
The loss of heterochromatin is associated with multiscale three-dimensional genome reorganization and aberrant transcription during cellular senescence
- Xianglin Zhang1,2,9,
- Xuehui Liu1,2,3,9,
- Zhenhai Du4,5,6,9,
- Lei Wei1,2,
- Huan Fang1,2,
- Qiongye Dong1,2,
- Jing Niu7,
- Yanda Li1,2,
- Juntao Gao1,2,
- Michael Q. Zhang8,
- Wei Xie4,5,6 and
- Xiaowo Wang1,2
- 1MOE Key Laboratory of Bioinformatics; Center for Synthetic and Systems Biology; Department of Automation, Tsinghua University, Beijing 100084, China;
- 2Bioinformatics Division, Beijing National Research Center for Information Science and Technology, Beijing 100084, China;
- 3State Key Laboratory of Medical Molecular Biology, Department of Pathophysiology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, China;
- 4Center for Stem Cell Biology and Regenerative Medicine, MOE Key Laboratory of Bioinformatics, Tsinghua University, Beijing 100084, China;
- 5THU-PKU Center for Life Sciences, Beijing 100084, China;
- 6School of Life Sciences, Tsinghua University, Beijing 100084, China;
- 7Department of Basic Medical Sciences, School of Medicine, Tsinghua University, Beijing 100084, China;
- 8Department of Molecular and Cell Biology, Center for Systems Biology, The University of Texas, Richardson, Texas 75080-3021, USA
-
↵9 These authors contributed equally to this work.
Abstract
Heterochromatin remodeling is critical for various cell processes. In particular, the “loss of heterochromatin” phenotype in cellular senescence is associated with the process of aging and age-related disorders. Although biological processes of senescent cells, including senescence-associated heterochromatin foci (SAHF) formation, chromosome compaction, and redistribution of key proteins, have been closely associated with high-order chromatin structure, the relationship between the high-order chromatin reorganization and the loss of heterochromatin phenotype during senescence has not been fully understood. By using senescent and deep senescent fibroblasts induced by DNA damage harboring the “loss of heterochromatin” phenotype, we observed progressive 3D reorganization of heterochromatin during senescence. Facultative and constitutive heterochromatin marked by H3K27me3 and H3K9me3, respectively, show different alterations. Facultative heterochromatin tends to switch from the repressive B-compartment to the active A-compartment, whereas constitutive heterochromatin shows no significant changes at the compartment level but enhanced interactions between themselves. Both types of heterochromatin show increased chromatin accessibility and gene expression leakage during senescence. Furthermore, increased chromatin accessibility in potential CTCF binding sites accompanies the establishment of novel loops in constitutive heterochromatin. Finally, we also observed aberrant expression of repetitive elements, including LTR (long terminal repeat) and satellite classes. Overall, facultative and constitutive heterochromatin show both similar and distinct multiscale alterations in the 3D map, chromatin accessibility, and gene expression leakage. This study provides an epigenomic map of heterochromatin reorganization during senescence.
Cellular senescence is stable cell cycle arrest that can be triggered by exogenous or endogenous stresses (van Deursen 2014) and contributes to tissue remodeling and age-related diseases, including cancer (Campisi 2013; Muñoz-Espín et al. 2013; van Deursen 2014). The occurrence of cellular senescence has been causally linked to persistent DNA damage response (DDR). According to the dependence on telomere length, cellular senescence can be categorized into replicative senescence and stress-induced premature senescence (SIPS) (Fridlyanskaya et al. 2015). Replicative senescence is triggered when telomere shortening leads to persistent DNA double-strand breaks after successive cell divisions, whereas SIPS is independent of telomere erosion and can be induced by oncogenic, oxidative, and genotoxic stress. SIPS induced by overexpression of oncogenes is commonly called oncogene-induced senescence (OIS), whereas premature senescence induced by genotoxic drugs (such as bleomycin, used for anticancer chemotherapy and capable of producing DNA damage) is another type of SIPS different from OIS (Robles and Adami 1998; Bhaumik et al. 2009; Schafer et al. 2017), also employed on BJ fibroblasts in this study.
Senescent cells are characterized by profound alterations in morphology, gene expression, and chromosome organization (Criscione et al. 2016b; Gorgoulis et al. 2019). In particular, heterochromatin alterations have been characterized in different types of senescence and age-related disorders (Cruickshanks et al. 2013; De Cecco et al. 2013; Chojnowski et al. 2020), and the “global heterochromatin loss and redistribution” model has been summarized (Tsurumi and Li 2012; Sen et al. 2016; Sidler et al. 2017). In OIS, constitutive lamina-associated domains detach from the nuclear lamina and form highly condensed senescence-associated heterochromatin foci (SAHF) (Chandra et al. 2012; Chandra and Kirschner 2016; Lenain et al. 2017; Boumendil et al. 2019). However, heterochromatin becomes hypomethylated and more accessible in other senescence types (Cruickshanks et al. 2013; De Cecco et al. 2013; Lowe et al. 2015), and pericentromeric satellites repressed in normal cells distend drastically during senescence (Swanson et al. 2013; Criscione et al. 2016b; Dillinger et al. 2017). Notably, genotoxic drug-induced senescence of BJ cell lines being used in this study rarely show SAHF (Kosar et al. 2011). Furthermore, after extended periods of culture, senescent cells will enter into deep (late) senescence and show continued heterochromatin remodeling. The alterations include the release of cytoplasmic chromatin fragments (CCFs) due to the loss of Lamin B1 protein (Freund et al. 2012; Ivanov et al. 2013), the increased openness of gene poor regions, and the up-regulation of retrotransposable elements expression in heterochromatin (van Deursen 2014). Heterochromatin loss has been associated with the process of premature aging (Zhang et al. 2015; Chojnowski et al. 2020), and stabilizing heterochromatin can attenuate cellular senescence (Deng et al. 2019; Hu et al. 2020). Therefore, dissecting heterochromatin reorganization and its relationship with gene expression is critical for understanding senescence and developing antiaging methods (Sen et al. 2016).
