
The mode of expression divergence in Drosophila fat body is infection-specific
Abstract
Transcription is controlled by interactions of cis-acting DNA elements with diffusible trans-acting factors. Changes in cis or trans factors can drive expression divergence within and between species, and their relative prevalence can reveal the evolutionary history and pressures that drive expression variation. Previous work delineating the mode of expression divergence in animals has largely used whole-body expression measurements in one condition. Because cis-acting elements often drive expression in a subset of cell types or conditions, these measurements may not capture the complete contribution of cis-acting changes. Here, we quantify the mode of expression divergence in the Drosophila fat body, the primary immune organ, in several conditions, using two geographically distinct lines of D. melanogaster and their F1 hybrids. We measured expression in the absence of infection and in infections with Gram-negative S. marcescens or Gram-positive E. faecalis bacteria, which trigger the two primary signaling pathways in the Drosophila innate immune response. The mode of expression divergence strongly depends on the condition, with trans-acting effects dominating in response to Gram-negative infection and cis-acting effects dominating in Gram-positive and preinfection conditions. Expression divergence in several receptor proteins may underlie the infection-specific trans effects. Before infection, when the fat body has a metabolic role, there are many compensatory effects, changes in cis and trans that counteract each other to maintain expression levels. This work shows that within a single tissue, the mode of expression divergence varies between conditions and suggests that these differences reflect the diverse evolutionary histories of host–pathogen interactions.
Differences in gene expression are drivers of phenotypic divergence in closely related species (King and Wilson 1975). These expression differences can arise through sequence changes in cis-regulatory elements, such as enhancers, or in the coding regions of trans-acting factors, such as transcription factors. These two types of changes differ in their impact. Changes in cis are local, typically affecting the expression of one gene at a time, whereas changes in trans can be broad, affecting all downstream targets of a gene. The relative prevalence of each of these types of changes may give insight into how expression divergence arises in a particular setting: through the accumulation of many fine-tuning cis-acting changes, by a smaller number of large impact trans-acting changes, or by both.
The prevalence and relative contributions of cis and trans changes are being explored in various model systems (Signor and Nuzhdin 2018). For example, within individual Drosophila melanogaster lines or between Drosophila species, the contributions of cis-acting changes generally increase with phylogenetic distance, and the precise balance of cis versus trans effects depends on the phylogenetic relationships and demographics of the genotypes being compared (Wittkopp et al. 2004, 2008; McManus et al. 2010; Coolon et al. 2014; Osada et al. 2017). These studies have elucidated the mode and tempo of the changes driving expression divergence; however, most studies use whole-body measurements of expression, thus averaging signal across multiple tissues. Therefore, these studies cannot examine the prevalence of cis and trans changes in specific biological processes, which may be subject to different types of selection pressure. In addition, given that many cis-regulatory elements act in a tissue-specific manner, studies that measure cis and trans effects with tissue-specific resolution may reveal effects undetectable in heterogenous samples.
Drosophila have an innate, but not adaptative, immune response, and this response is a powerful system for measuring the contributions of cis and trans changes for several reasons. First, the immune response is inducible, with active and inactive states. This allows for the clear delineation of the transcriptional response of the immune system from that of other processes. Second, the fat body within the immune system is an optimal tissue for study. Although other tissues participate in the immune system, the fat body is a primary driver of the humoral response (Buchon et al. 2014), and it is relatively easy to isolate. Last, there is ample variation in the resistance, survival, and transcriptional response to infection between individual D. melanogaster lines (Lazzaro et al. 2004, 2006; Sackton et al. 2010; Hotson and Schneider 2015), suggesting there are many sequence changes driving these differences.
To quantify changes in cis and trans that drive transcriptional divergence in the immune response, we use allele-specific expression analysis of RNA-seq data (Wittkopp et al. 2004; Signor and Nuzhdin 2018; Frochaux et al. 2020). In this approach, we compare a gene's expression levels in two parental lines to the expression levels of each parental allele in the resulting F1 hybrids. Differences in expression owing to changes in cis, for example, a sequence change in a promoter or enhancer, will only affect the expression of the corresponding parental allele. Thus, changes in cis are independent of cellular environment and will be observed as an allelic imbalance between the parents that is maintained in the hybrids. Differences in trans, for example, a coding sequence change in a transcription factor, will affect the expression of both alleles in the F1 hybrids and thus will be observed as a differential expression in the parental lines that is not maintained in the F1 hybrids. Combining allele-specific expression analysis with RNA-seq allows us to determine the prevalence of cis and trans changes genome-wide.
When comparing the innate immune response of different D. melanogaster lines, it is not clear whether cis or trans changes will dominate. Changes in cis generally affect a single gene's expression and thus may be easily tolerated, as they only introduce small amounts of phenotypic variation. Changes in trans can affect the expression of many genes at once and efficiently introduce a large amount of phenotypic variation, but changes in trans may be harder for the organism to tolerate, as they also increase the likelihood of deleterious effects. However, the specific biology of the innate immune response may temper this expectation. Antimicrobial peptides (AMPs) are among the most highly up-regulated genes in response to infection, but the deletion of individual AMP genes often has little to no measurable effect on infection survival (Hanson et al. 2019). This suggests that to get an appreciable phenotypic effect, synchronous changes in gene expression are required, which can result from a trans-acting change. In addition, within D. melanogaster lines, trans changes are typically more prevalent (Wittkopp et al. 2008; Coolon et al. 2014). In this setting, the observation of a large number of cis-acting changes would imply that immune-responsive expression divergence is achieved through the divergence of one gene at a time, suggesting a fine-tuning process. Conversely, a preponderance of trans-acting changes would imply that expression divergence is achieved through changes in upstream factors that can simultaneously modulate the expression of many target genes.
