
Non-Mendelian inheritance patterns and extreme deviation rates of CGG repeats in autism
- 1Department of Medical Genetics, University of Antwerp, 2600 Antwerp, Belgium;
- 2NIHR BioResource, Cambridge University Hospitals NHS Foundation Trust, Cambridge Biomedical Campus, Cambridge, CB2 0QQ, United Kingdom;
- 3Department of Haematology, University of Cambridge, NHS Blood and Transplant Centre, Cambridge, CB2 0PT, United Kingdom;
- 4Department of Medical Genetics, Cambridge Institute for Medical Research, University of Cambridge, Cambridge, CB2 0XY, United Kingdom
Abstract
As expansions of CGG short tandem repeats (STRs) are established as the genetic etiology of many neurodevelopmental disorders, we aimed to elucidate the inheritance patterns and role of CGG STRs in autism-spectrum disorder (ASD). By genotyping 6063 CGG STR loci in a large cohort of trios and quads with an ASD-affected proband, we determined an unprecedented rate of CGG repeat length deviation across a single generation. Although the concept of repeat length being linked to deviation rate was solidified, we show how shorter STRs display greater degrees of size variation. We observed that CGG STRs did not segregate by Mendelian principles but with a bias against longer repeats, which appeared to magnify as repeat length increased. Through logistic regression, we identified 19 genes that displayed significantly higher rates and degrees of CGG STR expansion within the ASD-affected probands (P < 1 × 10−5). This study not only highlights novel repeat expansions that may play a role in ASD but also reinforces the hypothesis that CGG STRs are specifically linked to human cognition.
CGG trinucleotide short tandem repeats (STRs) are tracts of DNA where the trinucleotide cytosine–guanine–guanine units are repeated in a head-to-tail fashion. CGG STRs are known as the cause of cytogenetically visible folate-sensitive fragile sites (FSFSs) (Kooy 2009). At this stage, CGG STRs have been linked to over 10 different genetic disorders, most being neurodevelopmental or neurodegenerative disorders (Depienne and Mandel 2021).
The primary pathogenic mechanism associated with CGG STRs is repeat expansion. When a CGG STR expands beyond its full mutation breakpoint (typically more than 200 repeat units), it initiates an epigenetic event that results in hypermethylation of the repeat and the surrounding CpG islands. This, in turn, leads to a hypercondensation of the surrounding chromatin. At this point, the condensed chromatin appears cytogenetically as a fragile site (Heitz et al. 1991; Oberlé et al. 1991; Pieretti et al. 1991). If the hypercondensation occurs in or within proximity to a gene, this prevents the molecular transcription machinery from accessing the gene in question, and the gene is effectively silenced (Curradi et al. 2002). CGG STRs can also be disease causing beyond the premutation breakpoint (typically more than 50 repeat units), with the classical example being the neurodegenerative Fragile X-associated tremor/ataxia syndrome (FXTAS) (Jacquemont et al. 2003). Furthermore, the pathogenic mechanism at play here differs from the full mutation, which involves transcription and RAN translation of the premutation-lengthened CGG STR (Tassone et al. 2004; Todd et al. 2013).
The majority of cloned CGG STRs are implicated in either autism, neurodevelopmental delay, or severe neurological syndromes. At this point, only seven of the observed 22 FSFSs have been experimentally validated and have been linked to a specific CGG STR locus (Debacker and Kooy 2007). However, several recent studies have identified strong candidate CGG STRs, which occur at the other 15 FSFSs (Garg et al. 2020; Trost et al. 2020; Annear et al. 2021). Furthermore, other neurological disorders are linked to CGG repeats that occur elsewhere in the human genome, where no fragile sites have been previously reported (Ishiura et al. 2019; Sone et al. 2019; Tian et al. 2019; Yuan et al. 2020; Yu et al. 2021).
We have previously shown how most CGG STR loci display at least some degree of length variation across the general population (Annear et al. 2021). Furthermore, it is understood that STRs show mutation rates magnitudes higher than unique DNA sequences, 10−6 to 10−2 versus 10−9 nucleotides per generation (Ellegren 2000; Fan and Chu 2007). Although STRs display higher rates of DNA mutation, it appears that repeat motif is typically preserved past repeat expansion or contraction mutations. For instance, although the FMR1 CGG repeat often contains repeat interruptions (AGG most typically), the CGG structure of the repeat is maintained (Eichler et al. 1994). Certain aspects of CGG STR inheritance have been closely examined. This is especially true where the FMR1 gene is concerned. For example, it is known how larger repeat sizes negatively affect repeat stability during parental transmission, leading to a greater chance of a full mutation event at the FMR1 locus (Willemsen et al. 2011). Furthermore, it is known that full mutation length FMR1 repeats are actively selected against in spermatozoa production (Reyniers et al. 1993; Malter et al. 1997). However, to date, very little is known about whether the same inheritance and mutation patterns seen in the disease-associated CGG STRs are replicated among all or any of the other known CGG STRs spread throughout the human genome.
In a previous study, we cataloged and categorized more than 6000 unique CGG STR loci across the human genome. The CGG repeats were shown as being enriched in genes related to neurological development and disease (Annear et al. 2021). Within this study, we aimed to elucidate the inheritance and length variability patterns of all known CGG STRs and establish if differences are present in the CGG STR landscape between autism-spectrum disorder (ASD)–affected and non-ASD-affected individuals. To achieve this, we conducted a genome-wide segregation analysis across a total study cohort of 1978 trios, with an affected proband, and 114 quads, with an affected proband and an unaffected sibling. This provided an intergenerational perspective of the variability and instability of the human CGG STR genetic landscape. Whole-genome sequencing (WGS) was applied to samples taken from all individuals, and bioinformatic STR genotyping tools were applied to the resulting WGS alignments.