With the development of chromosome conformation capture (3C) technology (Dekker et al. 2002), researchers have been able to investigate 3D genome folding on a genome-wide scale. The high-order structure of the mammalian genome is organized at the local (topologically associated domains [TADs] and loops) and global (interchromosome contacts, A/B compartments) levels (Zhang et al. 2018). Loops are usually mediated by CTCF and cohesin and contribute to structural maintenance and proper gene regulation (Fudenberg et al. 2016). TADs are conserved regulatory units of the 3D genome and their disruption can lead to gene dysregulation (Dixon et al. 2012; Lupiáñez et al. 2016). A/B compartments and interchromosomal contacts show interaction preferences for different genomic regions. A and B compartments comprise primarily transcriptionally active and inactive regions, respectively (Lieberman-Aiden et al. 2009). With 3C technology, especially Hi-C, researchers have begun to gain insight into the 3D genome of senescent cells.
In Hutchinson-Gilford progeria syndrome (HGPS), a type of premature aging disease, poor compartmentalization has been observed in the late passages (McCord et al. 2013). In contrast, OIS and replicative senescence maintain genome-scale A/B compartment patterns. OIS is characterized by decreased short-range interactions and increased long-range interactions in low GC content regions, which is associated with condensed SAHF formation (Chandra et al. 2015; Chiang et al. 2019), whereas replicative senescence shows a global increase in local contacts accompanied by chromosome compaction and globally decreased chromatin accessibility (Criscione et al. 2016a). Researchers also found that redistribution of proteins or protein complexes contributes to genome reorganization of senescent cells (Zirkel et al. 2018; Iwasaki et al. 2019; Olan et al. 2020). Although researchers have separately assayed heterochromatin remodeling and 3D genome reorganization in different studies, the relationship between the 3D reorganization and the “loss of heterochromatin” phenotype in senescent cells is poorly understood.
This study aims at dissecting chromosome folding alterations on different scales and its relationship with gene expression upon the loss of heterochromatin in senescent cells. To achieve this, we leveraged a SIPS model in human diploid BJ fibroblasts induced by bleomycin which exhibits the loss of heterochromatin phenotype. By carrying out multi-omics profiling including Hi-C, ChIP-seq, ATAC-seq, and RNA-seq, we characterized 3D genome reorganization, chromatin remodeling, and gene expression changes during the process of senescence, and dissected the relationship between the loss of heterochromatin phenotype and multiscale 3D genome reorganization and transcription alterations. We expect that these findings will expand our knowledge on heterochromatin reorganization during senescence.
Results
The loss of heterochromatin phenotype in bleomycin-induced senescence
A SIPS model for cellular senescence (S) and deep senescence (DS) in human diploid fibroblasts (BJ cell line) induced by bleomycin was used (Fig. 1A; Bhaumik et al. 2009). Quiescent cells (Q), induced by 0.1% fetal bovine serum (FBS) and with temporary cell cycle arrest, were used as an additional control besides normal growing cells (G). Senescent cells showed temporary up-regulation of TP53 and CDKN1A and increased senescence-associated β-galactosidase (SA-β-gal) activity (Supplemental Fig. S1A,B), which are well-characterized senescence features (Beauséjour et al. 2003; Mijit et al. 2020). Next, we performed in situ Hi-C, ATAC-seq, ChIP-seq (H3K9me3 and H3K27me3), and RNA-seq for each cell state (Fig. 1A; Supplemental Table S1). Biological replicates showed high reproducibility for these experiments (Supplemental Figs. S2–S4). Differentially expressed genes between G and S or G and DS were enriched for senescence-associated terms (Supplemental Table S2) by DAVID (Huang et al. 2009), further confirming the senescent state of S and DS.
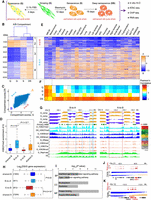
Compartment switching of H3K27me3-associated heterochromatin. (A) The study design. Each cell state has two biological replicates. (B) Hierarchical clustering of the differential compartment bins. There are four clusters, out of which clusters 1 and 2 correspond to the A-to-B category and clusters 3 and 4 correspond to the B-to-A category. (C) Scatterplot of compartment scores in G versus DS. Each point represents a 40-kbp bin, and B-to-A regions occupy a higher proportion than A-to-B regions. (D) Box plots of H3K27me3 enrichment of G for four compartment categories—shared-B, B-to-A, A-to-B, and shared-A. Shared-B and shared-A were used as a control group. B-to-A regions show significantly higher enrichment of H3K27me3. A Wilcoxon test was used to identify the statistical significance. (E) Compartment scores of 25 published Hi-C profiles in the differential compartment bins obtained from panel B. (F) Pearson's correlation between four cell states and 25 published Hi-C profiles in terms of the differential compartment bins. (G) WashU Epigenome Browser views of compartment switching. B-to-A regions are overlapped with H3K27me3 and Polycomb chromatin state, while A-to-B regions are depleted in measured histone modifications. (H) Box plots of fold changes of gene expression in four categories. The numbers of genes included in each category are labeled. Statistical analysis was performed by Wilcoxon test. (I) Gene Ontology of derepressed genes in B-to-A regions. (BP) Biological process, (MF) molecular function, (CC) cellular component. Top two terms in each annotation category are shown. (J,K) Browser views of some derepressed genes in B-to-A regions.
Western blot showed that there was a global loss of H3K9me3 (Supplemental Fig. S1A), and ChIP-seq analysis showed the loss was enriched for H3K9me3-rich regions of G (Supplemental Fig. S1C), which is consistent with the heterochromatin loss phenotype. ChIP-seq used here was not absolutely quantitative and could only reflect relative changes across different genomic regions. Notably, the genomic regions showing relative loss of H3K9me3 were accompanied by relative increased H3K27me3 (Supplemental Fig. S1D), suggesting possible compensation mechanisms as also observed for H3K27me3 and DNA methylation (Reddington et al. 2013). Next, we investigated the 3D genome alterations at both global (A/B compartment, long-range, and interchromosome interactions) and local (intra-TAD interaction and loop) levels upon heterochromatin loss.