To measure the contributions of cis- and trans-acting changes in the Drosophila innate immune response, we measured fat body gene expression in two sequenced inbred D. melanogaster lines and their F1 hybrids, in control and infection conditions. To find signaling pathway–specific effects, we separately infected the animals with either Gram-positive Enterococcus faecalis (Efae) or the Gram-negative Serratia marcescens (Smar). These bacteria have different strengths of virulence and separately trigger the two primary immune-signaling pathways in the fly. We quantified the contribution of cis and trans effects in the control and in each infection condition. This approach enabled us to examine the evolutionary changes that drive expression divergence in response to a stimulus, while minimizing the confounding effects of multiple tissue types.
Results
Two geographically distinct lines show genotype-specific immune response
To measure the relative contributions of cis- and trans-acting effects in the innate immune response, we needed two inbred, sequenced strains of D. melanogaster with abundant genetic variation and phenotypic differences in the immune response. The founder lines of the Drosophila Synthetic Population Resource fit these requirements, making them ideal candidates (King et al. 2012). To maximize the likelihood of finding variation in these lines, we selected two lines from different continents: the A4 line, also known as KSA2, collected from the Koriba Dam in South Africa, and the B6 line, collected from Ica, Peru. By using the available SNP data, we found 462,548 SNPs between A4 and B6, with about half of them falling into exonic regions, indicating that 0.9% of exonic bases varied between the genotypes, with an average of 25.3 variants per gene. The extensive variation in the coding regions allowed us to map, on average, 11.2% (±1.3%) of RNA-seq reads in an allele-specific manner.
To assess the divergence in the A4 and B6 immune responses, we measured gene expression pre- and postinfection in the abdominal fat body, the primary site of immune response. To do so, we performed RNA-seq on the dissected fat bodies of 4-d-old males from both lines that had been infected with either Gram-positive E. faecalis (Efae) or Gram-negative S. marcescens (Smar). We selected these bacteria because in D. melanogaster, Gram-positive infections generally stimulate the Toll pathway, and Gram-negative infections generally stimulate the IMD pathway, although there is additional nuance owing to signaling cross talk and the contributions of other signaling pathways (Busse et al. 2007; Lemaitre and Hoffmann 2007; Tanji et al. 2010; Buchon et al. 2014; Troha et al. 2018). We measured expression before infection and 3 h post infection to capture the early transcriptional response before the complicating effects of feedback. As a control, we performed RNA-seq on the fat bodies of uninfected, unwounded animals from each genotype (see Methods). This choice means that, compared with that of the control, the expression response observed in the infected samples includes both wound healing and infection responses.
In response to Efae infection, we found sizable genotype-specific effects in the immune response. To detect these effects, we performed two types of differential gene expression analysis: We compared control and infected samples to find Efae-responsive genes, and then within this group, we looked for genes differentially expressed between the A4 and B6 genotypes. We found 1165 differentially expressed genes between the control and infected samples regardless of genotype (Fig. 1A). We categorized these Efae-responsive genes into four groups based on their differential expression between genotypes. Group 1 genes showed no genotype-specific expression, Group 2 genes are differentially expressed only in the control samples, Group 3 genes are differentially expressed only in the infected samples, and Group 4 genes are differentially expressed in both the control and infected samples. Of the 500 Efae-responsive genes showing genotype effects, 87% (433 genes) are in Group 3, whereas only 10 genes are in Group 1 and 57 genes are in Group 4 (Fig. 1B). This indicates that many Efae-responsive genes show genotype-specific expression, and these differences are typically only revealed in response to infection.
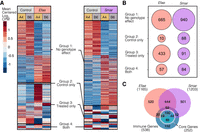
The A4 and B6 D. melanogaster lines have variation in their response to Gram-positive E. faecalis (Efae) infection and Gram-negative S. marcescens (Smar). (A) We measured expression in the fat bodies of the A4 and B6 lines infected with Efae or with Smar, 3 h post infection. We found 1165 and 1205 differentially expressed genes in response to infection to Efae and Smar, respectively, relative to control samples. Mean centered log2 average CPM values for each condition are displayed. We categorized the infection-responsive genes into four groups, based on their differential expression between the two fly genotypes: genes showing no genotype-specific expression (Group 1), genes showing genotype-specific expression only in the control condition (Group 2), genes showing genotype-specific expression only in the infected condition (Group 3), and genes showing genotype-specific expression in both the control and infected conditions (Group 4). (B) Among genes showing genotype effects, the majority of genes in Efae fell into the Group 2 classification, indicating a large amount of genotype-specific expression variation is revealed upon infection with Efae. Among Smar-responsive genes, roughly equal numbers show expression differences between the genotypes before (Group 2), after (Group 3), and both before and after infection (Group 4). (C) We intersected the genes we identified as differentially expressed in response to infection and a list of previously published immune-responsive genes. More than half of the verified immune genes were identified as differentially expressed in the abdominal fat body, with half of these immune genes being shared between conditions. Among these previously identified immune genes, core genes are differentially expressed across all infections. We detected ∼40% of the core set as differentially expressed in both our infection conditions, despite differences in the genetic background, tissue type, and time point used in our study versus previous work.
In response to the Smar infection, we found 1203 differentially expressed genes between the control and infected samples (Fig. 1A). To look for genotype-specific expression, we categorized the 1203 Smar-responsive genes into the four previously mentioned groups. For this infection, we found roughly equal numbers of genes in Groups 2–4, with 88, 91, and 84 genes, respectively (Fig. 1B). This indicates that a higher fraction of Smar-responsive genes shows genotype effects before infection than that of Efae-responsive genes (P = 1.7 × 10−11, chi-square test, Bonferroni-corrected), whereas a higher fraction of Efae-responsive genes shows genotype effects after infection (P = 9.5 × 10−67, chi-square test, Bonferroni-corrected).