Results
Genome-wide determination of monogenic, informative, and deviating CGG STR genotypes
The study population consisted of three distinct cohorts consisting of 167 and 1811 trios and 114 quartets, for a total of 6390 individuals. All trios consisted of both biological parents and a neurodevelopmentally or ASD-affected proband, and the quartets consisted of an ASD-affected proband, both parents, and an unaffected sibling. The 6390 individuals were genotyped across the 6063 previously categorized CGG repeat loci. Overall, 11,885,983 complete parent-to-child CGG repeat genotype comparisons were collected, which translated to a total of 23,391,708 CGG repeat inheritance transmissions (22,814,576 autosome and 577,132 sex chromosome transfers) (Table 1). Among the compared genotypes, 80% (n = 9,511,223/11,885,983) were monogenic; namely, the repeat lengths at the locus in question were equal across all members of the trio. Also, 15.6% (n = 1,853,704/11,885,983) were informative; namely, repeat genotypes at the locus in question varied across the proband, mother, and father; however, the genotype observed in the proband was reconciled by the repeat lengths observed in the mother and father. Finally, in 4.4% (n = 521,030/11,885,983) of the compared genotypes, either one or both of the observed proband alleles were divergent from the repeat lengths observed in the parents; this resulted in a total of 2.6% (n = 597,844/23,391,708) of the observed transmissions deviating from the genotypes of the parents. Although some technological constraints exist when examining STRs in short-read-based data, we showed in previous work how different STR genotypers produced concordant results for the same STRs investigated here (Annear et al. 2021). Furthermore, many studies are available on the accuracy, validation, and comparison of STR genotyping tools (Dolzhenko et al. 2019; Mousavi et al. 2019; Halman and Oshlack 2020).
Breakdown of the complete parents-to-proband genotype comparisons across all 6063 known CGG STR loci per study cohort
CGG STR distribution and stability
Across all 6390 individuals, we observed 521,030 deviating proband–parent genotype comparisons, accounting for a total of 597,844 deviation events (potential mutations), occurring at 5881 of the known CGG repeat loci. Furthermore, 5989 of the CGG STR loci displayed varying genotypes within the parents. Therefore, in 97% of known CGG STRs, a repeat deviation event was observed from the parental repeat length within one generational transfer, and 99% of CGG STR loci were observed to display some degree of polymorphism throughout the tested population, which is in line with our previous findings (Annear et al. 2021). Although CGG STRs occur ubiquitously throughout the human genome (localized primarily within or upstream of genes [Annear et al. 2021]), the most highly deviating repeats appear to cluster together. These clusters are illustrated in Figure 1A on Chromosomes 1, 2, 7, 9, 16, 17, 19, and 22.
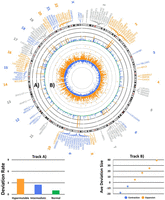
Distribution and deviation rate of known CGG short tandem repeats (STRs) throughout the human genome. Genomic positioning (GRCh38) and HGNC gene symbols where the hypermutable CGG STRs are localized. (A) Deviation rate of all known polymorphic CGG repeats: (hypermutable) ≥3 × 10−1 deviations per gamete per generation, (intermediate) ≥1 × 10−2 deviations per gamete per generation, and (normal) more than zero deviations per gamete per generation. (B) Average repeat length deviation for both expansions and contractions of each polymorphic CGG STR locus (CGG repeat units) (Gu et al. 2014).
We observed a total of 372,071 expansion deviations versus 225,773 contraction deviations; this was a ratio of 1.65 in favor of expansions. This indicates that although contractions of CGG repeats do readily take place, the CGG repeats tend toward expansion at a significantly higher rate (Pearson's chi-squared test; P < 0.001) (Fig. 1B). Although we observed that CGG STRs were capable of both expanding and contracting in size, Figure 2 illustrates how repeats of smaller repeat lengths can expand by large degrees across a single generation. Distribution of the repeat deviation size and number can be observed in Supplemental Figure S1.
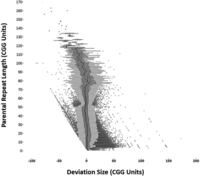
Distribution of proband repeat length deviations versus the corresponding parental repeat length. The distribution and outliers of the deviating repeat length genotypes observed in each proband compared with that of the transferred repeat of their parent. Mean and interquartile range mutation sizes appear to be uncoupled from parental repeat length, at least for the repeats outside of the pathogenic range.
The mutation rate was strongly dependent on the genetic region of the repeat (Fig. 3). Although the largest portion of CGG repeats was observed in the 5′-UTR gene regions, the region that displayed the highest overall rates of CGG repeat deviation was the intergenic region. The 5′ (1-kb) upstream, intronic, and ncRNA regions displayed similar rates of repeat deviation. The lowest rates of repeat deviation were observed in the 3′-UTR, 3′ (1-kb) downstream, and exonic regions. Although this is not surprising for the 3′ regions as CGG repeats here are generally small and limited in number, it was interesting to observe the relatively low repeat change rate in the protein-coding regions. The most variable CGG repeats appeared clustered on Chromosomes 2 and 9. Among the 200 most mutable CGG loci observed, 42 and 40 were located on Chromosomes 2 and 9, respectively, together accounting for 41% of the most mutable repeats. For context, the next most frequent number of highly mutable loci was detected on Chromosome 1, which displayed 15 of such loci. We investigated CGG STR rate and size deviation as a function of the genetic region and localized gene. Regarding genetic region, we observed two major differences. First, the intergenic repeats showed significantly larger deviation rates compared with the other genetic regions (model coefficient = 0.055 ± 0.025, z = 2.170, P = 0.03) (Fig. 3A). Second, the average size deviation of the 3′-UTR repeats was significantly larger than the repeats of the other genetic regions (model coefficient = 0.150 ± 0.063, z = 2.377, P = 0.018) (Fig. 3B). This is likely affected by the low sample size of CGG STRs present within the 3′-UTR region.
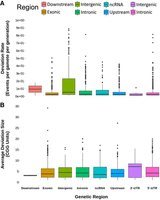
A comparison of CGG short tandem repeat characteristics between the specified genetic regions. (A) Distribution of repeat deviation rate of all unusually mutable CGG STR loci within the different genetic regions and (B) distribution of the mean deviation size for each CGG repeat per given genetic region. (A) Intergenic regions display, by far, the largest rate of CGG repeat variation than any given region. (B) Despite differences in the rate of deviation, on average, CGG repeats show comparable behaviors at the level of genetic regions. The one difference can be seen in the downstream region. Note that outliers have been removed from the display in order to improve figure resolution.