Senescent cells lose cell type–specific compartmentalization at H3K27me3 enriched regions
First, we calculated A/B compartment scores across the whole genome and identified differential compartments by pairwise comparisons across four cell states (see Methods). Hierarchical clustering of these differential compartments showed that compartment switching was progressive from growing to senescence and further to deep senescence (Fig. 1B). G shared similar A/B compartment patterns with Q, whereas S and DS were more similar (Fig. 1B; Supplemental Fig. S2B), suggesting that compartment switching was not attributed to cell cycle arrest but instead to the senescence process. Differential compartment bins (12.5% of the whole genome) (Supplemental Table S3) were clustered into four clusters, corresponding to A-to-B regions (3.1% of the whole genome) and B-to-A regions (9.4% of the whole genome) according to the directions of compartment score changes. B-to-A regions in deep senescence occupied the majority of the differential compartment bins (75% of the differential bins) (Fig. 1C), with more peaks detected in DS by ATAC-seq performed in this study (Supplemental Fig. S5A,B). It suggested that active A compartments occupied a higher proportion of the genome of DS (Fig. 1C). In-house ATAC-seq profiles in senescent cells suggested that there was globally increased accessibility in heterochromatin, as there were more ATAC-seq peaks locating at B compartments in deep senescent cells (Supplemental Fig. S5A).
To fully understand the relationship between genomic features and the differential compartments, we annotated the differential compartments with histone modifications and ChromHMM states of growing cells from the Roadmap Epigenomics Project (Roadmap Epigenomics Consortium et al. 2015) and in-house H3K27me3 profiles across all cell states. Compared with shared-A compartment and shared-B compartment, B-to-A regions were enriched for H3K27me3 in both growing and deep senescent cells (Fig. 1D; Supplemental Fig. S5E–G) and the bivalent regulatory or repressed Polycomb ChromHMM state (Fig. 1G; Supplemental Fig. S5D), whereas A-to-B regions were depleted of any measured histone modification signals (H3K4me1, H3K4me3, H3K9me3, H3K27me3, H3K27ac, H3K36me3) (Fig. 1G; Supplemental Fig. S5D). Compared with shared-B regions, B-to-A and A-to-B regions were not enriched for H3K9me3 (Supplemental Fig. S5C), implying heterochromatin in G did not obviously alter A/B compartment structure. By comparing with other cell types or tissues in terms of the differential compartments (Barutcu et al. 2015; Schmitt et al. 2016; Wu et al. 2017), we found that G showed negative correlations to them, except for mesenchymal stem cells and IMR-90 cells, which have been reported to share similar epigenomic profiles with G (Fig. 1E,F; Roadmap Epigenomics Consortium et al. 2015). However, A/B compartment scores of DS showed positive correlation with that of other cell types or tissues (Fig. 1E,F), suggesting that the differential compartments were cell type–specific compartments. Overall, senescent cells lost cell type–specific compartmentalization at H3K27me3 enriched regions.
Moreover, we found weak but significant association between A/B compartment switching and gene expression (Fig. 1H). The derepressed genes were enriched for the interferon response (Fig. 1I; Supplemental Table S4) by DAVID (Huang et al. 2009), which has recently been defined as a novel phenotype of late senescence (De Cecco et al. 2019). For example, some interferon-stimulated genes (ISGs) located in B-to-A regions, such as MX1, OAS2, and OAS3, were derepressed during deep senescence (Fig. 1J,K). Similarly, deactivated genes residing in A-to-B regions were enriched for extracellular matrix-associated terms (Supplemental Table S4), another important feature in senescence (Mavrogonatou et al. 2019). For example, the expression of deactivated gene COL11A1, a collagen gene, declined accompanied by compartment switching (Supplemental Fig. S5H) during senescence. It suggested that compartment changes were associated with senescence-specific gene expression.
Increased long-range interactions between heterochromatin losing H3K9me3 during senescence
Although heterochromatin became more accessible during senescence, global A/B compartment organization did not change much (87.5% of the whole genome). We wondered how interactions of heterochromatin changed during senescence. We addressed this issue from two aspects—intrachromosome and interchromosome interactions. Globally, S and DS shared more similar interaction patterns but showed differences with G or Q at the chromosome arm level (Fig. 2A), suggesting that there were alterations of chromatin structure associated with cellular senescence. First, to characterize the changes of interactions across different distances, we plotted power-law decay curves (Supplemental Fig. S6A,B). Both A-A and B-B interactions showed increased local interactions and decreased long-range interactions in S and DS. More specifically, A-A interactions of <400 kbp increased, while B-B interactions of <1 Mbp increased. Notably, B-B interaction still occupied the dominant proportion of long-range interactions in S and DS. A saddle plot of A/B compartment interactions also showed this trend (Supplemental Fig. S6C). It suggested that interactions between B compartments were maintained during cellular senescence. However, power-law decay curves could not clearly reflect relative changes between A-A and B-B interactions of the same distances and the exact positions of differential interactions.
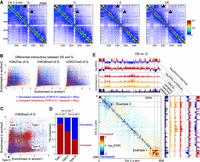
H3K9me3-associated heterochromatin of G gains long-range interactions. (A) Contact maps of the p arm of Chr 3 across four cell states. The arrows on the plot point out positions of some differences. (B) Scatterplots of histone modification (H3K27ac, H3K36me3, and H3K27me3) enrichments of G for two genomic anchors of differential interactions. Each point represents a significantly differential interaction (FDR < 0.01, distance > 1 Mbp). Red represents increased interactions in DS, and blue denotes decreased interactions. Each interaction has two genomic anchors, whose histone modification enrichments of G are shown as x and y coordinates, respectively. These plots could inform the relationship between the enrichment of histone modifications in two genomic anchors and the directions of differential interactions. (C) Scatterplots of H3K9me3 occupation of G in anchors of differential interactions. Interactions are classified into three types (Type I, Type II, and Type III). Type I represents interactions of two H3K9me3-enriched genomic regions; Type II represents interactions between H3K9me3-enriched regions and H3K9me3-depleted regions; Type III represents interactions of two H3K9me3-depleted regions. (D) The composition of significantly increased and decreased interactions for three interaction types. P-values were calculated by a χ2 test. (E) Differential interactions of the p arm of Chr 3. Examples 1 and 2 show increased interactions between heterochromatin and decreased interactions between euchromatin, respectively. The tracks of histone modifications of G and fold changes of H3K9me3 and H3K27me3 between DS and G are shown. Although Example 1 resides in pericentromeric regions, increased heterochromatin interactions also occupy other genomic regions.
