95% confidence interval: 0.371–0.743), respectively (Fig. 2B). This is because the nB2 embryos carried about 75% of the C. nigoni genome but only 25% of the C. briggsae genome. The unexpected correlation in the TE expression between bB2 and C. nigoni suggested an elevated TE dysregulation in the bB2 but not in the nB2 embryos.
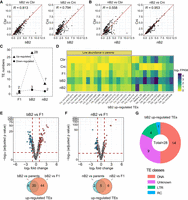
TEs show a disproportionately higher level of misregulation in the bB2 embryos than in the nB2 embryos. (A,B) Pairwise comparison of normalized counts of read mapped to individual TE families (abbreviated as TEs, see Methods) between bB2 (A) or nB2 embryos (B) and parental embryos. Each dot represents a single TE family. Also shown is Pearson's correlation coefficient (R). (C) Number of significantly up- (triangle) and down-regulated TE families (circle) in F1, nB2, and bB2 embryos as indicated. (D) A heatmap showing the normalized count of the reads mapped to the 28 TEs that are significantly up-regulated in the bB2 embryos in the parental and hybrid embryos. TEs are divided into four categories (see Methods). Scale of expression intensity is shown on the right. (E,F) Top: Volcano plots of log2 fold changes (FC) of TE expression in the bB2 (E) or nB2 (F) embryos compared with that in the F1 embryos. The significantly up-, down-regulated TEs, or those without significant change TEs, are differentially colored in red, blue, and gray, respectively. Bottom: Venn diagrams show the number of significantly up-regulated TE numbers in bB2 or nB2 embryos compared with parental or F1 embryos. (G) Pie chart showing the breakdown of the 28 significantly up-regulated TEs in the bB2 hybrid at class level.
To determine whether specific TEs were significantly dysregulated in bB2 embryos, we first examined all TE families that produced detectable RNA transcripts (referred to as expressed TEs). We identified a total of 267 and 281 expressed TE families in C. briggsae and C. nigoni, respectively (Supplemental Fig. S4A). The comparable number of expressed TEs was surprising because the repeat contents in the two species diverged substantially from each other (Ren et al. 2018; Yin et al. 2018). We detected a higher number of expressed TEs in all types of hybrid embryos relative to the number in both parents. For example, we observed 385, 347, and 362 expressed TEs in bB2, nB2, and F1 embryos, respectively (Supplemental Fig. S4A; Supplemental Table S2), indicating both C. briggsae- and C. nigoni-originated TEs were expressed in the hybrids. The slightly elevated number of expressed TEs in bB2 hybrid relative to other types of hybrids was mainly attributed to the Class II (DNA) TEs. We next examined all TE families that underwent significant derepression/up-regulation, defined as those with at least a twofold increase in expression level and false discovery rate (FDR) corrected P value < 0.05 relative to the TE expression level in both parents (see Methods). We observed significant derepression of TE families in all three types of hybrids, but the bB2 embryos showed a significantly higher number of derepressed TEs, that is, 28, relative to seven and two in the nB2 and F1 embryos, respectively (Fisher's exact test, P < 0.0001), despite the similar number of down-regulated TE families among the three types of hybrid embryos (Fisher's exact test, P = 0.876) (Fig. 2C,D; Supplemental Table S3). Of the 28 derepressed TE families in bB2 embryos, 17 showed low expression levels in both parents (FPKM less than 1), indicating that these TEs were silenced in C. briggsae or C. nigoni but derepressed in the bB2 embryos (Fig. 2D). Finally, we contrasted the expression of TE families between F1 hybrid embryos and bB2 or nB2 embryos. Again, the number of up-regulated TE families in the bB2 embryos was significantly higher than that in the nB2 embryos (Fig. 2E,F). Specifically, 64 TE families were significantly up-regulated in the bB2 embryos, of which 20 were a subset in the 28 TE families mentioned above (Fig. 2E). In contrast, only 11 TE families were significantly up-regulated in the nB2 embryos relative to the F1 embryos (Fig. 2F). These results support the notion that bB2 embryos were more vulnerable to TE derepression than nB2 embryos, which is consistent with the WISO hypothesis.
To gain an initial idea of the function of the significantly derepressed TE families in the bB2 embryos, we grouped the 28 TE families into different TE classes. The results showed that half of the 28 TE families were Class II (DNA) TEs, nine were classified as Unknown, four were long terminal repeats (LTRs) and one was classified as Rolling Circle (RC) (Fig. 2G). It is worth noting that the C. briggsae parents expressed a significantly smaller number of unknown types of TEs compared with the number expressed by C. nigoni (Supplemental Fig. S4A), which is consistent with its lower repeat contents relative to C. nigoni. However, TE expression did not show a significant change at the class level in the bB2 embryos relative to other classes of embryos (Supplemental Fig. S4B), suggesting that differential expression took place mainly at the TE family level.