Intergenerational CGG STR length deviation
Previous studies place a typical STR variation rate between 1 × 10−3 and 10 × 10−3 mutations per gamete per generation (Jeffreys et al. 1988; Weber and Wong 1993; Huang et al. 2002). However, this seems to vary with both the length and the nucleotide composition of the repeat motif (Fan and Chu 2007). In contrast, the mutation rate of “unique” DNA is far lower, with rates of 0.15 × 10−7 to 1 × 10−7 per gamete per generation (Fan and Chu 2007; Turner et al. 2017). Although previous studies provided insight into the mutation rates of STRs, they were limited by the number of repeats they could practically interrogate. In this study, we were able to focus on all known CGG repeats throughout the human genome. Overall, we observed repeat length deviations at 5881 of the 6063 investigated CGG STR loci. The majority of the categorized repeats’ deviation rate fell within the previously reported mutation rates, with 59% (n = 3577/6063) of CGG repeats falling under a rate of 1 × 10−2 events per gamete per generation. As illustrated in Figure 4, many CGG repeats displayed deviation rates that far exceeded the established norm of STR mutation. Although these highly length-deviating repeats were primarily localized within the intronic and intergenic genetic regions, they were also observed within the other genetic regions. An interesting observation was the trend of the exonic localized repeats to show one of the lowest rates of repeat deviation and displaying significant difference in average mutation rate in comparison to the intronic and intergenic localized CGG STRs (exonic vs. intronic, z = −2.56, p.adj = 0.03; exonic vs. intergenic, z = −8.56, p.adj = 1.61 × 10−16).
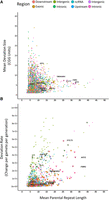
Illustration of the deviation characteristics of the CGG short tandem repeats that were determined to display polymorphism within the screened cohorts. (A) Generational repeat length deviation rate versus the average parental repeat length and (B) mean repeat length deviation size versus the average parental repeat length of all CGG repeat loci that displayed at least one deviating proband–parent genotype. Each point represents a unique CGG repeat locus. Color represents the genetic region in which the repeat is localized. Triangles represent the repeats that are localized within genes associated with neurodevelopmental/neurodegenerative disorders. Several known highly mutable and disease-causing repeats are highlighted for comparison. The absolute value was reported regarding deviation size, and contractions and expansions were not differentiated from each other.
We examined both the rate (Fig. 4A) and degree (Fig. 4B) of which the proband repeat genotypes deviated from their respective parental genotypes. First, as indicated in literature, the repeat deviation rate was proportional to the mean parental repeat size (r2 = 0.577), as both the mean mutation rate and rate variance increased along with parental repeat length. This was expected, as it has long been documented with the FMR1 repeat that a larger repeat length comes with a larger instability (Morton and Macpherson 1992; Nolin et al. 2003, 2019). However, what was not expected was that many repeats that showed smaller parental genotypes far exceeded the deviation rate and degree of that observed in the larger repeats. Furthermore, we observed that the average size variation decreased as parental repeat length increased (r2 = 0.557). This may suggest, that although a larger parental repeat may result in a higher risk of size deviation, a pathogenic repeat expansion may still be a rare event and not necessarily coupled to the initial parental size. This observation was further supported by the large number of the smaller repeats (mean parental repeat length < 10) that displayed some of the highest average deviation sizes.
Through genetic annotation, it was determined that the 6063 known CGG STRs were localized within 3380 genes, 267 ncRNAs, and 374 intergenic regions. We determined that 996 of the loci stood out as containing repeats that displayed a deviation rate or average deviation size that differed significantly from the entire set of CGG repeat–containing genes/loci (Supplemental Table S1). Many of the variable CGG STRs were localized within or next to genes that have been associated with or directly linked to genetic disease. In total, of the 5989 STR loci that displayed deviation, 844 repeats were linked to 481 genes that may or are known to contribute toward autism, intellectual disability, neurodevelopmental delay, and neurodegenerative phenotypes.
The largest expansion deviation observed was 245 CGG repeat units at repeat locus Chr 2: 86,913,985, which is located on intron 1 of the RGPD1 gene. The largest contraction deviation was 92 CGG repeat units at repeat locus Chr 1: 148,679,544, which is located in the 5′-UTR of NOTCH2NLB, a paralog of NOTCH2NLC that contains a CGG repeat that has previously been linked to neuronal intranuclear inclusion disease (NIID) and is a prime candidate for the causative repeat of the FRA1M fragile site (Ishiura et al. 2019; Sone et al. 2019).
Non-Mendelian CGG STR inheritance patterns
The presence of informative reads for loci where parents displayed a heterozygous repeat length genotype provided a unique opportunity to track the repeat that was transferred. This led to some interesting data being collected regarding the effect of repeat length on repeat inheritance. First, we looked at the tendency of repeats to undergo a contraction or expansion event based on the parent whose repeat allele underwent change. In general, we observed no significant difference in repeat change events whether the repeat was inherited from the mother or the father. The lack of difference between parents appeared to be consistent when we looked specifically at expansions, contractions, and the degree of change in the size-changing event. In the case of fragile X syndrome (FXS), it is known that the repeat expansion is maternally inherited. However, although we observed predictions of pathogenic-sized repeat mutations within this study, both mothers and fathers were represented as the transferring parent within this small subset. At the FMR1 locus, we observed 32 predictions where the FMR1 repeat changed to a premutation length in the proband. In 85% of these cases, the transferring parent was the mother. Although we observed that ∼3% of FMR1 CGG repeat alleles were present beyond the premutation breakpoint, comparable to rates seen elsewhere, it was rare for an FMR1 locus of this size to be transferred from parent to child. In the context of the FMR1 repeat, we observed a total of five transfers from a potential 42 premutation-sized parental repeats. All other cases of probands showing FMR1 repeats of premutation-sized and larger appeared to be caused by repeat expansion (Supplemental Fig. S4). This effect could be owing to large premutation-sized repeats being highly unstable.
We looked at allele transfer from heterozygous parents (Fig. 5). We observed across all three cohorts, that when parents displayed a heterozygous genotype, there was a preference for the smaller allele to be transferred to the child (Pearson's chi-squared test; P = 0.003) (Fig. 5A–D). Overall, a ratio of 1.38 for smaller:larger repeat alleles was observed for fathers and a ratio of 1.39 was observed for mothers. No parent-of-origin effect was detected, and the same trend was observed across all three cohorts.