To assess whether there is also phenotypic divergence on the organismal level, we performed the Efae and Smar infections and measured survival and bacterial load. In response to Efae infection, we found differences in the ability to survive infection between genotypes, with B6 surviving infection longer than A4 (Supplemental Fig. S1A). In response to Smar, we found there were no significant differences in survival, but bacterial load was lower in A4 than in B6 (Supplemental Fig. S1B,C). Together, these data show that there are differences between the two lines in their ability to resist or survive infection.
To compare our tissue-specific measurements to previous work, we intersected our Efae- and Smar-responsive genes to an existing list of immune-responsive genes. This list is an expanded version of the Drosophila immune-responsive genes set (DIRGS) and constitutes the summation of more than two decades of work in Drosophila (De Gregorio et al. 2001; Lemaitre and Hoffmann 2007; Troha et al. 2018). Of 538 genes on this list, we found more than half of these (297 genes) were identified as immune-responsive in our data (Fig. 1C). Troha and colleagues (2018) identified a subset of immune-responsive genes as core, that is, the genes that are differentially expressed regardless of the type of bacterial infection. Of these 252 core genes, ∼40% were found to be both Smar- and Efae-responsive in our data. Therefore, despite differences in the genetic background, tissue (previous studies were typically performed with whole-body sampling), and time points, our findings show concordance with previous studies of gene expression in response to infection. We also show that the A4 and B6 lines have divergence in immune-responsive expression, making them suitable for subsequent F1 hybrid experiments.
Cis-acting effects dominate expression variation in the uninfected fat body
To effectively quantify cis and trans effects, we needed to accurately analyze the allelic expression in F1 hybrids. By using the allele-specific alignment pipeline (ASAP) (F Krueger, https://www.bioinformatics.babraham.ac.uk/projects/ASAP/), we quantified allele-specific expression in our samples. Because we are working with males, we were able to use the fraction of misassigned X Chromosome reads as a metric of our pipeline's accuracy (Supplemental Methods). On average, 0.5% of X Chromosome reads were misassigned (SD = 3%) (Supplemental Table S1). The consistent, low level of misassigned reads verifies our ability to accurately quantify allelic expression.
We next sought to quantify cis and trans effects in the control samples. We used the complete set of parental RNA-seq reads and the subset of the F1 hybrid reads that could be assigned to a specific allele. By using three separate generalized linear models, we tested for differential expression in the parents, allelic imbalance in the F1 hybrids, and trans effects between the parents and F1 hybrids (see Methods) (Davidson and Balakrishnan 2016; Osada et al. 2017; Takada et al. 2017). We then categorized each gene into one of six categories (Fig. 2A). Genes showing no differential expression in the parents or F1 hybrids are conserved. Genes showing differential expression in both the parents and F1 hybrids and no trans signal are cis-only. Genes showing differential expression in the parents and not the F1 hybrids are trans-only. Some genes show evidence of both cis and trans effects and are either compensatory (if the changes on expression are in opposite directions) or cis+trans (if the changes on expression are coherent). Genes that do not fall into any of these categories are undetermined.
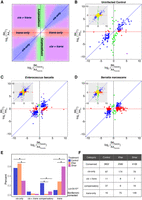
The relative contributions of cis and trans effects to expression divergence are condition specific. (A) Here we show a schematic of the expected locations for genes falling into four classifications of causes of expression divergence in plots that show the expression ratio of a gene in the parental lines (x-axis) against the allele expression ratio in the F1 hybrids (y-axis). (B) In the uninfected control condition, of 4960 genes that could be detected in an allele-specific manner, 153 genes showed cis or trans signal. Of these 153 genes, most showed cis-acting effects. Panel F displays the precise numbers of genes in each category. (C) In response to Efae infection, expression divergence is driven predominantly by changes in cis. (D) In response to Smar infection, expression divergence is dominated by changes in trans. (E) We compared the fraction of genes categorized into each divergence class in the three conditions and found that the modes of expression divergence were condition specific.
Of the 4959 genes that were expressed in the preinfection fat body and could be detected in an allele-specific manner, 77% were conserved (3802 genes) (Fig. 2B,F). We found 151 genes showing unambiguous cis or trans effects. Among these 151 genes, cis effects dominated the signal: 90% of genes (135 genes) showed cis signal (including cis-only, cis + trans, and compensatory genes), and 57% (87 genes) showed cis-only effects. Forty-two percent of genes (64 genes) showed trans signal, and only 10% of genes (16 genes) showed trans-only effects. One-quarter of genes (37 genes) were compensatory, even when using an experimental design to avoid the artificial inflation of compensatory signal (Methods) (Fraser 2019; Zhang and Emerson 2019). Additionally, to ensure that any differences in the quality of our in-house A4 and B6 transcriptomes do not affect our conclusions, we quantified cis and trans effects using sets of high-confidence genes at multiple levels of stringency and found that this had negligible effects on the detected signal (Methods) (Supplemental Fig. S2; Supplemental Table S2). From these data, we can conclude that in the unstimulated state, most genes have conserved expression levels in the fat body, and among those genes that diverge, cis effects dominate, with a sizable number of genes showing compensatory cis and trans changes.
More cis than trans effects are found in Efae-infected fat body expression
We quantified cis and trans effects in Efae-infected samples following the same methodology. We found ∼52% of genes (2580 genes) are conserved, and 379 genes showed unambiguous cis or trans effects (Fig. 2C). To identify genes whose expression divergence is specific to the immune response, we eliminated genes that show cis or trans signal in the control sample. After this filtering, ∼69% of the genes showing cis or trans effects (263 genes) remained; 66% of these genes (174 genes) show cis-only signal, and 28% (75 genes) show trans-only signal. Only eight genes (3%) show concordant cis + trans effects, and only six genes show compensatory effects. Of the genes that show cis-only signal, roughly even numbers of genes show higher expression in each genotype, consistent with the idea that cis-acting changes affect a single gene at a time. In contrast, of the genes showing trans-only signal, nearly twice as many were expressed more highly in the B6 genotype (48 genes) than in the A4 genotype (27 genes; P = 0.0105, chi-square test). This suggests that one or a few changes in upstream regulatory factors are responsible for this observation, and below, we identify candidate genes. Because we do not observe this trend toward higher B6 expression in the control samples and have removed genes that showed any evidence of mapping bias (Methods), we are confident this trend reflects true biological differences in the immune response. In sum, we find both cis and trans effects drive Efae-responsive expression divergence, with cis effects dominating.