A markedly higher number of coding genes with transgressive expression are observed in the bB2 embryos than in other types of hybrid embryos
We speculated that the differential phenotypes of the bB2 and nB2 embryos may be associated with differential dysregulation of one-to-one orthologous protein-coding genes between the two hybrid embryos (Supplemental Table S4). To test this, we performed a pairwise comparison of normalized read counts between parental and hybrid embryos. Because the genomic contents of the bB2 and nB2 embryos were expected to be dominated by the C. briggsae and C. nigoni genome, respectively, and the similarity in the expression of one-to-one orthologs was modest between C. briggsae and C. nigoni (R = 0.866, Supplemental Fig. S5A), we expected that the expression levels of orthologs in bB2 and nB2 embryos would show a biased correlation with their expression levels in C. briggsae and C. nigoni embryos, respectively. However, this was the case only for nB2 but not bB2 embryos (Fig. 3A,B). Specifically, the Pearson's correlation coefficient of the expression levels between bB2 and C. briggsae embryos was comparable to that between nB2 and C. briggsae embryos (0.914 [95% confidence interval: 0.909–0.918] vs. 0.913 [95% confidence interval: 0.908–0.918]), whereas the correlation coefficient of expression levels between nB2 and C. nigoni embryos was significantly higher than that between bB2 and C. nigoni embryos (0.960 [95% confidence interval: 0.957–0.963] vs. 0.902 [95% confidence interval: 0.897–0.907]). These results suggested that bB2 embryos suffered from a higher degree of dysregulation of one-to-one orthologs than nB2 embryos, supporting the notion that the C. briggsae gene regulation is more vulnerable to genetic perturbation than the C. nigoni genome. A higher degree of similarity in the expression levels of one-to-one orthologs between F1 and C. nigoni or nB2 embryos than between F1 and C. briggsae or bB2 embryos was expected at least partially caused by maternal effect (Supplemental Fig. S5B–E).
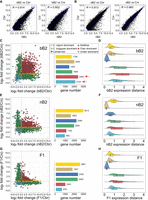
bB2 embryos show a higher degree of transgressivity in expression inheritance profile of coding genes relative to other types of hybrid embryos. (A,B) Pairwise comparisons of the normalized expression levels (read counts) of the one-to-one orthologs between bB2 (A) or nB2 (B) and parental embryos. Also shown is Pearson's correlation coefficient (R). Linear regression lines are shown in blue. (C,E,G) Scatter (left) and bar plots (right) showing the expression level change of the orthologs in the bB2 (C), nB2 (E), or F1 (G) embryos compared with that of each parental species, that is, Caenorhabditis briggsae (x-axis) and Caenorhabditis nigoni (y-axis). Inheritance categories are differentially color coded as indicated. Red dashed lines indicate the “0” of log2 fold change for each axis. The total gene number of each inheritance category in the three hybrids is shown. The numbers of over- and under-dominant are highlighted with arrows. (D,F,H) Density plots with overlaid box plots showing the expression distance (see Methods) of one-to-one orthologs within each inheritance category for bB2 (D), nB2 (F), and F1 (H) embryos.
To identify the sources of the differential correlations between bB2 or nB2 hybrid embryos and parental embryos, we examined the inheritance profiles of each individual orthologous gene pair by comparing their expression levels in each type of hybrid embryos with those in both parental species. Based on the inheritance pattern of gene expression, the orthologs were grouped into the following categories as previously described (McManus et al. 2010; Sánchez-Ramírez et al. 2021): conserved (expression level comparable with that in both parents), additive (expression level higher than that in one parent, but lower than that in the other parent), species-specific dominant (C. briggsae- or C. nigoni-dominant, defined as an expression level comparable with one parent but significantly deviating from the level in the other parent), and transgressive (over- or under-dominant, defined as an expression level simultaneously deviating from the level in both parents in the same direction) (see Methods). We observed the highest number of transgressively expressed genes in bB2 embryos, with 2452 (21%) and 2134 (18%) classified as over-dominant and under-dominant, respectively (Fig. 3C). In contrast, we only observed 1162 (10.5%) and 1043 (9.4%) genes classified as over-dominant, and 993 (8.9%) and 678 (6.1%) as under-dominant, in the nB2 and F1 embryos, respectively (Fig. 3E,G; Supplemental Tables S5–S7). However, we observed a reduction in the number of conserved genes in the bB2 embryos. For example, bB2 embryos showed a total of 1483 (12.7%) conserved genes, whereas nB2 and F1 embryos showed 2269 (20.4%) and 2442 (20.1%) conserved genes (Fig. 3C,E,G), respectively, which is consistent with their arrested phenotypes (Fig. 1C,D). A recent study showed that, in F1 hybrid adults, additive and transgressive expressed genes maintained a larger magnitude of expression distances than genes of other inheritance types (Sánchez-Ramírez et al. 2021). We showed that bB2 embryos demonstrated the longest distances for over- and under-dominant genes, whereas the distances in nB2 and F1 embryos were comparable (Fig. 3D,F,H). Taken together, these results showed that the bB2 embryos suffered from more severe dysregulation in coding genes than nB2 embryos.