To address this problem, we made use of a binomial distribution and biological replicates to call differential interactions (Dixon et al. 2012) with 200-kbp resolution contact maps between G and DS (see Methods). We annotated significantly increased or decreased interactions (FDR < 0.01, distance > 1 Mbp) with histone modifications of G to fully understand the relationship between genomic features and differential interactions (Fig. 2B,C). Compared with H3K27ac-, H3K36me3-, or H3K27me3-marked regions, H3K9me3-associated heterochromatin of G occupied a higher percentage of increased interactions with themselves (Fig. 2B–D). These increased interactions did not preferentially localize to pericentromeric regions (Supplemental Fig. S7). To further clarify the relationship between interaction changes and H3K9me3 enrichment of G, we split differential interactions into three types, representing interactions between H3K9me3-enriched regions, interactions between H3K9me3-enriched regions and H3K9me3-depleted regions, and interactions between H3K9me3-depleted regions, respectively (Fig. 2D). The compositions of increased and decreased interactions across different types (the proportions of increased interactions were 0.836, 0.635, and 0.407, respectively, in the three interaction types) further confirmed increased interactions of heterochromatin (Fig. 2D). Thus, compared with interactions between H3K9me3-depleted regions, heterochromatin regions gained long-range (1 Mbp–40 Mbp) interactions, especially with themselves. However, these heterochromatin regions did not show dramatic A/B compartmentalization changes (Supplemental Figs. S8, S9). During senescence, these heterochromatin regions tended to lose H3K9me3 and gain H3K27me3, as the increased interactions were enriched for relative decreased H3K9me3 and increased H3K27me3 (Fig. 2E; Supplemental Fig. S10).
Interchromosome interactions provide a good measurement for interaction preferences (Yaffe and Tanay 2011; Rao et al. 2014). So, we examined whether we could observe the same tendency of heterochromatin interactions on the interchromosomal level. Due to the sparsity of interchromosome interactions, we called differential interchromosome interactions at 1-Mbp resolution (Fig. 3A,B; Supplemental Fig. S11; Yaffe and Tanay 2011). Based on the numbers of significantly increased and decreased interactions (FDR < 0.01) of the same bins, we divided the 1-Mbp resolution bins into three categories—increased-, decreased-, and unchanged-interaction bins (Fig. 3C). Again, the increased-interaction bins were enriched for H3K9me3-associated heterochromatin of G, while the decreased-interaction bins were enriched for H3K27ac-, H3K36me3-, and H3K27me3-associated regions of G (Fig. 3D–G). Furthermore, differential interactions of increased- and decreased-interaction bins showed that H3K9me3-depleted regions of G tended to lose contacts with themselves, whereas H3K9me3-enriched regions of G tended to gain contacts with themselves (Fig. 3H). In line with intrachromosomal changes, these bins largely maintained A/B compartmentalization (Supplemental Fig. S12). Integrating H3K9me3 and H3K27me3 profiles in G and DS, we also found that increased interchromosome interactions were also enriched for decreased H3K9me3 and increased H3K27me3 during senescence (Fig. 3H). In summary, compared with interactions between euchromatin regions, heterochromatin regions gained interactions with themselves in terms of long-range and interchromosome interactions.
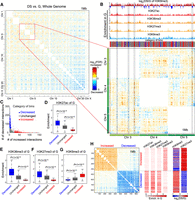
Differential interchromosome interactions. (A) Differential interactions of interchromosome between DS versus G. Intrachromosome interactions were removed before calculating differential interchromosome interactions. Colors represent −log10(FDR); red represents increased interactions, blue denotes decreased interactions. The unbalanced distribution of decreased and increased interactions in large and small chromosomes might be due to the differences of gene density and chromosome length for different chromosomes. Small chromosomes interact more frequently with each other than other chromosomes in G, and these interactions globally decrease during senescence, which was also verified by direct subtraction of contact maps (Supplemental Fig. S11). (B) Zoomed-in view of the interchromosome interactions of Chr 3, Chr 4, and Chr 5. Regions within boxes are examples of genomic regions harboring consistent decreased or increased interactions. Histone modification tracks of G, fold changes of H3K9me3 between DS and G, and bin categories were added to the top of heatmap. Increased-interaction bins were enriched for H3K9me3 of G and associated with loss of H3K9me3 in DS. (C) Scatterplot of the numbers of increased interactions and decreased interactions (FDR < 0.01) for 1-Mbp bins. Points represent 1-Mbp bins. According to the distribution of two numbers, bins were classified into three categories: increased-, decreased-, and unchanged-interaction bins. (D–G) Box plots of histone modification enrichments of G for three categories of bins. P-values were calculated with a Wilcoxon test. (H) Differential interactions within and between increased- and decreased-interaction bins. This map was generated by extracting differential interactions of the increased- and decreased-interaction bins from the whole-genome differential interaction map (panel A). Right panel shows the enrichments of histone modifications of G and fold changes of H3K9me3 and H3K27me3 between DS and G. The heatmap verifies the tendency of increased interactions between heterochromatin and decreased interactions between euchromatin. Bins showing increased interactions had relative increased H3K27me3 and decreased H3K9me3.
H3K9me3-associated heterochromatin gains local intra-TAD contacts
We have shown that DS maintained, or even gained, long-range interactions between heterochromatin; it is unclear how local interactions (intra-TAD interaction and loops) change. First, we called TAD boundaries for all cell states. Although TAD boundaries were largely conserved across all states, S and DS shared more TAD boundaries than G/Q (Fig. 4A; Supplemental Fig. S2C), implying there were local interaction changes associated with cellular senescence.