Trans effects dominate expression variation in the Smar-infected fat body
Last, we quantified cis and trans effects in response to Smar infection. We found ∼82% of genes (4106 genes) are conserved, and 355 genes showed unambiguous cis or trans signal (Fig. 2D). We again filtered out genes that show cis or trans effects in the control samples and were left with 251 genes that have immune-specific signal. Of these, 31% (79 genes) showed cis-only signal, and roughly equal numbers of cis-only genes showed higher expression in each genotype. Seven genes showed cis + trans effects, and 16 genes had compensatory signal. Fifty-nine percent of genes (149 genes) showed trans-only signal. Within trans-only genes, we found that 71% (106 genes) showed greater expression in B6. In summary, in response to Smar infection, trans effects drive the majority of expression divergence between the two genotypes, and few genes show compensatory effects.
Comparisons of cis and trans signals in different conditions reveal both infection-specific and shared divergence
To systematically assess modes of expression variance under different conditions, we compared the proportion of genes falling into the different categories (Fig. 2E). The control and Efae-infected samples had a greater proportion of cis-only genes than the Smar samples (control vs. Smar P = 4.0 × 10−6, Efae vs. Smar P = 6.8 × 10−14, chi-square test, Bonferroni-corrected). All three groups differ in the proportion of trans-only genes, with Smar-infected samples showing the highest proportion of genes with trans-only signal, followed by Efae, and then the control samples (control vs. Efae P = 3.5 × 10−4, control vs. Smar P < 1.5 × 10−16, Efae vs. Smar P = 3.1 × 10−11, chi-square test, Bonferroni-corrected). We also found that the uninfected fat body showed significantly more compensatory signal than either infected sample (control vs. Efae P < 1.5 × 10−16, control vs. Smar P = 1.8 × 10−6, chi-square test, Bonferroni-corrected). Taken together, this suggests one of two possibilities. One possibility is that before infection, when the fat body is carrying out its metabolic functions, there is less pressure for expression divergence. An alternative interpretation is that immune-responsive genes are more tolerant of expression divergence and subject to less pressure to maintain expression levels. In response to infection, there is ample expression divergence, which is driven by both cis and trans effects. The extent to which each type of effect contributes is dependent on the particular pathogen, suggesting that the relative importance of local and pleiotropic changes is specific to different infection pressures.
Although we generally expect the two infections to regulate gene expression via distinct signaling pathways, we also anticipated some genes would be regulated in both infections, owing either to cross talk between the IMD and Toll pathways (Busse et al. 2007; Tanji et al. 2010) or to more general infection and wound responses. We found 86 genes with unambiguous cis and/or trans signal in response to both Efae and Smar infection (Supplemental Data S1). Of these genes, 71 showed concordant classification. Therefore, in the majority of genes shared between these two infections, the same genetic differences are likely driving the expression divergence in both infection conditions.
Differential expression of detection genes is a likely source for genotype expression bias in observed trans effects
Because we observed that genes with trans-only effects tended to be more highly expressed in B6 than in A4 in both infection conditions, we hypothesized that changes in a handful of upstream immune factors are responsible for this phenomenon. The changes in upstream regulators could either be infection specific or shared. Out of 202 genes showing trans-only signal in either infection, only 17 genes were shared, indicating that the bulk of trans-acting changes are likely infection specific.
Immune-detection genes, immune-signaling genes, or transcription factors differentially expressed between genotypes are likely sources of trans-acting changes, because these genes have the ability to affect the expression of many downstream targets. We posited that these genotype-specific differences had to be present in the control to have the effects at the 3-h post-infection timepoint. Of the 295 genes that are differentially expressed between genotypes in the control samples, we found 22 genes that are prime candidates, which we will refer to as trans-source candidates (Table 1).
Transcription factors and immune genes identified as potential sources of trans effects in infection
Five peptidoglycan recognition protein (PGRP) genes are potential mediators of the large number of trans effects observed in the Smar infection. Four of these PGRPs (PGRP-SC1a, PGRP-SC1b, PGRP-SC2, PGRP-LB) are negative regulators of the IMD response, and the last gene, PGRP-SD is a positive Toll and IMD regulator (Bischoff et al. 2006; Zaidman-Rémy et al. 2006; Iatsenko et al. 2016; Charroux et al. 2018; Lu et al. 2020). All of the negative regulators, PGRP-SC1a, PGRP-SC1b, PGRP-SC2, PGRP-LB, are more highly expressed in A4. Given that these are negative regulators of the IMD pathway, this finding is congruent with the observation that genes showing a trans-only signal tend to show greater expression in B6. PGRP-SD is more highly expressed in A4, and given its role as a positive regulator of the IMD response, it is not consistent with the trend of a higher B6 expression of genes showing trans-only signal. Because the four negative regulators are more highly expressed in A4, it is possible this balance between negative and positive IMD regulators can account for the expression trend seen in Smar trans-only genes.
Although there were fewer trans effects in the Efae-infected samples than in the Smar-infected samples, the pattern wherein most trans-only genes showed greater expression in B6 than A4 was maintained. Of the 22 trans-source candidates, we found two Toll-specific genes: Spatzle-Processing Enzyme (SPE) and spatzle (spz), which are both more highly expressed in A4. spz encodes the Toll receptor ligand and SPE is required to generate the active form of Spz, so differential expression of these genes can drive a large number of downstream changes. In addition, the PGRP-SD protein can act as a positive regulator of both the Toll and IMD responses and is also found to have higher expression in the A4 line. Given these observations, it is likely that other sources are responsible for the trans effects observed in Smar infections.