A greater number of significantly dysregulated coding genes are observed in bB2 embryos than nB2 embryos
To examine the number and function of genes with significant dysregulation of expression levels in hybrids compared with the expression levels in both parents, we identified all orthologs that were significantly up- or down-regulated in F1, bB2, and nB2 hybrid embryos relative to the parental embryos. We defined a significantly dysregulated gene as one in which the transcript abundance was significantly higher or lower than the abundance in both parents, with at least a twofold change (FDR corrected P <0.05) (see Methods). We observed that bB2 embryos showed a significantly higher number of dysregulated genes than the other types of hybrid embryos, with about fourfold more up-regulated genes (χ2 test, P < 0.0001) and about ninefold more down-regulated genes in bB2 relative to nB2 (χ2 test, P < 0.0001) (Fig. 4A; Supplemental Tables S5–S7). To further compare the expression intensities of dysregulated genes, we generated a heatmap using the normalized transcript abundances of all misregulated genes in the parental species and the three types of hybrid embryos. The results showed a markedly higher level of gene dysregulation in bB2 embryos compared with that in nB2 and F1 embryos (Fig. 4B), consistent with the WISO hypothesis.
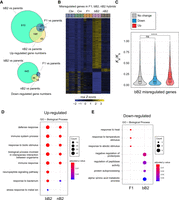
Characterization of the significantly misregulated coding genes in the F1, bB2, and nB2 embryos. (A) Venn diagrams showing the number of up- (top) and down-regulated genes (bottom) in the bB2, F1, or nB2 embryos compared with both parental embryos. (B) Heatmap showing the expression intensity of all the misregulated genes in the parental, F1, bB2, and nB2 embryos. Note the difference between the bB2 and the remaining categories of embryos. Row Z-score scale bar is shown at the bottom. (C) Violin plots with overlaid box plots showing the Ka/Ks values of the genes that are up-regulated, down-regulated, and those not showing a significant change in the bB2 embryos. (****) P < 0.0001; (ns) not significant compared with those without significant change (Wilcoxon ranked-sum test). (D,E) Gene Ontology (GO) analysis for biological processes of up- (D) and down-regulated genes (E) in each category of hybrid embryos as indicated.
Given that genes involved in hybrid incompatibility usually evolve faster than the highly conserved essential genes (Maheshwari and Barbash 2011), we examined whether the dysregulated genes evolved faster than those that were not dysregulated. To this end, we calculated the molecular evolution ratio (Ka/Ks) of all one-to-one orthologs between C. briggsae and C. nigoni. We found that the Ka/Ks values were significantly higher for the up-regulated genes than those without significant dysregulation in bB2, F1, and nB2 embryos (P < 0.0001, Wilcoxon rank-sum test, Fig. 4C; Supplemental Fig. S6A,B; Supplemental Table S8), whereas the Ka/Ks values of the down-regulated genes were significantly higher only in F1 and nB2 embryos but not in bB2 embryos (Supplemental Fig. S6A,B). These results suggest that the up-regulated genes in hybrid embryos tend to undergo faster evolution, which may have implications for the development of hybrid compatibility. It remains unclear why only the down-regulated genes in bB2 embryos showed no significant change in evolution speed relative to all genes without dysregulation.
To gain an initial idea of which biological pathways were vulnerable to perturbation in the hybrid background, we performed Gene Ontology analysis for all genes that showed significant up- or down-regulation in all types of hybrid embryos relative to parental embryos. We found that most of the enriched pathways from the up-regulated genes were common to the bB2 and nB2 embryos, including those involved in the defense and immune responses. bB2 embryos also showed unique pathways, including those involved in neuropeptide signaling and the stress response to metal ion (Fig. 4D). These results suggest that up-regulation of these genes may contribute to the unique arrest phenotypes in the bB2 embryos. Notably, no biological process was enriched in the up-regulated genes in the F1 embryos. The down-regulated genes were not shared between the three categories of hybrid embryos. For example, those showing significant down-regulation in bB2 embryos were mainly enriched in the processes including protein degradation and amino acid metabolism, whereas those significantly down-regulated in the F1 embryos were mostly enriched in the processes including heat shock responses. It is worth noting that no biological process was significantly enriched for down-regulated genes in the nB2 embryos (Fig. 4E). These results suggest that down-regulated genes in the hybrid background are highly dependent on the genetic makeup.