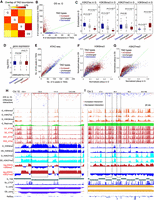
Local contact alterations during senescence. (A) The proportions of overlapped TAD boundaries between four cell states. (B) Scatterplot of the numbers of increased and decreased interactions (FDR < 0.01) for each TAD. Each point represents a TAD; x and y coordinates represent the numbers of increased and decreased interactions within the same TADs. Based on two numbers, TADs were classified into three types—increased-, decreased-, and unchanged-interactions TADs. (C) Box plots of the enrichment of histone modifications of G for the three TAD types. A Wilcoxon test was used for calculating P-values. (D) Box plots of fold changes of gene expression between DS and G. Genes whose maximum FPKM of G and DS > 1 were used. P-values were calculated by t-test. (E) Scatterplot of the ATAC-seq peak numbers in G and DS for the three TAD types. TADs are the units for counting ATAC-seq peaks. The mean ratios of DS to G for peak numbers are 1.08, 1.16, and 1.83, corresponding to decreased-, unchanged-, and increased-interaction TADs, respectively. Wilcoxon test, P = 4.00 × 10−298 for increased versus decreased; P = 6.11 × 10−271 for increased versus unchanged. (F) Scatterplot of H3K9me3 pileup in G and DS. Each point represents a 10-kbp bin and color represents the types of TADs to which it belongs. Increased-interaction TADs are enriched for relative decreased H3K9me3 than the two other TAD types. (G) Scatterplot of H3K27me3 pileup in G and DS. Increased-interaction TADs are enriched for relative increased H3K27me3, whereas decreased-interaction TADs are enriched for relative decreased H3K27me3. (H,I) WashU Epigenome Browser views of two examples of increased- and decreased-interaction TADs. Boxes in panel H shows increased interactions within TADs, which are enriched for H3K9me3 of G and novel ATAC-seq peaks in DS. The regions were also accompanied by decreased H3K9me3 and increased H3K27me3 during senescence. Panel I shows two decreased-interaction TADs showing opposite features.
To further investigate local interaction alterations during senescence, we called differential interactions with 20-kbp resolution contact maps. Due to the sparsity of long-range interactions for high-resolution maps, we only considered intra-TAD differential interactions (FDR < 0.01). We classified TADs into three categories (Supplemental Table S5) based on the ratio of increased to decreased interaction numbers within TADs: increased-, decreased-, and unchanged-interaction TADs (Fig. 4B). Associating histone modifications of growing cells with these TADs showed that increased TADs were enriched for H3K9me3 and depleted of H3K27ac, H3K36me3, and H3K27me3 (Fig. 4C,H), whereas decreased TADs showed opposite enrichments (Fig. 4C,I). However, this local interaction changes had no significant association with expression alterations for genes with FPKM > 1 (Fig. 4D).
Then, we investigated the relationship between chromatin remodeling and the local interaction changes. In-house ATAC-seq profiles showed that TADs with increased intra-TAD interactions gained more peaks than decreased or unchanged TADs in DS (the mean ratios of DS to G for peak numbers are 1.08, 1.16, and 1.83, corresponding to decreased-, unchanged-, and increased-interaction TADs, respectively; Wilcoxon test, P = 4.00 × 10−298 for increased vs. decreased; P = 6.11 × 10−271 for increased vs. unchanged; P = 4.94 × 10−48 for unchanged vs. decreased) (Fig. 4E,H,I). Excluding TADs overlapping with compartment boundaries (Beagan and Phillips-Cremins 2020) did not change this trend (Supplemental Fig. S13), and further H3K9me3 and H3K27me3 ChIP-seq across cell states showed that increased-interaction TADs were enriched for relative decreased H3K9me3 and increased H3K27me3 (Fig. 4F,G; Supplemental Fig. S14). These findings suggested that the loss of H3K9me3 and the gain of accessibility in heterochromatin were associated with increased intra-TAD interactions during cellular senescence.
Novel loops in heterochromatin are associated with increased accessibility of CTCF anchor regions
To further investigate local interaction changes in heterochromatin, we called loops by HiCCUPS (Supplemental Table S6; Supplemental Fig. S15; Durand et al. 2016). Through the comparison of loops in G and DS and annotation with histone modifications of growing cells, we found that the DS-specific loops were enriched for H3K9me3-associated heterochromatin, whereas the G-specific loops were enriched for H3K27ac- and H3K36me3-associated euchromatin (Fig. 5A). This suggested that heterochromatin in G tended to form new loops in DS, whereas euchromatin in G tended to lose loops in DS.
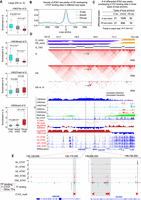
Novel looping within heterochromatin of G during senescence. (A) Box plots of histone modification enrichments of G for G-specific, DS-specific, and shared loops. The y-axis is the average enrichment of histone modifications of G for genomic regions between two loop anchors. The numbers of loops for three categories are listed in the bottom. (B) The aggregated plots of ATAC-seq peak density of DS around loop anchors. (C) The numbers of differential ATAC-seq peaks overlapping CTCF binding sites in anchors of different loop types. We called differential ATAC-seq peaks in DS versus G and calculated the numbers of peaks residing in anchors of three different loop types. DS-specific loops occupy significantly more DS-specific peaks than G-specific peaks. (D) WashU Epigenome Browser view of novel looping during senescence. Tracks include compartment scores, TAD, normalized interactions of G and DS, differential interactions between G and DS (red indicates increased interactions in DS and blue indicates decreased interactions in DS), histone modifications, loops, and ATAC-seq signals. (E) Zoomed-in view of two anchors of the novel loop. The orientations of CTCF motifs are labeled. Novel peaks overlapping potential CTCF binding sites within loop anchors are established in DS.
CTCF has been considered as a critical transcription factor for looping; thus, we examined the enrichment of CTCF binding sites for three types of loops. We found that both G-specific, DS-specific, and shared loops were enriched for ATAC-seq peaks overlapping potential CTCF binding sites (Fig. 5B), and DS-specific loops are significantly more enriched for DS-specific ATAC-seq peaks than G-specific or shared loops (Fisher's exact test, P = 7.44 × 10−7) (Fig. 5C). For example, in a heterochromatin locus around FAM135B and COL22A1 (Fig. 5D,E), loop anchors harboring potential CTCF binding sites showed drastically intensified ATAC-seq signals accompanied by novel looping in DS. This loop is absent in IMR-90 but appears in GM12878 (Supplemental Fig. S16), further verifying the formation of novel loops. Furthermore, the CTCF motif pairs of the two anchors were forward-reverse orientated (Fig. 5E; Supplemental Fig. S17), consistent with the model of loop extrusion (Guo et al. 2015; Nuebler et al. 2018). The novel loop-associated genes FAM135B and COL22A1 showed insignificant up-regulation or “leakage,” as FPKM (fragments per kilobase of exon model per million mapped fragments) of the two genes increased from <0.001 to >0.01 (adjusted P > 0.05 with DESeq2). These results suggested that increased accessibility in potential CTCF binding sites might help to form novel loops in the heterochromatin of G which loses H3K9me3 in DS.