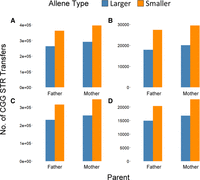
Based on the informative genotypes, when parents displayed heterozygous repeat length genotypes at given CGG locus the times the smaller and larger repeats were transferred to the child for cohorts total (A), NGC trios (B), MSSNG trios (C), and MSSNG quads (D). Although total observations differed from cohort to cohort, the same proportional trend of larger to smaller allele transfers was consistent across all trios, as was the trend of no difference between the total transfer observations of mothers and fathers.
Next, we investigated the trends of generational repeat transfer based on the parental repeat allele difference (Fig. 6A–D). Based on Mendelian principles, when parents have a heterozygous repeat length at a given locus, the representation of the larger and smaller repeat alleles should be equal in their children. However, this appeared to not be the case for the CGG repeats. As illustrated in Figures 6 and 7, we determined that when the parental repeat alleles at a given locus displayed a small difference (between one and five CGG units) both the larger and smaller allele were represented in the proband at similar rates. However, as the repeat length difference increased in the parents (between six and 14 CGG units), there was a dramatic increase in the representation of the smaller allele in the proband. The trend seemed to plateau at a high representation (∼90%) for the smaller allele at repeat differences between 16 and 40 CGG units. As the difference increased further (41–65 CGG units), we observed that the representation of the smaller allele lessened and became far more variable; however, it was far from 50:50 as expected. Finally, as repeat differences moved toward the extreme (more than 70 CGG units), we observed an almost total exclusion of the larger alleles from the proband's genotype. Furthermore, these trends were observed in all three separate cohorts.
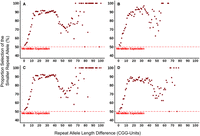
Illustration of the non-Mendelian representation of the smaller repeat allele in the proband by repeat length size difference in the parent for the cohorts total (A), NGC trios (B), MSSNG trios (C), and MSSNG quads (D). Although the representation of both the smaller and larger parental allele appeared in equilibrium at lower differences, there was an exponential increase in representation of the smaller allele from five repeat units onward. A final plateau of ∼90% in favor of the shorter allele was reached by a difference of about 15 CGG units. This effect is further illustrated by the ratio of the smaller versus larger repeat allele in Supplemental Figure S2.
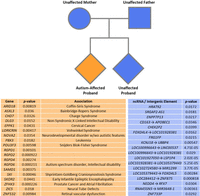
Pedigree chart representation of all families within the MSSNG quads cohort. The cohort consisted of 114 families made up of an autism-affected proband and unaffected mother, father, and sibling. Overall, through logistic regression, 37 distinct genetic regions were determined to show significantly greater rates and degrees of CGG repeat variation in the probands. These 37 regions included 19 genes, three ncRNAs, and 15 intergenic elements. Thirteen of the 19 genes have been previously linked to autism, neurological, or neurodevelopmental disorders. Four genes have been previously linked to cancers. Two genes had not been linked to any disorder; however, both are paralogs of genes implicated in neurological conditions. Furthermore, several genetic loci remained significant even after application of the most stringent multiple testing correction; see Supplemental Table S3.
Comparison of CGG STR inheritance between autism-affected and nonaffected siblings
The data obtained from the Quad cohort present an opportunity to observe the differences in CGG patterns between autism-affected individuals and their unaffected siblings. Across the cohort of 116 quads, 649,314 and 650,064 complete parent-to-child CGG STR genotypes were determined for the probands and siblings, respectively, with a near-identical result (99.99% similarity). Overall, 37,426 proband–sibling deviation genotype comparisons could be made. We observed a greater rate of repeat deviation (more than 1200 additional deviation events) among the affected group versus the unaffected group. This suggests an ∼3% greater number of repeat deviations in probands versus siblings. As outlined in the previous section, the CGG STRs were localized around 4021 specific genetic loci. Across this cohort, variation was observed at 2916 of 4021 loci. Furthermore, differences in repeat deviation between the affected and unaffected individuals were observed in 1790 of the genetic loci.
We used multiple logistic regression to predict the probability that specific CGG STR loci presented a greater rate and degree of length deviation based on whether the individual was autism-affected versus unaffected. Overall, it appeared that both deviation size (model coefficient = 0.021 ± 0.008, z = 2.56, P = 0.01) and rate (model coefficient = 0.065 ± 0.014, z = 4.40, P = 1 × 10−5) were significantly different between probands and siblings, with deviation rate being the major contributor. This resulted in the identification of 37 distinct genetic regions (Supplemental Table S1) displaying a significant increase (rate and degree) of expansion deviation among the affected probands. Additionally, we observed 23 genetic loci that displayed a tentative association toward greater STR deviation rate and size within the autism-affected probands (Supplemental Table S2). Furthermore, no CGG STR–containing regions displayed significantly increased rates or size deviation within the unaffected sibling group versus the probands.
The 37 genetic regions with altered deviation rates included 19 genes and three ncRNAs. A number of these genes (ARID1B, ZIC5, ZFHX3, SKI, PBX3, CHD7, POU3F3, and SKOR2) displayed a 9.3- and eightfold GO term enrichment in the brain and central nervous system development pathways, respectively. Genes RGPD1, RGPD2, RGPD4, and RGPD6, which are involved in NLS-bearing protein import into the nucleus, displayed an exceptionally high 181-fold enrichment and were among the most variable CGG STRs within the human genome. SKI and CHD7 are involved in both nose and olfactory bulb and lobe development and showed a 158- and 79-fold enrichment (Supplemental Fig. S3), respectively. Finally, >50% of these genes were implicated as being involved in nucleic acid binding and regulation. Furthermore, 15 of the 37 significantly deviating repeat regions were localized within intergenic space. This observation highlights how intergenic elements may play an as-of-yet-unelucidated role in genetic expression and human cognition.