Given that both bB2 and nB2 embryos show embryonic lethality, we sought to determine whether essential genes were significantly enriched in the dysregulated genes of hybrid embryos. We, therefore, performed enrichment analysis of the up- and down-regulated genes in terms of essential genes in the F1, bB2, and nB2 embryos, as previously described (Supplemental Fig. S7; Li et al. 2016b). These results show that, except for the down-regulated genes in F1 hybrids, both the up- and down-regulated genes in all categories of hybrid embryos were significantly depleted for essential genes (Supplemental Fig. S7D). The results showed that the essential genes were less susceptible to dysregulation than other genes in the hybrids.
piRNAs and 22G small RNAs are significantly up-regulated in bB2 embryos compared with nB2 embryos
We previously reported that increased levels of 22G RNAs in sterile C. nigoni strains carrying an introgression fragment from the C. briggsae X Chromosome is associated with the down-regulation of spermatogenesis genes (Li et al. 2016b), demonstrating that 22G RNAs play an important role in the regulation of TEs and coding genes in the hybrid background. 22G RNAs can be generated from many different primary small RNA triggers, including both piRNA-dependent and piRNA-independent mechanisms. They mediate the silencing of protein-coding genes, transposons, pseudogenes, and cryptic loci through both transcriptional and post-transcriptional mechanisms (Gu et al. 2009; de Albuquerque et al. 2015). To examine whether small (18–30 nt) regulatory RNAs, including 22G RNAs, were differentially dysregulated between bB2 and nB2 embryos, we performed small RNA sequencing after 5′ polyphosphatase treatment of the extracted RNAs from each of the parental and hybrid embryos as described previously (Supplemental Fig. S1; Supplemental Table S1; Li et al. 2016b). To evaluate the quality of our sequencing data, we calculated the proportion of small RNA reads ranging from 18 to 30 nucleotides in length with regard to their first nucleotide at the 5′ end. We observed a large proportion of reads that were 22 nucleotides long and bore a guanine at their 5′ end, which was indicative of the successful retrieval of this specific type of small RNAs (Supplemental Fig. S8; Pak and Fire 2007; Pak et al. 2012).
We compared the abundance of piRNAs between the bB2 and nB2 embryos. We found that both C. briggsae- and C. nigoni-originating piRNAs were significantly up-regulated in the bB2 embryos compared with the nB2 embryos (P < 2.2 × 10−16 for both categories of RNAs, Wilcoxon ranked-sum test; Fig. 5A,B; Supplemental Tables S9, S10), suggesting a role of piRNAs in mediating the differential arresting phenotypes of the bB2 embryos. We next compared the expression abundances of 22G RNAs targeting TEs between bB2 and nB2 embryos. We found that the expression levels of TE-targeting 22G RNAs, including those that were dysregulated in the bB2 embryos, were highly similar between bB2 and nB2 embryos (Supplemental Fig. S9A,B; Supplemental Table S11). We finally examined whether 22G RNAs that target coding genes showed a biased expression profile toward their parent-of-origin. We found that 22G RNAs in both types of hybrid embryos barely showed any biased correlation with either of their parents (Supplemental Fig. S9C,D; Supplemental Table S12). This may be because the majority of 22G RNAs were maternally deposited in the embryos, even though paternal contribution of 22G RNAs in C. elegans was also reported (Stoeckius et al. 2014).
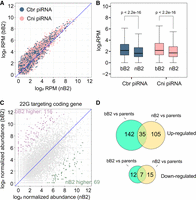
The abundances of 21U RNAs and 22G RNAs targeting coding genes are significantly higher in the bB2 embryos than in the nB2 embryos. (A) Scatter plot showing pairwise comparison of normalized read counts of the Caenorhabditis briggsae- (blue) or Caenorhabditis nigoni-specific 21U RNAs (pink) between bB2 and nB2 embryos. Note that the overall abundances of 21U RNAs are apparently higher in the bB2 than in the nB2 regardless of their parent-of-origin. (B) Box plots showing the normalized read counts of C. briggsae- (blue) or C. nigoni-specific 21U RNAs (pink) in the bB2 and nB2 embryos. P values of pairwise comparison with Wilcoxon ranked-sum test is indicated. (C) Scatter plot showing the pairwise comparison of normalized read count of 22G RNAs targeting orthologous coding genes between the bB2 and nB2 embryos. The numbers of genes with a significantly higher abundance of targeting 22G RNAs in the bB2 relative to the nB2 embryos (purple) or vice versa (green) are indicated. (D) Venn diagram showing the number of coding genes targeted by the 22G RNAs that were significantly up- (top) and down-regulated (bottom) in the bB2 or nB2 embryos relative to both parents.