In addition to differences in expression between genotypes, function-altering differences in the coding sequences of immune genes may also be the source of trans-acting changes. As a first approach, we analyzed the coding sequence differences between A4 and B6 in the 22 trans-source candidates identified above using the Ensembl Variant Effect Predictor (VEP) (McLaren et al. 2016). There are a number of nonsynonymous changes, some of which fall into functional domains (Supplemental Fig. S3; Supplemental Tables S3, S4). Predicting the effect of these mutations on individual protein function, however, remains a challenge.
As an alternative approach, we analyzed the proportions of synonymous to nonsynonymous coding changes between A4 and B6 in several larger gene sets. Previous work has shown that immune-related genes have a higher average rate of adaptive evolution than other gene classes (Sackton et al. 2007; Obbard et al. 2009). We wanted to see if, for our particular genotypes and genes of interest, the same held true. We considered all genes expressed in the fat body above a threshold of one count per million (CPM) and then sorted them into two groups: genes that are differentially expressed in response to either or both infections (DE infection) and those that are not (fat body detected). We then intersected each of these gene lists with our curated immune-responsive gene set to generate both a list of differentially and nondifferentially expressed immune genes (DE immune and non-DE immune, respectively) (Fig. 3A). We posited that, given the large number of trans effects in response to infection, differentially expressed immune-related genes may have a greater proportion of nonsynonymous changes compared with the fat body–detected gene set. We found that DE immune genes have a significantly higher fraction of nonsynonymous sequence changes (24%) compared with the fat body–detected genes (21%) (P = 0.01, chi-square test, Bonferroni-corrected), suggesting that some of these changes may be under selection and possibly the source of our trans-acting signal (Fig. 3A,B). In comparison, the non-DE immune genes had a lower proportion of nonsynonymous changes (19%, P = 1.6 × 10−4, chi-square test, Bonferroni-corrected), suggesting that the elevated rate of nonsynonymous changes in DE immune genes is not simply reflective of their immune status. In summary, we find that differentially expressed immune genes have a larger proportion of nonsynonymous changes between our genomes of interest than did fat body–detected or nondifferentially expressed immune genes. Some of these nonsynonymous changes may be capable of altering the function of these proteins and therefore drive expression divergence of downstream genes in a trans-acting fashion.
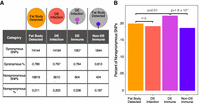
There is a greater proportion of nonsynonymous SNPs in previously identified immune-responsive genes. (A) To look for the prevalence of nonsynonymous SNPs in our genotypes and genes of interest, we defined four gene sets. Among genes detected in the fat body samples, we separated genes into those that were differentially expressed in response to either infection (DE infection) and those that were not (fat body detected). Within the fat body–detected genes, we defined previously identified immune genes showing no differential expression in response to infection (non-DE immune), and among the DE infection genes, we refined the gene list to include previously identified immune genes (DE immune). The numbers indicate the total number of SNPs found in each gene set and the percentages of synonymous and nonsynonymous SNPs. (B) DE immune genes have a higher proportion of nonsynonymous SNPs than the fat body–expressed genes, which suggests they may carry function-altering SNPs at a higher rate than the fat body–expressed genes. P-values are Bonferroni-corrected from chi-square tests with the proportion of nonsynonymous SNPS relative to the fat body–expressed gene set.
Genes with cis effects have greater transcription factor binding site divergence than genes with trans effects
The above analysis sought to identify changes in expression or protein sequence that may drive the observed trans effects; cis-acting changes also drive expression divergence of a large number of genes. These changes encompass mutations in several types of DNA features, including promoters, enhancers, and untranslated regions. We analyzed the patterns of divergence in immune-responsive transcription factor binding sites (TFBSs) to see if they were consistent with our delineation of cis- and trans-acting effects. We hypothesized that genes whose divergence was owing to cis-acting effects would show more divergence in the associated TFBSs than those without them.
We scanned potential regulatory regions of our genes of interest for TFBSs in the A4 and B6 genomes. There are relatively few characterized immune-responsive enhancers in the fat body, so instead we extracted 1-kb regions upstream of the transcription start site of genes showing any cis- or trans-acting changes in infected conditions. We searched these regions for binding sites corresponding to four known immune-responsive transcription factors: dorsal (Dl), Relish (Rel), serpent (Srp), and CrebA (Shazman et al. 2014). CrebA modulates transcription in response to both Gram-positive and Gram-negative bacteria (Troha et al. 2018). Srp binding sites have been previously used to identify immune-responsive enhancers (Senger et al. 2004). Relish is a NF-kB transcription factor downstream from the IMD pathway, and Dl and its paralog Dorsal-related immunity factor (Dif) are downstream from the Toll signaling pathway. For this analysis, however, only Dl was considered because Dif homodimers have fewer specific binding preferences than Dl and because Dif/Rel heterodimers bind sequences similar to Rel homodimers (Senger et al. 2004). Given the cross talk between the Toll and IMD pathways, we searched both Efae- and Smar-responsive genes for both Dl and Rel binding sites. For each gene, we calculated the difference in the total number of TFBSs in the A4 and B6 genomes. We then compared the genotype differences between genes showing any cis effects and genes showing exclusively trans effects (see Methods). We hypothesized that genes showing cis effects would have more differences in TFBSs than the trans-effected genes, which would be observed as a broader distribution in TFBS differences.
For all transcription factors except Dl (Fig. 4A–E), the genes with cis effects did indeed show a broader distribution of difference than those with trans effects (all TFs: P = 8.8 × 10−13; Rel: P = 2.9 × 10−2; Srp: P = 7.1 × 10−10; CrebA: P = 1.5 × 10−7; F-test to compare distribution variances, Bonferroni-corrected). Although most genes do not differ in TFBS numbers, 22% of genes with cis changes differed, as opposed to only 18% of trans-affected genes, but this difference was not significant (Fig. 4F). As the number of characterized immune-responsive enhancers and transcription factors increases, we will be able to refine this analysis to more accurately identify potential causative mutations of cis effects.