Additionally, of the further 23 tentatively CGG-associated regions (Supplemental Table S2), 15 were localized within genes. Among these 15 genes, 10 have been linked to either congenital, neurodevelopmental, or ASDs. These include Baratela–Scott syndrome (XYLT1), congenital microphthalmia (HMGB3), Gabriele–de Vries syndrome (YY1), hand–foot–genital syndrome and preaxial deficiency (HOXA13), intellectual developmental disorder, X-linked, with panhypopituitarism (SOX1), Lambert–Eaton myasthenic syndrome and developmental and epileptic encephalopathy (SOX21), immunodeficiency-centromeric instability-facial anomalies syndrome (ZBTB4), neuroocular syndrome (PRR12), autism (SMIM10L2B), and mental retardation syndrome (CAMK2B).
Discussion
We conducted an STR genome-wide analysis across three different cohorts, accounting for a total of 6390 individuals. Six thousand sixty-three unique CGG STR loci (previously categorized) were analyzed (Annear et al. 2021). We interrogated the CGG repeat class using ExpansionHunter using whole-genome sequence data of the entire study population consisting of 1978 trios (neurodevelopmentally affected proband, mother, and father) and 114 quartets (neurodevelopmentally affected proband, unaffected sibling, mother, and father). Overall, the repeat allele size deviation of the CGG STRs is striking. We found that, within one generation, 97% of the CGG loci (n = 5881) show repeat length deviation, and overall, 99% of CGG repeat loci showed length heterogeneity to some degree, although the majority of CGG STR loci (n = 3577) fell within the previously reported range of STR mutation, 0.1–1 × 10−2 mutations per gamete per generation (Jeffreys et al. 1988; Weber and Wong 1993). However, in comparison to more recent reports of STR mutation rates, our observations tend to be higher. In two separate studies, mean mutation rate observations of 3.7 × 10−5 and 5.6 × 10−5 mutations per locus per generation were described (Mitra et al. 2021; Steely et al. 2021). However, these included repeats of 1- to 20-bp repeat motifs, regardless of nucleotide composition. These differences are likely owing to our specific interrogation of the CGG class of trinucleotide STRs. A considerable proportion of CGG loci (n = 2304) showed nontypical rates (more than 1 × 10−2 deviations per gamete per generation) of genotype deviation when the proband was compared to the parents. In some rare cases, the genotype deviation rate exceeded more than 3 × 10−1 deviations per gamete per generation. Although the true underlying reason for extreme STR variation currently eludes us, it has been implicated in gene expression, morphological variation, and evolution (Fondon and Garner 2004; Sperling and Li 2013; Fotsing et al. 2019). However, the mechanism behind repeat variation is typically attributed to slipped strand mispairing owing to the simple and highly repetitive nucleotide composition of STR tracts (Kornberg et al. 1964; Fan and Chu 2007).
CG-rich STRs stand out in comparison to other STRs of alternate nucleotide composition owing to their tendency to be localized within or in proximity to genes, primarily to the 5′ UTR of genes (Subramanian et al. 2003; Kozlowski et al. 2010; Annear et al. 2021). The role CGG STRs play in disease is being further understood as more CGG repeats are being identified as the etiological agents of neurodevelopmental and neurodegenerative diseases. By looking at the repeats showing nontypical deviation behavior, we determined that genetic location had quite an effect on repeat deviation rate but not necessarily on deviation size. From the perspective of repeat deviation length, we observed that the variation and distribution of repeat length changes were relatively equal, with the exception of the STRs localized within 3′ UTRs. However, the deviation rate was strongly dependent on the genomic region. Although the largest portion of CGG repeats is observed in the 5′-UTR gene regions, overall the regions that displayed the highest rates of CGG repeat mutation were the intergenic, intronic, and immediate (up to 1000-bp) 5′-upstream regions. The most mutable STRs were localized within the intergenic regions, many of which reached extreme rates of deviation, suggesting a two- to threefold increase in mutation rate for (likely) nontranslated versus translated repeats. Furthermore, excluding several outliers, the repeats localized within gene exons showed the lowest rates of repeat deviation alongside the repeats localized within the 3′ UTRs. This was predicted as one would expect the repeats localized within protein-coding regions to be better conserved. Nevertheless, multiple outliers were observed, and these repeats may be prime disease-causing candidates. An example is the repeat that occurs in exon 1 at amino acid position 60 of the YY1 gene that, in reference GRCh38, encodes for a five poly(G) tract (Verheul et al. 2020).
A characteristic of CGG STRs that was elucidated in this work was the effect of parental repeat length on both the repeat length deviation and repeat deviation. From the perspective of the FMR1 repeat, it is typically thought that the longer the CGG repeat becomes, the greater the chance that the repeat will expand into a full mutation in the child. Our data support this conclusion to a degree, as we observed that the larger parental repeats tended to present larger deviation rates, and smaller parental repeats typically presented smaller deviation rates. However, we observed the reverse trend occurring when it came to deviation size, in which the smaller repeats had a far larger variation in repeat length deviation compared with the larger repeats. This trend is similar to size-dependent mutational bias reported by Huang et al. (2002), as we, too, observed that the shorter repeat alleles more readily gained repeat units. A gene that shows this interesting trend is SOBP. Furthermore, it is a potential repeat-associated disease-causing gene, as it has been shown that truncating mutations in this gene results in both syndromic and nonsyndromic autosomal-recessive intellectual disability (Nolin et al. 2003). The gene contains three repeats, two within the 5′-UTR region and an additional repeat in exon 6 of the gene, encoding for a polyproline tract (Nolin et al. 2019; Trost et al. 2020). Within our cohort, 5′-UTR repeat 1, with an average parental repeat length of 32 CGG units, displayed a deviation rate of 1.75 × 10−1, and 5′-UTR repeat 2, with an average parent repeat length of 10, displayed a deviation rate of 0.19 × 10−1. However, the repeats displayed an average mutation size of 3.8 and 6.8, respectively. The exonic repeat displayed the lowest deviation rate of 0.07 × 10−1.