To determine whether there were any dysregulated 22G RNAs in the hybrid embryos, we directly contrasted the abundance of 22G RNAs targeting coding genes between bB2 and nB2 embryos. We observed 116 genes that were targeted by a significantly higher abundance of 22G RNAs in the bB2 embryos relative to the nB2 embryos, compared with 69 in the nB2 embryos (χ2 test, P < 0.001) (Fig. 5C). To further determine to what extent the 22G RNAs were dysregulated relative to both parents, we performed differential gene expression analysis of the 22G RNAs relative to both parents, similar to the analysis of coding genes. We found 177 genes were targeted by a significantly higher abundance of 22G RNAs in the bB2 embryos relative to both parents, compared with 140 in nB2 embryos (χ2 test, P < 0.05). However, the number of genes targeted by the significantly down-regulated 22G RNAs was slightly lower in bB2 than in nB2 embryos (Fig. 5D). Most of the coding genes targeted by the dysregulated 22G RNAs were not shared between nB2 and bB2 (Fig. 5D). Taken together, these results suggest a greater role of 22G RNAs in the dysregulation of their corresponding coding genes in bB2 embryos than in nB2 embryos.
Potential dysregulation of microRNAs in bB2 embryos
In addition to piRNAs and 22G RNAs, miRNAs are another category of small RNAs that frequently show differential expression in hybrids (Li et al. 2016a,b). We previously reported a significantly up-regulated miRNA, miR-237, in sterile males of a C. nigoni strain carrying a C. briggsae introgression fragment, which may affect spermatogenesis in the hybrid males (Li et al. 2016b). We sought to determine whether miRNAs were also dysregulated in the bB2 embryos relative to the nB2 embryos. To this end, we compared the number and expression intensity of dysregulated miRNAs between the two categories of hybrid embryos. We observed an increased number of up-regulated miRNAs in bB2 embryos compared with nB2 embryos. Specifically, we identified nine significantly up-regulated miRNAs in the bB2 embryos but only two in the nB2 embryos relative to the parental species. Notably, the two miRNAs that were significantly up-regulated in the nB2 embryos were also significantly up-regulated in the bB2 embryos (Fig. 6A). Similarly, we observed a total of three significantly down-regulated miRNAs in the bB2 embryos but only one in the nB2 embryos relative to both parental species. No miRNA was uniquely dysregulated in the nB2 embryos (Fig. 6A). In direct comparison between nB2 and bB2, we found seven and 10 miRNAs were up-regulated and down-regulated in bB2 relative to nB2, respectively (Fig. 6B). Three out of the seven miRNAs specifically up-regulated in the bB2 embryos, that is, Cbr-miR-77, Cbr-miR-239b, and Cbr-miR-786, also showed a significant increase in their abundance relative to that of both parents. Two out of the 10 miRNAs specifically down-regulated in the bB2 embryos, Cbr-miR-244 and Cbr-miR-245, also showed a significant decrease in their abundance in the bB2 embryos relative to both parents (Fig. 6B; Supplemental Table S13). Although the numbers of altered miRNAs are too small to conclude that this is a significant difference, this is indicative that dysregulation in miRNAs may be important in the phenotypic difference between the two.
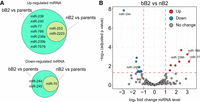
Number of up-regulated microRNAs is higher in the bB2 embryos than in the nB2 embryos. (A) Venn diagram showing the numbers of significantly up- (top) and down-regulated (bottom) miRNAs in bB2 or nB2 embryos relative to both parental embryos. (B) Volcano plot showing pairwise comparison of the abundances of miRNAs between the bB2 and nB2 hybrid embryos. Shown is log2 fold changes (FC) of miRNA expression in bB2 relative to nB2 embryos. miRNAs with significant up- or down-regulation, or no significant change relative to both parents, are highlighted in red, blue, and gray, respectively. Names of the miRNAs that are significantly up- or down-regulated specifically in the bB2 embryos compared with both parents are indicated.