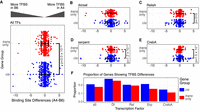
There are greater differences in TFBSs in cis-affected genes than trans-affected genes. (A) We identified TFBSs for the four immune-responsive transcription factors Dl, Rel, Srp, and CrebA in a 1-kb region upstream of 219 cis-affected genes and 199 trans-affected genes. Differences in total TFBS numbers between genotypes were calculated for each gene and plotted. We find that variance in the distribution of these differences is significantly greater in genes showing cis effects (F-test to compare distribution variances, Bonferroni-corrected). (B) For Dl TFBSs, there was not a significant difference in the width of the TFBS distribution between genes showing cis effects and trans effects. (C–E) For Rel, Srp, and CrebA TFBSs, there was a broader distribution of TFBS differences in genes with cis effects than in genes with trans effects. (F) A larger proportion of genes showing cis effects had a difference in total TFBSs than did genes showing trans effects, although the differences in these proportions were not significant.
Discussion
Here, we quantified the mode and extent of expression divergence in the Drosophila abdominal fat body, both in an uninfected control condition, in which it carries out a variety of metabolic roles, and in response to two types of infection. We found that two geographically isolated lines of D. melanogaster are phenotypically distinct in their immune responses, differing on both the organismal and transcriptional levels. By comparing gene expression in the fat body between these lines and their F1 hybrids, we quantified the contributions of cis and trans effects to expression divergence in the uninfected control, Efae-infected, and Smar-infected conditions. Both the control and Efae-infection conditions were dominated by cis effects, whereas the Smar-infection condition had an abundance of trans effects. The uninfected control also showed a greater proportion of compensatory effects, suggesting that there is stabilizing selection to maintain fat body expression levels of certain genes in the absence of an infection. Among the genes showing changes in trans, we found that expression of the B6 allele is typically higher, and we identified expression divergence in a group of proteins that may drive these trans effects. By analyzing the TFBS content of upstream regions of genes, we found that genes with cis effects show evidence of more TFBS divergence than do genes with trans effects. Overall, we find that the mode of evolution in expression divergence can vary between conditions in a single tissue and likely represents condition-specific selection pressures.
Our unique approach to measuring the mode of expression divergence gave rise to several novel observations about the relative contributions of cis and trans effects on expression variation. Although there have been a number of studies aimed at disentangling the contribution of cis and trans changes to gene expression in Drosophila, few have sought to answer this question using a single organ or with different physiological stimuli (Wittkopp et al. 2004, 2008; McManus et al. 2010; Coolon et al. 2014; Osada et al. 2017). Our approach allows us to examine evolutionary changes in response to perturbation while minimizing the confounding effects of multiple tissue types. A previous study by Juneja et al. (2016) found, among geographically distinct flies, a large number of cis-acting changes that cause whole-body expression divergence in response to an infection with mixture of bacteria. This is concordant with our finding of a large number of cis-acting changes in both infection conditions, but this study did not quantify trans-acting changes or distinguish between Toll- and IMD-specific responses. By measuring expression in the heads and abdomens of multiple D. melanogaster lines, another group reported the predominance of changes in cis over those in trans but did not measure these differences in different physiological states or attempt to dissect individual tissues in the head or abdomen (Osada et al. 2017). Most recently, two studies sought to uncover the underlying genetics of resistance to either Pseudomonas entomophila or E. faecalis infection, and each identified novel drivers of phenotypic divergence (Chapman et al. 2020; Frochaux et al. 2020). Here, we sought to directly assess the contribution of cis and trans sequence changes in a single tissue in the context of multiple treatment conditions, giving a unique high-resolution view of the evolutionary sequence changes underlying expression divergence.
With our approach, we were able to uncover two key trends. First, we found that compensatory mutations were more frequent in the control samples than in either of the infected conditions. Previous studies in several organisms had suggested that compensatory effects were very prevalent (McManus et al. 2010; Goncalves et al. 2012; Schaefke et al. 2013; Coolon et al. 2014). However, certain choices in experimental design can inflate estimates of compensatory effects (Fraser 2019; Zhang and Emerson 2019). Our study avoids this artifact and therefore yields a more accurate estimate of compensatory effects across multiple conditions. Additionally, a large proportion of studies addressing cis and trans effects in animals do so in “control” conditions, which may not reveal the full extent of selection forces that act on gene expression (Goncalves et al. 2012; Davidson and Balakrishnan 2016; Osada et al. 2017; Signor and Nuzhdin 2018). We find evidence that the genes involved in the maintenance of basic metabolic functions of the uninfected fat body are under different selective pressures than those involved in immune response. Unlike the immune-responsive genes, which must contend with a continuously evolving pathogen landscape, the genes carrying out metabolic functions may be subject to stabilizing selection, given relatively unchanging nutritional availability. In future studies, it will be interesting to further probe which systems and conditions show enrichment for these different patterns of expression divergence.
Second, we observe that the relative contribution of cis- and trans-acting changes are perturbation specific. In response to Efae infection, cis effects dominate expression changes, whereas in the Smar infection, trans changes are predominant. The prevalence of either cis or trans effects can be reasonably justified in our system, but we did not anticipate that the proportion of these effects would be infection specific. Because changes in trans factors have pleiotropic effects, it has been suggested that changes to these factors are under more selective constraint than are cis-acting elements, and thus, cis effects can more readily introduce small-scale variation into a system (Schaefke et al. 2013). In some cases, however, arriving at a more fit phenotype may require the coordinated alteration of expression of many genes, which may be more readily achieved by changes to trans-acting factors. In our D. melanogaster lines, S. marcescens is more virulent than E. faecalis: A higher dose of E. faecalis is needed to achieve similar levels of mortality to that of S. marcescens (Supplemental Fig. S1). It is possible that adaptation to highly virulent pathogens or rapidly evolving pathogens requires large-scale, synchronous changes to expression, whereas adaptation to less virulent pathogens is possible with smaller, localized mutations. Experiments with a wider range of pathogens, particularly those that trigger the same signaling pathway, will further illuminate the relationship between the mode of expression divergence and the host–pathogen relationship. In addition, expansion of the study to more D. melanogaster genotypes or to other time points will yield a more complete picture of the modes of expression divergence in the immune response.