CGG STRs are linked to multiple mechanisms of pathogenicity. These include transcription silencing through repeat-mediated hypermethylation, RNA gain of function, and toxic polypeptide production through RAN translation (Burman et al. 1999; Amiri et al. 2008; Kearse and Todd 2014). To our knowledge, our analysis detected and categorized all CGG STRs that have been previously reported in the literature and linked to disease (Table 2). It can be seen that disease-linked repeats show a deviation rate above that of the typical CGG mutation rate. Although the observed repeat lengths are not all the most extreme, they cluster among the CGG loci of the larger size. Furthermore, as illustrated in Figure 4, we observe many other CGG repeats that reflect the characteristics of known disease-causing repeats. Not only do these repeats show a similar nature, but many of the repeat-containing genes are implicated in ASD, neurodevelopmental, and neurodegenerative disorders. The repeat within FRA10AC1 is an excellent example of how disease caused by CGG repeats may be overlooked. The causative repeat at FRA10A has been identified for some time (Sarafidou et al. 2004); however, until recently no phenotype had been linked. von Elsner and colleagues (2022) have shown that biallelic loss-of-function variants in the FRA10AC1 gene lead to neurodevelopmental disorder with growth retardation (von Elsner et al. 2022). Likewise, if a patient were to harbor two expanded repeat alleles or an expanded repeat and a loss-of-function variant, as seen in XYLT1 and Baratela–Scott syndrome (LaCroix et al. 2019), we would expect to see the same phenotype. This illustrates how pathogenic CGG repeat expansions may be overlooked through a combination of limitations that NGS techniques have with repeat expansions and the focus on single variant detection. Regarding pathogenic and fragile sites linked to CGG STRs, it is the hypermethylation of the expanded repeat that produces the cytogenetically visible fragile site. However, it is unclear whether all CGG STRs share the same pathogenic breakpoint. For example, the FMR1 repeat hypermethylates at around 200 CGG repeat units, but this may not be representative of all CGG STRs in other genetic location contexts. At present, it is unknown if all CGG repeats described here behave in a similar manner.
Known and implicated disease-associated CGG repeats
The availability of quads containing ASD-affected probands accompanied by unaffected siblings allowed for direct comparisons to be made regarding the genetic landscape of CGG STRs in an ASD context. Several CGG-containing genetic regions displayed a significant increase in rate and size deviation within the ASD-affected group. The presence of longer STRs in ASD-affected individuals has been previously reported (Winnepenninckx et al. 2007; Metsu et al. 2014a,b), and it has been proposed that STR expansions may contribute up to 2.6% of the risk of ASD (Trost et al. 2020). However, this study specifically provides links between several novel CGG STR loci to ASD and highlights the genes that may be responsible. Additionally, the lack of CGG STRs with significantly increased variability in the unaffected group would suggest that this phenomenon is explicitly linked to ASD. The majority of the ASD-linked, gene-localized CGG STRs highlighted within this study are present within genes that are linked to neurological function or neurodevelopmental disorders. Genes including, but not limited to, SKI, NOVA2, ARID1B, DLG3, YY1, SKOR2, and ZIC5 are all examples of prime candidates in which a CGG STR expansion may lead to neurological disease. Evidence has emerged suggesting epigenetic changes as an etiology of ASD, with differential methylation being a primary contributor (Wang et al. 2022). The variable nature and tendency toward expansions of CGG STRs leading to epigenetic changes may explain many idiopathic ASD cases, with CGG STR expansion being the trigger of hypermethylation events. Furthermore, this, coupled with the fact that it is known that STRs affect gene expression and that STRs may be a class of expression quantitative trait loci (eQTL) (Willemsen et al. 2011; Fotsing et al. 2019; Bakhtiari et al. 2021), highlights how variable CGG STRs may be primary contributors to oligogenic or complex inheritance, especially in ASD and neurodevelopmental disorders. Past research also identified significant enrichment of CGG STRs within neurodevelopmental disorder genes (Annear et al. 2021). This provides a promising route for future research into how these highly variable CGG STR loci, especially those in the 5′ gene regions, may affect gene expression and subsequently affect phenotype.
From an evolutionary perspective, the RANBP2 like and GRIP domain-containing protein (RGPD) genes are a highly interesting family of genes regarding CGG STRs. There are eight segmentally duplicated RGPD genes, originating from RANBP2, located on Chromosome 2, and their gene family copy number has been correlated with increased brain volumes in humans and primates (Ciccarelli et al. 2005; Bekpen and Tautz 2019). The RGPD genes are enriched with CGG STRs, and many of the most mutable CGG STRs identified in our analysis are clustered within RGPD1, RGPD2, RGPD4, and RGPD6. Although mutations in RANBP2 are linked to acute necrotizing encephalopathy and RGPD6 deletion has been implicated in ASD and severe intellectual disability, very little is known about the RGPD genes in regard to both their biological function and role, if any, in disease (Neilson et al. 2009; Chen et al. 2017). However, these RANBP2 paralogs appear to be unique to the great apes and are lacking in other mammalian genomes (Bekpen and Tautz 2019). These genes and their highly variable CGG STRs are located in regions of Chromosome 2 that were produced through intrachromosomal segmental duplications despite Chromosome 2 overall being poor in segmental duplications (Bailey et al. 2002; Ciccarelli et al. 2005). It is intriguing that although Chromosome 2 contains many of the most variable CGG STRs, these STRs are then further localized within segmental duplications. Here, we raise the question of CGG STRs’ involvement in human evolution and the development of human cognition.
Regarding neurodegeneration, this research suggests that CGG STRs in the premutation range (more than 50 CGG units) may be far more common in presentation than previously thought. It is now known that although CGG repeats may occur at different genomic locations, such as the FMR1, GIPC1, LOC642361, LRP12, and NOTCH2NLC repeats, expansions of similar size result in similar clinical manifestations of neurodegenerative disease. Although a significant portion of neurodegenerative disease can currently be accounted for through genetic and familial factors, there remains missing heritability across a large portion of neurodegenerative diagnoses (Dilliott et al. 2021). However, although the discovery of new genetic causes focuses on the detection of variants through methods such as GWAS, STRs and other structural variants are receiving more interest as potential etiologies for neurodegenerative disease (Theunissen et al. 2020). This is well shown by STRs in C9orf72 and ATXN2 that cause amyotrophic lateral sclerosis (ALS), as well as the CGG STRs previously mentioned. As this work shows the presence of many highly variable CGG STRs localized within many neurodegenerative genes, we propose that CGG STRs may play a role within both the missing and complex heritability of neurodegenerative conditions.