Discussion
Caenorhabditis species have independently evolved androdioecy (hermaphrodites and males) from dioecy (females and males) multiple times, such that hermaphrodites are able to fertilize themselves in addition to being fertilized by their conspecific males (Brenner 1974; Kiontke et al. 2004, 2011). This change in reproductive mode is associated with a substantial reduction for genome size and an increase in the homozygosity of hermaphroditic species (Kanzaki et al. 2018; Ren et al. 2018; Yin et al. 2018). These two types of changes are likely to be coupled, because in hermaphroditic species, parental conflicts are expected to be reduced, and DNA elements that are necessary for dealing with incoming DNA sequences, including foreign repetitive elements such as TEs, are less likely to be under purifying selection and so may be subject to degeneration and ultimate loss. This takes place on top of the loss of coding genes that tend to be male-specific (Rödelsperger et al. 2018). Consistent with this, our data and data from previous studies show that numerous repetitive DNA sequences have been lost from the genome of the selfing species C. briggsae compared with the genome of the outcrossing species C. nigoni (Ren et al. 2018; Yin et al. 2018). Crossing and simulation data in C. elegans suggest that segregation distortion of chromosomes of different size may lead to genome size reduction in hermaphroditic species (Wang et al. 2010a), and such a trend appears to be the ancestral state in the Caenorhabditis lineage (Le et al. 2017). Given the significant size difference between C. briggsae and C. nigoni homologous chromosomes, it would be interesting to test whether such distortion is upheld in interspecies hybrids.
Here, we provide evidence that genome-wide gene expression regulation in the selfing species, C. briggsae, is strongly affected by introgressions from its outcrossing relative, C. nigoni, whereas the genome of outcrossing C. nigoni appears to be more robust at taking DNA elements from C. briggsae. These results are consistent with the WISO hypothesis—the selfing species is “overpowered” by introgressions, whereas the outcrossing species is less strongly affected. Our analysis of the expression of protein-coding genes, transposable elements, and small regulatory RNAs offer a potential explanation for the vulnerability of the C. briggsae genome. This is because we show a significantly greater genome-wide dysregulation in the bB2 hybrids, which contains ∼75% of the C. briggsae genome and 25% of the C. nigoni genome, compared with the nB2 hybrids with ∼75% and 25% of the C. nigoni and C. briggsae genomes, respectively. It is plausible that the elevated dysregulation is responsible for the full-penetrance of lethality in the bB2 hybrid embryos, which essentially prevents the subsequent crossing. We speculate that there might be some key regulatory elements in the C. briggsae outcrossing ancestor, present in either a heterozygous or homozygous form, that are essential for dealing with foreign DNA elements, such as repetitive TEs, and these regulatory elements were lost from the C. briggsae genome during the transition to hermaphroditism. This proposed hypothesis helps explain why there are no viable F2 progeny resulting from backcrossing with C. briggsae, although the F2 progeny from backcrossing with C. nigoni are viable. The loss of these elements made bB2 embryos incompetent to deal with DNA elements from their outcrossing sisters. This leads to the reinforcement of reproductive isolation between the two species, which may explain how the stability of the C. briggsae genome is able to be maintained, even when C. briggsae likely occupies a similar ecological niche as its sister species (Félix et al. 2014; Schulenburg and Félix 2017). Consequently, the nearly isogenic C. briggsae genome becomes a relatively closed system with a smaller genome, adopting a unique evolutionary trajectory that is independent of its outcrossing sister species once hermaphroditism is achieved. Multiple such key elements may evolve after the initial element to further reinforce species boundaries. The identification of such regulatory elements remains the central task for understanding how the stability of the isogenic genome is maintained. We speculate that a similar mechanism is used in the development of hermaphroditism in other species, including C. elegans and C. tropicalis. Taken together, our results show that regulation of the C. briggsae genome is vulnerable to genetic exchange with its outcrossing sister species, C. nigoni, whereas regulation of the C. nigoni genome is not so strongly affected. These results are consistent with the WISO hypothesis, which has previously been supported by evidence mostly from studies in plants (Brandvain and Haig 2005).
An important caveat to this interpretation is the application of Haldane's rule to our results. Haldane's rule states that the heterogametic sex is more likely to be sterile than the homogametic sex (Schilthuizen et al. 2011). This is consistent with our observations as judged in both F1 hybrid progeny and those in the subsequent generations of hybrids between C. briggsae and C. nigoni. It is possible therefore that the observed bias in gene dysregulation could be partially caused by a distorted ratio between the male and female embryos, which carry one and two X Chromosomes, respectively. However, it seems very unlikely that Haldane's rule could be fully responsible for the observed asymmetry in gene regulation. There must be some transcriptional dysregulation that is caused by the introgression rather than solely the result of distortion of the sex ratio. It is also worth noting that our hybrid F1 females were derived from the cross between C. briggsae males and C. nigoni females only. It would be interesting to repeat the experiments in the future using the hybrid F1 females derived from the cross between C. briggsae hermaphrodites and C. nigoni males, and contrast the results to those of this study.