Gene expression leakage in heterochromatin and aberrant expression of repetitive elements
Although there were increased accessibility and local interaction alteration in heterochromatin accompanied by the loss of H3K9me3, no significantly concordant global gene expression alterations were observed (Fig. 4D). We wondered whether there were alterations to the distribution of gene expression. We calculated the distribution of FPKM and found that S and DS had a significantly higher proportion of genes (Q: 24.5%, G: 25.6%, S: 30.3%, DS: 31.1%, t-test, P < 0.02) with low FPKM (0.001–1) (Fig. 6A; Supplemental Fig. S18). This observation was consistent across biological replicates and not attributed to sequencing depths (Fig. 6A). We named this phenomenon as “gene expression leakage,” which could also be observed in other senescence bulk and single-cell RNA-seq profiles (Supplemental Figs. S19, S20; Alspach et al. 2014; Hoare et al. 2016; Tang et al. 2019). To elucidate this phenomenon, we evaluated its relationship with compartment changes and local interaction alterations (Fig. 6B,C). Gene expression leakage was not specific for B-to-A switching but was highly enriched in unchanged B compartments (Fig. 6B; Supplemental Fig. S21). However, this phenomenon mainly existed in increased-interaction TADs which lost H3K9me3 and gained accessibility during senescence (Fig. 6C; Supplemental Figs. S21, S22). We proposed that increased accessibility and local interaction changes in heterochromatin were associated with gene expression leakage.
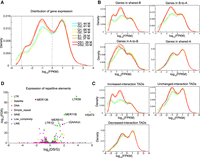
Gene expression leakage and aberrant repetitive element expression. (A) Distribution of gene expression (FPKM) for all biological replicates. Sequencing depths are labeled on the right. The sequencing depths are similar across all replicates, ruling out the effects of different sequencing depths. (B) Distribution of gene expression for different compartment categories. Gene expression leakage is enriched in shared-B and B-to-A categories. (C) Distribution of gene expression for different TAD types. Gene expression leakage is enriched in increased-interaction TADs. (D) Differential repetitive element expression during senescence. The colors represent the class to which repeat elements belong.
Because the majority of repetitive elements are located in heterochromatin, we also analyzed their expression levels with in-house RNA-seq through RepEnrich (Criscione et al. 2014). We found that retrotransposons and satellites were aberrantly expressed during senescence (Fig. 6D; Supplemental Table S7). Notably, retrotransposon LTR39 (ERV1 family, LTR class) transcripts were elevated by ∼14-fold in DS. Satellite elements, including HSATII (logFC = 5.76, FDR = 5.60 × 10−62), HSAT5 (logFC = 1.73, FDR = 1.38 × 10−19), alpha satellite (ALR/Alpha) (logFC = 0.86, FDR = 9.90 × 10−7), and simple satellite repeat (CATTC)n (logFC = 2.01, FDR = 1.80 × 10−16), showed significant up-regulation, which was also revealed in replicative senescence (De Cecco et al. 2013). Heterochromatin loss has been considered as the inducer of aberrant repetitive element expression (Andrenacci et al. 2020), which is consistent with our finding. Thus, heterochromatin loss, 3D reorganization, and aberrant transcription are interconnected to each other during senescence (Fig. 7).
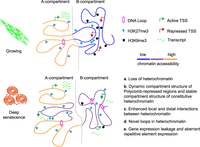
The model for heterochromatin reorganization in senescent cells. During senescence, there are multiscale reorganizations of heterochromatin. B-to-A regions are associated with H3K27me3. Compared with H3K9me3-depleted regions, H3K9me3-associated heterochromatin of G tended to increase both long and local interactions and form novel loops. These changes were accompanied with loss of H3K9me3, gain of H3K27me3, and increased chromatin accessibility. Both B-to-A regions and increased-interaction TADs showed gene expression leakage.
Discussion
We applied in situ Hi-C, ATAC-seq, ChIP-seq, and RNA-seq techniques to investigate 3D genome folding, chromatin remodeling, transcriptome alterations, and their relationship in a SIPS model without condensed SAHF but exhibiting the loss of heterochromatin phenotype. Chromatin reorganization was progressive from G to S and further to DS (Supplemental Fig. S23). Both H3K27me3- and H3K9me3-associated regions gained chromatin accessibility and showed gene expression leakage; however, they showed multiscale but different changes in the 3D genome architecture. Especially, the reorganization of heterochromatin was always associated with relative decreased H3K9me3, increased H3K27me3, and increased chromatin accessibility. This study revealed the relationship between 3D genome reorganization and the loss of heterochromatin phenotype in senescent cells.
We observed that heterochromatin lost H3K9me3 and became globally derepressed through in-house ATAC-seq, H3K9me3 ChIP-seq, and western blot of H3K9me3 (Fig. 4E,F; Supplemental Fig. S1A), consistent with the consensus for heterochromatin loss in senescent cells (Tsurumi and Li 2012). H3K9me3-associated heterochromatin showed relative enhanced long-range interactions between themselves (Figs. 2E, 3H) and retained the global A/B compartment patterns. This is in agreement with recent finding that the attraction between heterochromatic regions is essential for the phase separation of the active and inactive genome (Falk et al. 2019). However, how the genome maintains A/B compartments in DS upon the loss of heterochromatin is not clear. One hypothesis is that relatively increased occupation of H3K27me3 in heterochromatin (Polycomb repressor complex has been shown as mediator of chromatin organization) is responsible for the maintenance of A/B compartment structure (Penagos-Puig and Furlan-Magaril 2020). Although it is still unclear how heterochromatin promotes the formation of A/B compartments (Penagos-Puig and Furlan-Magaril 2020), drastic loss of H3K9me3 and globally increased accessibility in heterochromatin could not globally alter A/B compartment structure.
Heterochromatin in DS showed local interaction changes, including increased intra-TAD interactions and novel looping. These increased intra-TAD interactions were accompanied with increased chromatin accessibility, which is consistent with the previous findings (Dixon et al. 2015; Krijger et al. 2016). We further showed that increased chromatin accessibility in CTCF binding sites was associated with novel looping. However, increased intra-TAD interactions and novel loops were not associated with differential expression of highly expressed genes (Figs. 4D, 5D,E) but linked to aberrant transcription—gene expression leakage and up-regulation of repetitive elements.