In summary, we find that the mode of expression divergence, as represented by the proportion of cis and trans effects in a system, is condition specific in the D. melanogaster abdominal fat body. This specificity may be a result of the distinct selective pressures that different host–pathogen interactions exert on the D. melanogaster immune system. In the course of our study, we found several candidate genes that may be the sources of the observed trans effects, which are most prominent in Smar infection. In the future, we can combine the data sets presented here with other types of functional genomics experiments to identify the specific sequence changes that drive cis-acting divergence. Taken together, these studies will provide a more comprehensive view of how regulation of expression in this rapidly changing system is wired and evolves.
Methods
Animal genotypes, infection protocols, and survival analysis
The A4 and B6 D. melanogaster lines, SNP tables, and genomic reads were received from the Drosophila Synthetic Population Resource (King et al. 2012). Flies were reared at 25°C on standard cornmeal fly food (Brent and Oster 1974). For all RNA-seq experiments 4-d-old males were infected with ∼15 nL of A600 = 0.5 OD solution of either E. faecalis or S. marcescens via microinjection, yielding an infection of ∼10,000 CFU per fly (Khalil et al. 2015). Survival and bacterial load experiments were performed using a modified infection protocol (Supplemental Methods). Uninfected controls were placed on a carbon dioxide pad for 6 min to mimic the effects of anesthesia used for microinjection. Bacteria were grown in liquid culture on a shaker overnight at 37°C and then diluted 1:1000 in fresh media in the morning. Cultures were grown until exponential phase and then pelleted down and resuspended in PBS for OD measurement and injection. Injections took place between 3:00 p.m. and 5:00 p.m. to account for the impact of circadian rhythm on immune response (Scheiermann et al. 2013).
To determine the number of unique SNPs between A4 and B6, we downloaded published SNP tables from the DSPR website (King et al. 2012). We selected SNPs that were not shared between lines and that also showed a reference allele frequency of <0.05. We then calculated total SNP differences for exonic and nonexonic regions using exon coordinates from FlyBase (dm6/iso-1: FB2019_01) (Thurmond et al. 2019).
Preparation and sequencing of RNA-seq libraries
For sequencing experiments, abdominal filets with the attached fat bodies were prepared as previously described (Krupp and Levine 2010) 3 h post infection. Three fat bodies per sample were suspended in TRIzol on ice (Invitrogen) and immediately stored at −80°C for later extraction (Kono et al. 2016). To mitigate the impact of batch effects, injections and RNA extractions were performed in groupings of six to eight samples, with at least two treatment conditions and two genotypes (A4, B6, A4B6, or B6A4) represented in each batch. A minimum of three biological replicates were collected for each treatment condition/genotype combination. Both the order of treatment and the order of RNA extraction were randomized for each batch. RNA was extracted using Zymo Research Direct-zol RNA extraction kits. Library construction was completed by protocol outlined by Serra et al. (2018). Samples were then sequenced on a Illumina NextSeq platform with a NextSeq 500/550 high output kit v2.5 to generate 43-bp paired-end reads. Data were imported to the UCI high performance computational cluster for trimming and mapping of sequenced reads.
Differential expression analysis
Reads were trimmed and filtered using Trimmomatic 0.35 (Bolger et al. 2014), specifying the parameters ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:6 SLIDINGWINDOW:4:15 MINLEN:30. Count and TPM data for each sample were then calculated using Salmon 0.12.0 aligner (Patro et al. 2017) using the dm6/iso-1 transcriptome and the parameters -l A ‐‐validateMappings. Count matrices of gene-level data were then constructed in R using the Tximport 1.12.3 package (Soneson et al. 2015). To find genes either differentially expressed in response to each infection, compared with control, or differentially expressed between genotypes, we used the edgeR 3.26.5 package (Robinson et al. 2010; McCarthy et al. 2012). For this analysis, we excluded lowly expressed genes (CPM < 1), accounted for extraction batch in our model, and corrected P-values with false-discovery rate (FDR) (Benjamini and Yekutieli 2001). Genes with an FDR < 0.05 were considered differentially expressed. Additionally, we assessed the potential effect of absolute expression on our ability to call genotype effects, and we did not find any significant sources of bias (Supplemental Fig. S5). Code and accompanying files related to this section are in Supplemental Code as both R-notebooks and HTML documents (Script1_fig1).
Generation of A4 and B6 transcriptome annotations
To map RNA-seq reads in an allele-specific manner, we created two reference transcriptomes by lifting over iso-1 genome annotations to sequenced A4 and B6 genomes. By use of the UCSC liftOver suite, custom chain files were created by mapping iso-1 homologous sequences to the A4 or B6 genome using BLAT (parameters -tileSize = 12 -minScore = 100 -minIdentity = 98) (Salinas et al. 2016). A subset of 7654 high-confidence genes were used for the subsequent analysis (Supplemental Methods).
Allele-specific expression analysis
RNA reads were assigned parental alleles using ASAP (F Krueger, https://www.bioinformatics.babraham.ac.uk/projects/ASAP/) using the A4 and B6 genomes and allowing for no mismatches. Nonuniquely assignable reads were discarded. Count and TPM data were then generated by aligning allelic reads to the corresponding transcriptome. Count matrices of gene-level data were then constructed in R using the Tximport 1.12.3 package (Soneson et al. 2015).