A thorough understanding of hybrid incompatibility relies on establishing the molecular identity of the hybrid incompatible loci we mapped previously through systematic introgression (Bi et al. 2015). By crossing these introgression-bearing C. nigoni strains with C. briggsae (which is expected to lead to the homozygosity of the introgression fragment in the C. nigoni background), we showed that the homozygosity of multiple genomic fragments from C. briggsae chromosomes produced complete lethality in the hybrids (Bi et al. 2019). This result should provide a nice entry point for mapping and cloning the loci responsible for the lethality, owing to the simplicity of the phenotypes. For example, the hybrid F1 progeny between the two wild-type species only show incomplete inviability, whereas the hybrid F1 progeny between C. briggsae and a C. nigoni strain carrying a marked C. briggsae genomic fragment led to complete inviability. The difference between these two phenotypes can be readily scored during mapping. Furthermore, the complete lethality suggests that the homozygosity of the C. briggsae fragments led to the loss of the homologous regions from the C. nigoni genome, and that these regions are essential for the viability of the bB2 embryos. Mapping and molecular cloning of the loci responsible for this lethality should identify the key regulatory DNA elements responsible for maintaining the genome stability of the selfing species, and thus the species boundary between the two.
Methods
Worm maintenance, egg preparation, and microscopy
All worm strains were maintained on NGM plates at 25°C with pre-seeded OP50 Escherichia coli. Wild-type C. briggsae (AF16) and C. nigoni (JU1421) were used as the starting strains for all the RNA-seq analyses. F1 worms were obtained by crossing C. briggsae males with virgin C. nigoni females. F1 progeny from such crosses were used as the mothers to produce the two backcrossing hybrids, bB2 (progeny from the crossing of the F1 females and C. briggsae males) and nB2 (progeny from the crossing of the F1 females and C. nigoni males) (Fig. 1A; Supplemental Fig. S1). To collect the embryos of the parental and hybrid strains, the worms were allowed to cross for 5 h, after which the males were removed. The fertilized females were allowed to lay eggs for 12 h followed by egg harvest. To determine embryonic lethality, five females were first allowed to cross with five males for at least 5 h prior to removing the males. The successfully mated females were allowed to lay eggs for another 5 h, after which the mothers were removed. The number of laid eggs was counted and the unhatched eggs were counted again after 12 h. Worms entering the elongation stage of embryogenesis were judged by the presence of embryonic folding and a pharynx. For imaging hybrid embryos, a histone labeled (HIS-72::GFP) C. briggsae strain was used as the starting strain (Zhao et al. 2010). The differential interference contrast (DIC) and fluorescent micrographs were captured using a Leica LSM510 confocal laser microscope.
RNA extraction and sequencing
All collected embryos were pooled, then soaked in TRIzol reagent (Invitrogen) and subjected to 10 rounds of flash freeze–thaw cycles in liquid nitrogen to thoroughly disrupt the eggs. Released total RNAs were extracted according to the manufacturer's instructions. mRNA purification and sequencing were performed by Novogene using the Illumina TruSeq stranded mRNA library preparation method (paired-end, 150 bp) on a HiSeq sequencing instrument (Illumina). For small RNA sequencing, total RNAs were first treated with RNA 5′-polyphosphatase (Lucigen) to convert all of the 5′-triphosphates to monophosphates before purified again by TRIzol reagents. Small RNA extraction and the preparation of sequencing libraries were also performed by Novogene using the Illumina TruSeq small RNA library preparation kit.
TE consensus library compilation
A TE consensus library was first complied by the concatenation of all the Caenorhabditis repeat library from Repbase (Bao et al. 2015) with the C. briggsae and C. nigoni repeat families that we previously predicted (Li et al. 2016b). All rRNAs, tRNAs, satellite DNAs, and simple repeats were removed from the combined library. To further remove redundancy, we aligned the consensus library to itself using BLASTN and output all of the paired comparison entries with more than 90% identity along over 90% of the subject or query sequence. We then discarded the shorter sequences of the paired entries and retained the longer ones by a customized Python script. The resulting TE consensus, after the removal of redundancy, and the Python script used for the removal were deposited in GitHub (https://github.com/JeffreyXIE/TE_library) and as Supplemental Code.