Gene expression leakage discovered in this study should be a common phenomenon in senescent cells, as it could be observed when we analyzed other senescence RNA-seq profiles (Supplemental Figs. S19, S20). Because gene expression leakage was exclusively observed in heterochromatin losing H3K9me3, it might be regarded as the result of heterochromatin loss in senescent cells, similar to aberrant repetitive element expression. Only a small proportion (10.2%) of leakage genes gained new ATAC-seq peaks in their promoters (Supplemental Fig. S24), and the majority of DS-gained peaks (96.3%) are located in intergenic regions or introns, indicating gene expression leakage was not, presumably, driven by the gained openness in their promoters. Although we cannot fully rule out the possibility of heterogeneity across senescent cells contributing to the leakage phenomenon of a few genes in our bulk RNA-seq data, reanalyzing a single-cell RNA-seq data set (Tang et al. 2019) showed that the expression of leakage genes was low but widespread across SIPS cells (Supplemental Fig. S20).
Researchers have assayed the 3D genome in OIS and replicative senescence in previous studies (Chandra et al. 2015; Criscione et al. 2016a). Reanalysis of these data sets revealed that heterochromatin showed globally distinct 3D genome reorganization compared with euchromatin across different senescence types (Fig. 4B; Supplemental Fig. S25). However, heterochromatin would show different remodeling or conformation due to different stimuli, also revealed in a recent study (Sati et al. 2020). For example, heterochromatin tended to detach from nuclear lamina and form condensed SAHF in OIS, whereas heterochromatin tends to lose H3K9me3 and gain local interactions in SIPS induced by bleomycin. These studies together showed that cellular senescence was heterogeneous and was highly dependent on the stimuli (Hernandez-Segura et al. 2018). Overall, this study provided indispensable insights into the 3D genome organization in senescent cells harboring the “loss of heterochromatin” phenotype.
Methods
Cell culture and senescence assay
Human diploid BJ fibroblasts purchased from the American Type Culture Collection were cultured in DMEM supplemented with 10% fetal bovine serum and penicillin and streptomycin. Senescence was induced with bleomycin (40 μg/mL; Biorbyt) for 2 h. On the 11th day or the indicated day following treatment, the cells were assayed for senescence-associated β-galactosidase activity (Dimri et al. 1995). Each cell state had two independent biological replicates for the following experiments. Western blot was carried out to detect TP53, CDKN1A, and H3K9me3. Total proteins were extracted from cells using RIPA lysis buffer, and the concentration was determined by BCA Protein Assay Reagent. Twenty micrograms of protein were loaded for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and then transferred by electroblotting to a PVDF membrane (Millipore). The following antibodies were used: TP53 antibody (1:500, sc-126, Santa Cruz Biotechnology), CDKN1A antibody (1:500, sc-6246, Santa Cruz Biotechnology), H3K9me3 antibody (1:1000, ab8898, Abcam) and GAPDH antibody (1:5000, ab8245, Abcam).
RNA-seq and analysis
Total RNA was extracted using TRIzol (Invitrogen), and mRNA was isolated with the NEBNext Poly(A) mRNA Magnetic Isolation kit (New England Biolabs). Reverse transcription was carried out, and cDNA was fragmented, purified, and ligated with adapters. PCR was performed using an End Repair/dA-tail ligated DNA template to include unique barcodes to identify each sample. The samples were sequenced as 125-bp pair-end reads using a HiSeq 2500 instrument (Illumina) with two biological replicates. The reads were mapped to the hg19 reference genome using TopHat2 (Kim et al. 2013). Next, Cufflinks was used to estimate gene expression (FPKM) with alignments (Trapnell et al. 2010). Finally, DESeq2 was used to estimate the differential genes between two cell conditions with the count number of each gene (Love et al. 2014).
ATAC-seq and analysis
ATAC-seq was performed as described previously (Buenrostro et al. 2015). In brief, approximately 25,000 cells were harvested and then lysed in cold lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, 0.5% Igepal CA-630) for 10 min. Cells were centrifuged at 500g for 10 min at 4°C. The supernatant was discarded, and the pellet was resuspended in a 50 µL transposition mix (TruePrep DNA Library Prep kit V2 for Illumina, TD501). The transposition system was incubated for 1 h at 37°C, and transposed DNA was purified using a Zymo DNA Clean & Concentrator-5 kit. Finally, the ATAC-seq libraries were PCR-amplified for 10 cycles and then purified and sequenced on an Illumina HiSeq 2500 sequencer. ATAC-seq reads were mapped to the hg19 genome and processed using esATAC (Wei et al. 2018).
ChIP-seq and analysis
The ChIP experiment was carried out as previously described (Hawkins et al. 2010) with H3K9me3 (ab8898, Abcam) and H3K27m3 (9733s, Cell Signaling Technology) antibodies. ChIPed and input DNA were used for library preparation and then sequenced with an Illumina NovaSeq 6000 sequencer. ChIP-seq data were processed with MACS2 (Zhang et al. 2008). First, sequencing data was mapped to hg19 with Bowtie 2 (Langmead and Salzberg 2012) and BAM files were deduplicated by Picard (https://broadinstitute.github.io/picard/). Next, we used “macs2 callpeak -B ‐‐SPMR” to generate normalized a pileup signal file of “fragment pileup per million reads”. Then, in-house script was used to get normalized pileup enrichment for 10-kbp bins across the whole genome (Supplemental Code).
Hi-C library preparation
Hi-C libraries were constructed with MboI as described previously with small modifications to minimize the sample losses (Rao et al. 2014). Briefly, 1–2 million cells were fixed with 1% formaldehyde for 10 min at room temperature. Formaldehyde was quenched with glycine for 10 min at room temperature. Then, the cells were lysed and digested with MboI overnight with rotation. MboI was then inactivated for 20 min at 62°C. After filling with biotin-14-dCTP, the fragments were ligated for 5.5 h at room temperature with rotation. Then, reversal of crosslinking and DNA purification were performed. The biotin-labeled DNA was sheared with Covaris M220 and then pulled down with Dynabeads MyOne Streptavidin C1 (Invitrogen). Preparation of the sequencing library, including end repair, dATP tailing, and adaptor ligation, was performed on beads. Nine cycles of PCR amplification were performed with ExTaq (TaKaRa). Finally, size selection was performed with AMPure XP beads, and fragments ranging from 300 bp to 500 bp were selected. All libraries were sequenced on an Illumina HiSeq X Ten according to the manufacturer's instructions.