To characterize expression divergence into cis and trans categories, differential expression was determined with unparsed parental reads and allele-specific reads from the F1 hybrids, using edgeR and three distinct GLM structures. Lowly expressed genes (CPM < 1) and X Chromosome genes were excluded from the analysis. For each condition, we first tested for differential gene expression between parental samples (Murad et al. 2019). Next, we tested for allelic imbalance, taking into account parent of origin and maternal genotype effects as outlined previously (Osada et al. 2017; Takada et al. 2017). For this test, we used half of the F1 hybrid samples. Finally, we tested for trans effects using parental samples and the remaining F1 hybrid samples (Supplemental Code: Script2_fig2.rmd section 4; J Coolon, pers. comm.). In all three tests, we assigned significance after adjusted P-values for multiple comparisons using the FDR method (Benjamini and Yekutieli 2001). By using the results from each test, we categorized each gene into one of five classes using the logic outlined in Table 2, which is based on previous studies (Emerson and Li 2010; McManus et al. 2010). Any genes that did not fit into the described patterns were categorized as “undetermined” and were excluded from further analysis. A complete list of genes and their categories for each condition is available in the Supplemental Data S1. The code and accompanying files related to this section are available in the Supplemental Code in both R-notebook and HTML document form (Script2_fig2).
Logic for cis and trans effect gene categories
Identification of sources of trans effects
To investigate potential sources of observed trans effects, we looked for genes differentially expressed in uninfected samples. We selected genes that show differential expression between A4 and B6 in uninfected samples. These genes were then intersected with a list of known Drosophila transcription factors, as well as known immune genes (De Gregorio et al. 2001; Lemaitre and Hoffmann 2007; Hammonds et al. 2013; Troha et al. 2018). Only genes that were transcription factors, immune-detection genes, or immune-signaling genes were considered to be candidates.
Analysis of SNPs in coding sequences
To better understand the effects of sequence changes on coding regions between our lines, we used the Ensembl VEP to predict the effects of SNPs on the resulting amino acid sequence (McLaren et al. 2016). The fat body–expressed gene set consists of genes expressed in the unstimulated fat body above a CPM of one and excludes genes differentially expressed in response to infection. DE infection genes are those differentially expressed in response to infection with either Efae or Smar. DE immune genes are those differentially expressed genes that are also previously verified immune-response genes, and non-DE immune genes are previously verified immune genes in the fat body expression gene set. Unless otherwise stated, figures were generated using ggplot2 3.3.2 package in R 3.6.0 (Wickham 2016; R Core Team 2019). Code and accompanying files related to this section are available in the Supplemental Code in both R-notebook and HTML document form (Script3_figure3).
Analysis of TFBS variation
To investigate the effects of noncoding sequence changes on observed expression divergence, we identified differences in TFBSs in potential cis elements of genes showing evidence of expression divergence. We selected 1-kb regions upstream of the transcription start site of genes showing cis or trans effects in response to infection (421 genes). TFBSs for these regions were passed through the tool for finding individual motif occurrences (FIMO) from the MEME suite (v 5.1.0) at P-value thresholds of either P = 0.001 or P = 0.0001 and at default parameters (Bailey et al. 2009). MEME motif files were generated using the sites2meme utility and TFBS sequences from OnTheFly (Shazman et al. 2014). Binding site data were downloaded into R 3.6.0 for analysis and plotting (R Core Team 2019). Binding sites with a P-value < 0.001 were considered in downstream analysis. This threshold was selected based on the ability to call a majority of previously identified Rel and Srp binding sites in four immune-responsive enhancers (Senger et al. 2004; Supplemental Table S5). For comparison, we categorized genes into two groups: cis genes or trans genes. Cis genes were defined as genes showing any cis effect (cis-only, cis + trans, and compensatory categories) in response to either infection (219 genes). Trans genes were defined as genes that showed trans-only effects and no other effects in response to either infection (199 genes). Genes showing any combination of trans-only and any cis effects were excluded from this analysis (three genes). Differences in the number of TFBSs were calculated by subtracting the number of TFBSs for each gene's upstream region in B6 from A4, for all TFs combined as well as for each TF separately. We tested for significance in the distribution of these TFBS differences between the cis- and trans-affected genes using an F-test for variance with the R 3.6.0 function var.test. We also repeated this analysis using the TFBS score instead of number, and the results mirrored those found for the TFBS number (Supplemental Fig. S4). The code and accompanying files related to this section can be found in the Supplemental Code in both R-notebooks and HTML document form (Script4_fig4).
Description of statistical tests
P-values for all single and multiple proportion comparisons were calculated using the R 3.6.0 prop.test function, which performs a chi-square test with Yate's continuity correction. For data for which more than one statistical test was performed on the same set of data, P-values were Bonferroni-corrected for familywise type I error by multiplying the P-value by the number of tests performed.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE155033.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank J.J. Emerson and Xinwin Zhang for their thoughtful comments and suggestions on this work; Ali Mortazavi and Lorrayne Serra for their insight and access to sequencing equipment; Tom Schilling for access to microinjection equipment; Joseph Coolon, Carl de Boer, and Rabi Murad for sharing their scripts as well as general guidance with computational protocols; and Anthony Long and Mahul Chakraborty for their insight and helpful comments on the genomes used. This work was funded in part by the National Science Foundation (NSF), award 1953324 to Z.W. B.A.R.-C. was supported by the California LSAMP Bridge to the Doctorate Program, NSF award 1500284. S.F. was supported by a University of California, Irvine, UCI UROP award. O.O. was supported by National Institutes of Health (National Institute of General Medical Sciences) grant R25 GM055246, T34 GM136498, and an UCI UROP award.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.269597.120.
- Received July 30, 2020.
- Accepted April 7, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