Analysis of mRNA-seq reads for TE expression
The adaptor sequences of raw mRNA-seq reads were first trimmed using Trim Galore! (version 0.5.0) (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), and the low-quality reads (Phred quality score < 25) were filtered out. The processed reads were assessed using FastQC (v0.11.8) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) for quality controls. For TE expression analysis, clean mRNA-seq reads were mapped against the TE consensus library using Bowtie 2 (version 2.3.4.3) (Langmead and Salzberg 2012) with “–‐‐very-sensitive” mode. The number of reads mapped to each TE family was calculated by summation of the mapped reads recorded in the SAM files. Only those with counts per million (CPM) larger than 1 in at least one of the three replicates were considered as being expressed. To calculate the overall and individual TE expression levels, the length of each TE was also taken into consideration during normalization processes, and thus the fragment per kilobase of transcript per million mapped reads (FPKM) from the three replicates was adopted for the calculation of read abundance. The differential TE expression analyses were performed by DEGseq package (version 1.46.0) installed in R (v4.1.0) (Wang et al. 2010b; R Core Team 2021) as previously described (Kelleher et al. 2012). Unmapped reads were also included in the differential TE expression analysis in order to produce more accurate results. For comparisons between the F1, bB2, or nB2 hybrids and the parental species, the dysregulated TEs were defined as those that showed a significant change (up or down) in expression relative to both parents (Benjamini and Hochberg FDR corrected P value < 0.05) with the level of the change more than or equal to twofold. To plot read abundance of bB2 or nB2 versus C. briggsae embryos, the TEs that showed C. nigoni-specific expression were excluded. Similarly, to plot read abundance of bB2 or nB2 versus C. nigoni embryos, the TEs that showed C. briggsae-specific expression were excluded.
Quantification of RNA-seq reads for mRNAs and small RNAs
mRNA and small RNA quantifications were conducted similarly as we described previously (Li et al. 2016b). The details of the analyses were included as Supplemental Materials. Briefly, raw RNA-seq reads were trimmed and filtered as described for TE analysis. The read count for each member of a given ortholog pair was combined to represent the expression level of the pair, named after the C. briggsae gene. C. briggsae- and C. nigoni-specific genes were excluded from subsequent analysis. A change in the normalized read count <1.3-fold was treated as no change in the expression level. For differentially expressed genes (DEGs) analysis, the significantly dysregulated genes (up- or down-regulation) were defined as those that showed a change in the expression level of at least twofold with FDR corrected P value output by the edgeR being < 0.05. For small RNA quantification, piRNAs were normalized to the total piRNA size factors calculated using DESeq2 (Love et al. 2014). 22G-RNA counts were normalized to the total 22G-RNA size factors using DESeq2. miRNA counts were normalized to the total miRNA size factors using DESeq2.
Statistical analysis
One-way ANOVA followed by multiple pairwise t-test with multiple test correction (Benjamini and Hochberg) were performed for comparing the median of embryonic lethality and elongated embryos percentage. Wilcoxon ranked-sum test was performed when comparing the median of transcript abundance, gene expression distances, and Ka/Ks values. Fisher's exact test and chi-square (χ2) test were performed for comparing the significance of the numbers of dysregulated TEs, coding genes, and small RNAs. The 95% confidence intervals of Pearson's correlation coefficient for TE and coding gene analysis were obtained by bootstrapping analysis for 1000 times.
Data access
All RNA-seq data and small RNA-seq raw reads generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA849332.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Cindy Tan for logistic support and members of Z.Z.’s laboratory for helpful comments. This work was supported by General Research Funds (N_HKBU201/18, HKBU12100118, HKBU12101520, HKBU12101522) from Hong Kong Research Grant Council, and State Key Laboratory of Environmental and Biological Analysis grant SKLP_2223_P06 and Initiation Grant for Faculty Niche Research Areas RC-FNRA-IG /21-22/SCI/02 from Hong Kong Baptist University to Z.Z.
Author contributions: X.D. performed crossing, sample collections, RNA extractions, data analysis, and draft manuscript writing. Y.P., M.Y., and L.Y. participated in sample collection, experimental setup, imaging, and phenotyping. P.S. performed small RNA data analysis, provided guidance for RNA-seq analysis, and contributed to manuscript writing. L.X. provided the guidance and resources to the project. Z.Z. and X.D. conceived the project. Z.Z. coordinated the project, provided guidance and resources, and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277205.122.
-
Freely available online through the Genome Research Open Access option.
- Received August 14, 2022.
- Accepted November 4, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