Obtaining Hi-C interaction matrices
Sequencing reads were aligned, filtered, and iteratively corrected as described previously using HiC-Pro (Servant et al. 2015). Briefly, paired-end reads were mapped to the human reference genome hg19 with default parameters. We are confident that realigning reads to hg38/GRCh38 would not alter our conclusions, as the analysis in this manuscript does not particularly focus on a specific region of the human genome or genome annotations that were updated between hg19 and hg38. After all alignment and filtering steps, the data were binned at different resolutions followed by matrix balancing. HiCRep was used to examine the reproducibility (Yang et al. 2017). Biological replicates showed high reproducibility. Therefore, for the downstream analysis, reads obtained from biological replicates were pooled except for special note. Interaction matrices and other genomic data were uploaded to the WashU Epigenome Browser for visualization (Zhou et al. 2013). DNA replication timing (wgEncodeUwRepliSeqBjWaveSignal) and CTCF binding sites (wgEncodeUwTfbsBjInputStdRaw) of the BJ cell line and TF binding regions data (wgEncodeRegTfbsClusteredV3) from the ENCODE Project (Rosenbloom et al. 2013) were downloaded from the UCSC Table Browser (Karolchik et al. 2004). Histone modifications of the BJ cell line were obtained from the Roadmap Epigenomics Project (Roadmap Epigenomics Consortium et al. 2015).
Analysis of the A/B compartment and identification of A/B compartment changes
Identification of the A and B compartments was performed similarly to previous reports with some modifications (Lieberman-Aiden et al. 2009; Dixon et al. 2015). The expected interaction matrices were calculated with normalized interaction matrices for each chromosome arm at 40-kbp resolution. We smoothed the observed matrices and expected matrices with a window of 120 kbp to generate smoothed observed/expected matrices. Then, Pearson's correlation matrices were calculated for the observed/expected matrices. Eigenvectors corresponding to the largest eigenvalue were obtained by performing PCA on the correlation matrices. However, the absolute values of the eigenvectors were biased by the size of the correlation matrices. We normalized the eigenvectors by multiplying the square root of the size of the interaction matrices. The normalized eigenvectors were called compartment scores. The interaction bins without interaction signals were removed before calculating the expected matrices. Finally, the direction of the eigenvectors was determined for the A or B compartment according to gene density.
We defined compartment changes according to changes of eigenvector signs or the absolute value of compartment score changes and found that the two methods had similar results (Supplemental Fig. S26). Here, we used the latter one to show the dynamic changes across four cell states. We set the threshold at 0.75, which was higher than the differences between the biological replicates.
Identification of TAD boundaries
TAD calling was performed by calculating the “insulation” score of each bin using 20-kbp resolution contact matrices (Crane et al. 2015). However, the insulation scores were biased by the choice of insulation square sizes. We chose different square sizes to calculate the insulation scores and call TAD boundaries. The boundaries shown under different parameters were treated as the stable boundaries, which we finally used. We introduced the false discovery rate (FDR) to set the threshold of boundary strengths. We shuffled contacts of the same distance to build a background contact matrix for each chromosome and call TAD boundaries with the same parameter. The boundaries called in shuffled maps were considered as false positives. We chose FDR = 0.05 to set the threshold. For different samples, two boundaries were treated as the same if their distance was smaller than 60 kbp (three bins).
Dynamic interaction calling
In order to confidently identify the differential interactions, we made use of the independent biological replicates and constructed the false positive background as previously reported (Dixon et al. 2012). We chose a binomial distribution to characterize dynamic interactions in two different samples. This analysis was performed on a raw interaction matrix. Differential interactions were called in both intrachromosome (20-kbp and 200-kbp resolution) and interchromosome (1-Mbp resolution) contact maps. For intrachromosome interactions, we calculated the mean contacts of different linear genomic distances (here, we used DS and G as an example). The fractions of DS contacts of the same distances were the expected probability of the binomial distribution. For a certain interaction, the number of contacts within two DS biological replicates was the observed success, and the number of contacts within G and DS biological replicates was the total trials. Then, we could calculate the probability of observing more extreme results with a binomial distribution, which was the P-value. Then, we combined replicates from different samples to construct the background and calculated the FDR. For interchromosome interactions, we just calculated one mean contact across all interchromosome interactions because of no differences of genomic distances. For 20-kbp resolution contact maps, we calculated dynamic interactions <6 Mbp. For 200-kbp resolution contact maps, we calculated dynamic interactions <40 Mbp.
Loop calling
We used HiCCUPS (Durand et al. 2016) to call loops with -r 20000 -k KR -f 0.1 -p 2 -i 8 -t 0.02, 1.5, 1.75, 2 -d 40000 with merged contacts from two replicates. We merged all loops called in G and DS and identified differential loops if loops overlapped with concordantly differential interactions (FDR < 0.001).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE133292. Scripts in calling A/B compartments, TADs, and differential interactions are available in Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank all members of the Xiaowo Wang laboratory for providing helpful advice and support for this project. The work was supported by the National Natural Science Foundation of China grants (nos. 62050152, 61773230, and 61721003 to X.W., 31830047 and 31725018 to W.X., 81890991 to J.G.), the State Key Research Development Program of China (2020YFA0906900 to X.W., 2019YFA0508900 to W.X., 2017YFA0505503 to J.G.), CAS Interdisciplinary Innovation Team (JCTD-2020-04 to J.G.), Beijing Municipal Natural Science Foundation (Z200021 to J.G.), the THU-PKU Center for Life Sciences (W.X.) and the THU-PKU Center for Life Sciences postdoctoral fellowships (Z.D.). W.X. is an HHMI international research scholar.
Author contributions: X.W., W.X., X.L., and X.Z. designed the experiments. X.L. and L.W. cultured and treated the cells. X.L. performed western blot and SA-β-gal staining. Z.D. performed Hi-C and ChIP-seq experiments. H.F. performed and analyzed ATAC-seq. X.Z. analyzed the Hi-C, RNA-seq, and ChIP-seq data. X.Z., X.L., X.W., and Z.D. wrote the manuscript, with all authors contributing to writing and providing feedback. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275235.121.
- Received January 8, 2021.
- Accepted April 27, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