Across all three cohorts we observed the trend that, from a repeat length perspective, CGG STRs did not segregate following Mendel's laws. In parents with heterozygous repeat genotypes, it appears, in general, that the smaller repeat allele is far more often reflected in their progeny than the larger repeat allele. Furthermore, the smaller allele is not only seemingly preferred, but as the length difference between the two parental alleles increases, the larger allele seems to be completely excluded from the inheriting child. From a Mendelian perspective, this is unexpected as one would predict both the smaller and larger allele to be reflected in the offspring in an approximately equal fashion. However, although not the norm, some non-Mendelian inheritance patterns are well documented when it comes to reduced penetrance and imprinting disorders (Van Heyningen and Yeyati 2004). Furthermore, it may seem that non-Mendelian inheritance and STRs are linked in some way. First, anticipation is known to warp Mendelian segregation and result in unusual inheritance patterns, with the trinucleotide disorders FXS and Huntington's disease being the primary examples (Van Heyningen and Yeyati 2004). Second, non-Mendelian inheritance patterns were reported in telomeric localized STRs in children displaying idiopathic mental disability (Colleaux et al. 2001; Van Heyningen and Yeyati 2004). More recently, Khamse and colleagues (2022) have reported a deviation from the Hardy–Weinberg equilibrium that results in a significant selection against the heterozygous repeat length genotype of the 5′-UTR CGG STRs within the SBF1 gene (Khamse et al. 2022).
The informative genotype data then confront us with the following question: Why the apparent bias against the larger CGG STR alleles? The primary limitation is that of the technical capabilities of the ExpansionHunter and short-read sequencing when handling low complexity and CG-rich STRs beyond read-length. In the cases in which the parental allele size differences increase beyond the sequencing read-length, it is difficult to maintain confidence that the smaller repeat allele bias is maintained. This is well illustrated in Figure 6 and Supplemental Figure S2, in which the patterns become chaotic once the read-length boundary has been reached. However, we are confident in the trends seen in the genotype predictions that are well within the read-length. We observe the trend of smaller repeat transfer bias emerging at differences as low as six repeat units. The vast majority of repeats genotyped within this study displayed a mean cohort size of four to 10 repeat units, again well within the read-length boundaries. Furthermore, as illustrated in Table 1, >80% of our genotype predictions were monogenic across trios and in line with the reference genome repeat lengths, therefore providing further confidence in our repeat length predictions. Another possibility is that the parental repeat genotypes determined from blood samples are not representative of the repeat genotype present in the germline and gametes. This would imply that the larger repeat sizes observed are the result of somatic mutation. However, although somatic mutation is likely a contributor, in these cases, the other allele is present in the child, meaning that if a somatic mutation has occurred, the germline genotype must be homozygous. Alternatively, we may be describing a novel mechanism underlying the inheritance of STRs. Could the larger repeats be actively selected against, as observed in the case of the exclusion of expanded FMR1 in the spermatozoa (Reyniers et al. 1993; Malter et al. 1997)? Additionally, could there be some form of repeat-mediated DNA repair, where expanded STRs are returned to their “normal” size or decreased in length? Elimination of DNA loops through the mismatch repair system, and recent work on the FAN1 DNA repair nuclease may suggest this (Fan and Chu 2007; Deshmukh et al. 2021). Classically, it is thought that STRs tend toward expansion, and to some degree, that would appear to be the case. However, it is known that repeat lengths in humans are not expanding out of control; therefore, a compensatory mechanism should be present. It may be that these trends that we are observing are the results of the biological mechanism that compensates for the effects of anticipation.
In conclusion, this research not only solidifies previous findings surrounding STRs but specifically shows new characteristics of CGG trinucleotide STRs. First, we solidify the idea that repeat variation rate is proportional to repeat length; however, we refine this concept and show how smaller repeats more readily show a greater degree of size variation and take on additional repeat units. Second, although 99% of CGG STRs were observed as polymorphic, we see how most repeats fall within the known “normal” variation rates. Although there is a continuum of increasingly variable repeats, there is a distinct subset of hypermutable CGG STRs that cluster in specific genetic regions and locations. Many of these locations harbor genes linked to neurological function and development. Third, in the case of heterozygous genotypes, CGG STR loci do not segregate by Mendelian principles. The shorter repeat allele length is typically selected and reflected in offspring. Furthermore, this trend appears to magnify as the repeat length difference increases between the two parental repeat alleles. Finally, there appears to be involvement of CGG STRs in ASD. In autism-affected individuals compared with their unaffected siblings, we observe significantly higher rates and degrees of CGG repeat variation in multiple genetic areas related to neurological function and development. Furthermore, the inverse is not observed in any other CGG STR–containing region. This may suggest that CGG STRs are explicitly linked to neurodevelopmental function and disorders.
Methods
Study cohorts and WGS
Within this project, WGS data were obtained from 5889 samples from the MSSNG project and 501 from the NGC project (Yuen et al. 2017; French et al. 2019). For the MSSNG cohort, PCR-free WGS was conducted on DNA samples obtained from blood or patient-derived cell lines of 1811 trios of an autism-affected proband and unaffected parents (5433 individuals) and 114 quads of an autism-affected proband, an unaffected sibling, and unaffected parents (456 individuals). Library preparation and sequencing were conducted on the Illumina HiSeq X platform at a read-length of 150 bp. All samples were aligned to the GRCh38/hg38 reference genome using BWA-MEM (Li and Durbin 2009). Full details on the MSSNG data pipelines can be obtained from their website (via MSSNG, https://research.mss.ng/). For the NGC cohort, PCR-free WGS was conducted on DNA samples obtained from blood of 167 trios of an affected proband (young children admitted to neonatal and pediatric intensive care units) and unaffected parents (French et al. 2019). Inclusion criteria were for any cases in which the child displayed a possible single gene disorder, whereas exclusion criteria were short stay admittance, prematurity without additional features, clear antenatal or history suggestive of a nongenetic cause, and where a genetic diagnosis was already present (French et al. 2019). DNA samples were shipped to Illumina for sequencing and were prepared with the Illumina TruSeq DNA PCR-free sample preparation kit (Illumina) as previously described (Karczewski et al. 2017; French et al. 2019). Samples were sequenced on the Illumina HiSeq 2500 at a read-length of 100 bp, and quality control and read alignment to the human reference genome GRCh38 were performed by Illumina as previously described (Carss et al. 2017; French et al. 2019). The average coverage obtained was 30×–40× for the nuclear genome and 800×–1000× for the mitochondrial genome. All WGS data used in this project are contained within and available through agreement with the NIHR Rare Disease Bioresource (https://bioresource.nihr.ac.uk/using-our-bioresource/our-cohorts/rare-diseases-bioresource/) and MSSNG Project (https://research.mss.ng/) databases.
Genome-wide CGG STR genotyping through ExpansionHunter
Genome-wide CGG repeat genotyping was conducted on the CRAM files, aligned to GRCh38, generated by the WGS described in the previous section. The STR genotyping algorithm ExpansionHunter (version 5.0.0) was used, developed by Dolzhenko et al. (2019). The default parameters were used, and the GRCh38 FASTA file was used for the genome reference argument. For the “-variant-catalog” argument, a custom CGG repeat catalog JSON file (Supplemental Methods S1) was used as developed and described previously by Annear et al. (2021) but was updated for the GRCh38 reference assembly. The resultant output VCF and JSON files were processed through a bioinformatic pipeline, STaRparse (https://github.com/CognitiveGenetics/STaRparse), in order to automatically parse, filter, analyze, and annotate the extracted CGG STR data. STaRparse was built using Python (3.6.8) and R (3.6.3) environments (Ihaka and Gentleman 1996; Van Rossum and Drake 2009). It is compatible with and was used in R 4.2.0 in this work (R Core Team 2022). To ensure the accuracy of repeat length predictions, loci were excluded based on sequence coverage and the presence of only flanking reads. Data were analyzed and summarized by repeat locus, genetic region, chromosome, and sample. The PyVCF (version 0.6.8) library and ANNOVAR were used by STaRparse for the parsing of the VCF data and the gene-based annotation of the CGG STR data, respectively (Wang et al. 2010).
Segregation analysis of CGG STR inheritance and mutation
The extracted CGG STR data were then put through a segregation analysis using a series of custom scripts through the bioinformatic pipeline AncSTR (https://github.com/CognitiveGenetics/AnceSTR). AncSTR compares the CGG STR length genotypes across the provided families and predicts the inheritance patterns for informative and deviating CGG repeat genotypes. Comparisons are based upon a smallest-difference-most-likely paradigm. AncSTR was built using Python (3.6.8) and R (3.6.3) environments (Ihaka and Gentleman 1996; Van Rossum and Drake 2009). It is compatible with and was used in R 4.2.0 in this work (R Core Team 2022).
Gene annotation and enrichment
Gene-based annotation of the detected CGG STR loci was performed using the software tool ANNOVAR and the refGene hg38 gene database (Wang et al. 2010). If a repeat fell outside of the gene body, it was annotated as being “upstream” or “downstream” if it was located within 1 kb of the start of the 5′ UTR or the end of the 3′ UTR of the gene in question. Otherwise, genetic regions were separated into the different functional elements: 5′ UTR, exon, intron, 3′ UTR, ncRNA, and intergenic. If a repeat was located >1 kb from a gene, it was defined as “intergenic” and was defined by the two genes, pseudogenes, or ncRNAs between which the repeat was localized. The PANTHER classification system (v17.0; Gene Ontology Phylogenetic Annotation Project) and ShinyGO (v0.75) were used to facilitate a high-throughput Gene Ontology analysis of the genetic regions identified as significantly represented within the autism-affected cohort (Ge et al. 2020; Mi et al. 2021).
Statistical comparison of CGG STRs
To compare the deviation rate and size of CGG STR repeat deviates across the different genetic regions, the data were tested for normality (Shapiro–Wilks test). The Kruskal–Wallis test was used to determine if there was a significant difference between the medians of the region groups. Further, post hoc testing was conducted using Dunn's test with the Benjamini–Hochberg procedure for P-value adjustment to compare which exact genetic regions differed. To compare the rate and degree of trinucleotide CGG tandem repeat expansions between autism-affected individuals and their unaffected siblings, we used a multiple logistic regression. Prediction of the probability of class membership (autism-affected probands = 1 vs. unaffected siblings = 0) was conducted on multiple predictor variables (genetic localization, rate of genotype deviation, and length of repeat deviation size). As we were predicting a binary outcome from a set of continuous predictor variables, the binomial link function was used. The Bonferroni correction and the Benjamini–Hochberg procedure were applied to the results of the logistic regression to correct for multiple testing. Furthermore, significant genetic regions were compared with a list of 1295 ID genes used at our center (Centre of Medical Genetics, Universitair Ziekenhuis Antwerpen, Universiteit Antwerpen) for routine screening for autism, intellectual disability, and related neurodevelopmental disease.
Data access
All ExpansionHunter CGG STR genotype prediction data are publicly available through the Dryad Digital Repository (https://doi.org/10.5061/dryad.8931zcrt3). The CGG STR catalog files (GRCh38/hg38) used in the ExpansionHunter analysis and the analysis scripts used in the study are accessible via GitHub (https://github.com/CognitiveGenetics/STaRparse and https:// github.com/CognitiveGenetics/AnceSTR). These scripts can also be found accompanying the article within Supplemental Code File S1 and Supplemental Code File S2.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the managers and curators of the MSSNG project and database (Autism Speaks) for both their cooperation and access to the whole-genome sequencing data stored within the MSSNG project database. This research was supported by the Steunfonds Marguerite-Marie Delacroix, National Institute for Health and Care Research (NIHR) Cambridge Biomedical Research Centre (BRC-1215-20014), NIHR Rare Disease Bioresource, The Rosetrees Trust, and the Isaac Newton Trust. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. We also acknowledge the support of the Research Fund of the University of Antwerp OEC-Methusalem grant “GENOMED.”
Author contributions: The study was conceptualized by R.F.K., D.J.A., and G.V. The methodology was optimized by D.J.A. and G.V. and the analyses were conducted by D.J.A. A.S.-J. and F.L.R. provided the whole genome data and assisted in the analyses. Together, D.J.A., G.V., and R.F.K. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277011.122.
-
Freely available online through the Genome Research Open Access option.
- Received June 9, 2022.
- Accepted October 14, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








